First, describe a biological engineering application or tool you want to develop and why. I want to optimize a strain of cyanobacteria for biomanufacturing. Cyanobacteria can be engineered to produce many useful things from atmospheric carbon dioxide, from commodity chemicals to bioactive compounds for pharmaceuticals, but harvesting the products is often energy intensive and expensive, especially at an industrial scale. I am particularly interested in cyanobacterial bioplastics, such as polyhydroxyalkanoates, because this would be a closed-loop carbon cycle for biodegradable plastic.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Goal: Prevent accidental release that could harm native ecosystems through microbial community shifts or production of commodity chemicals in the natural environment.
Subgoal: Include biocontainment systems in all commercially used industrial bioproduction strains.
Subgoal: Institute testing standards and protocols to notice any accidental release when it occurs.
Goal: Increase access to the genetic tools and strains used for cyanobacterial bioproduction to allow more chemicals to be manufactured in this carbon-neutral way.
Subgoal: Publish cyanobacterial genetic engineering research (such as new tools, etc.) in open access journals or make PDFs available on personal/lab websites.
Subgoal: Enable strain sharing.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Policy to require specific risk mitigation and demonstration of effectiveness under realistic application conditions for engineered bacteria approval.
Purpose: Currently, engineered bacteria that might affect environment and public health need to be approved by the EPA, FDA, or USDA for commercial use. This new policy would enact specific requirements for approvals for engineered bacteria. Additionally, many publications about genetic biocontainment discuss it as potential risk mitigation, but the effectiveness of the biocontainment is only demonstrated under specific laboratory conditions (i.e. axenic, optimized media, etc.).
Design: This would be a change in current federal standards and approval processes. The EPA, FDA, and USDA would need to write and implement new policies, potentially train risk assessors and application managers, and develop testing procedures to ensure compliance. With the overturning of the Chevron doctrine, likely this sort of new policy would require the buy-in of either the companies trying to get their products approved or US Congress to pass new legislation.
Assumptions: Companies and reseachers abide by federal regulations regarding testing and approval. Risk assessment is done in good faith, rather than by companies prioritizing profit over safety. Risk assessment is done by trained ecological and biological risk assessors who know what to look for or be aware of.
Risks of Failure and Success: This could fail if the requirement is too stringent to allow any new products to be approved. This could also fail if the requirements are too lax, and not all risks are accounted for and mitigated. If experimental conditions do not properly reflect application conditions, what appeared to be effective mitigation in the lab might not be effective mitigation in application.
Researchers and inventors could also implement relevant and effective genetic biocontainment in any engineered bacteria used for commercial biomanufacturing.
Purpose: For risks around the unintended spread of engineered bacteria or their synthetic genetic constructs, genetic biocontainment can mitigate these risks by preventing proliferation and/or degrading the relevant DNA. By tying the biocontainment system to the intended use of the bacterium, researchers manage risk in a relevant manner, thus ensuring that the bacterium is specific to the intended application and minimizing spread thereby reducing risks.
Design: Any developer of an engineered bacteria that could be intentionally or unintentionally released would need to research biocontainment and engineer a system into their bacteria. This would require a change in the current culture of the field, where the risks of engineered bacteria spread and mitigation through biocontainment are sometimes discussed, but mostly considered somewhat niche. If it became common practice to consider application and risks thereof for the products of synthetic biology, I think the design of these sorts of safeguards would be more widespread. Any sort of research requires funding and incentive, so universities, grant funders, and biotech companies would need to start looking for these considerations in proposals to motivate it.
Assumptions: Genetic biocontainment is a good strategy to mitigate the potential ecological and public health risks of new synthetic biology products. These risks are limited to ones we think to test (i.e. microbial community shifts, horizontal gene transfer of antiobiotic resistance genes or other functions, proliferation of engineered bacteria in unintended location, local specific bacterial extinction event in the case of a particularly robust engineered bacterium).
Risks of Failure and Success: If we rely too heavily on genetic biocontainment, a failure of the genetic system could result in losing that protection against risk. It’s also possible risks would not be seriously considered because we too easily trust biocontainment to minimize the risk.
Establish professional society for cyanobacteria-specific or general photosynthetic-organism research to promote resesarch and tool sharing.
Purpose: Currently, microalgae research is generally lumped along with all other non-model microbes in synthetic biology. A professional association or conference specific to photobiocatalysis could be a gathering place to collect all relevant tools, protocols, and standards, as well as potentially institute a shared ethics or goal to include improving access to the research and its products.
Design: Perhaps a starting point would be to invite cyanobacteria, eukaryotic microalgae, macro-algae, and plant synthetic biologists to a conference on photobiocatalysis, along with industry representatives from companies using or creating engineered phototrophs. This might be best done under the banner of an existing synthetic biology or metabolic engineering professional association (such as the Society for Biological Engineering in the American Institute of Chemical Engineers). If there is enough interest at the conference, attendees could work together to establish a more specific sub-association, or just resolve to discuss access and research sharing at the conference itself.
Assumptions: This is a large enough field to host such a specific conference. It might be too niche, but I don’t think so; it might be a conference on the smaller side at first though probably.
Risks of Failure and Success: It’s possible industry and start-ups might not want to popularly share their research as there is an economic disincentive.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Risk Mitigation for Approval
Biocontainment in Practice
Photobiomanufacturing Professional Society
Enhance Biosecurity
• By preventing incidents
1
1
3
• By helping respond
2
3
3
Foster Lab Safety
• By preventing incident
2
n/a
2
• By helping respond
2
n/a
2
Protect the environment
• By preventing incidents
1
1
2
• By helping respond
1
2
2
Other considerations
• Minimizing costs and burdens to stakeholders
3
3
3
• Feasibility?
2
3
2
• Not impede research
3
2
1
• Promote constructive applications
1
2
1
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. I would prioritize the requirement of risk assessment and mitigation strategies for federal approval of engineered bacteria. I believe this would have the biggest impact in terms of allowing engineered bacteria to be used for public good (such as biomanufacturing) while preventing potential harm (such as ecosystem destabilization by permanently altering native microbiome in instances of escape). The development of genetic biocontainment tools and implementation thereof becoming regular practice in the field of engineered microbes would be awesome, but I think would be harder to bring about and would take longer - although it might actually have more impact. The establishment of a professional society could help institute such norms. Starting a new conference would probably be easiest in terms of discovering feasibility - proposing it to a handful of host organizations would rapidly identify whether this is currently worth pursuing or if it would need to be worked on for a while first.
References:
Chemla, Y; Sweeney, CJ; Wozniak, CA; et al. Engineering Bacteria for Environmental Release: Regulatory Challenges and Design Strategies. Authorea. July 05, 2024. DOI: 10.22541/au.171933709.97462270/v2
George, DR; Danciu, M; Davenport, PW; et al. A bumpy road ahead for genetic biocontainment. Nature Communications, 15(650). January 20, 2024. DOI: 10.1038/s41467-023-44531-1
Schmelling, NM; Bross, M. What is holding back cyanobacterial research and applications? A survey of the cyanobacterial research community. Nat Commun 15, 6758. August, 8, 2024. DOI: 10.1038/s41467-024-50828-6
Week2 Lecture Prep
Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? Polymerase error rate: $1 : 10^{6}$. The human genome is around 3.2 Gb, or $3.2 * 10^{9}$ basepairs. Biological polymerases are error-correcting; they have have proofreading mechanisms. There are also mutation repair mechanisms.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? The average human protein is encoded within 1036bp. This might be answerable based on the last slide titled “Fabricational Complexity”, but I couldn’t quite figure out what these formulas are supposed to be calculating without explanation. So instead, we can do some back-of-the-napkin math together. 1036bp is $1036/3 \approx 345$ codons, or 344 amino acids (because of the stop codon at the end), assuming that the 1036bp figure doesn’t include introns. Most amino acids have either 4 or 2 codons that can encode for it, although a couple have more or less. We’ll average it out to approximately 3 codons per amino acid. I imagine that not all amino acids are used at the same frequency in human proteins, but I don’t actually know what it is off the top of my head, so we’re just going to go with what we have. Each possible DNA sequence for an amino acid sequence includes every combination with all possible codons for each amino acid. So assuming an average human protein has 344 amino acids, and the average number of codons per amino acid is 3, then there are $3^{344} = 1.3 E164$ different ways to code for an average human protein. In practice, not all tRNAs are synthesized at the same frequency, so it might take unreasonably long for certain codons to be recognized during chain extension; and during DNA replication, errors can be made and some errors will be more tolerable than others due to codon wobble.
LeProust:
What’s the most commonly used method for oligo synthesis currently? Phosphoramidite synthesis.
Why is it difficult to make oligos longer than 200nt via direct synthesis? There are side reactions that occur, causing the accumulation of errors (incorrect bases).
Why can’t you make a 2000bp gene via direct oligo synthesis? I think this is because of the side reactions in Q2, right? Like, the accumulation of errors limits oligo synthesis to around 200 bases in practice. Also, oligos are single-stranded DNA; a 2000bp gene is double-stranded, and therefore you’d either need to synthesize both strands and ligate them together, or synthesize one strand and use it as a template for PCR or something.
Church:
Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own: BioStabilization Systems - ARPA-H \
Biologic therapeutics are critically important for a number of diseases, but require careful and specific conditions at all points on the supply chain to maintain efficacy. Specifically, cell therapies and biologics require extreme cold to prevent degradation, thus making biologics inaccessible to people who don’t live near a specialized medical center. To solve this problem, we propose to express biologic therapeutics in extremophiles from abyssal marine sediment, which demonstrated little cell proliferation in low-oxygen environments but regained metabolic activity when incubated with oxygen. We predict that the faster cell turnover period at warmer temperature, oxygen-rich, and high-nutrient conditions will allow us to engineer these bacteria to produce the biologic therapeutic molecules. Once production is achieved, we will seal the cells into low-oxygen capsules for transport, which we predict will slow their metabolic rate enough to preserve the goal product until oxygen is provided again. If successful, this research could expand access to biologic therapeutics to anywhere that can aseptically incubate microbes at room temperature and purify the molecules therein.
References:
Morono, Y; Ito, M; Hoshino, T; et al. Aerobic microbial life persists in oxic marine sediment as old as 101.5 million years. Nat Commun 11, 3626. 2020. DOI: 10.1038/s41467-020-17330-1
Suzuki, Y; Webb, SJ; Kouduka, M; et al. Subsurface Microbial Colonization at Mineral-Filled Veins in 2-Billion-Year-Old Mafic Rock from the Bushveld Igneous Complex, South Africa. Microb Ecol 87, 116. 2024. DOI: 10.1007/s00248-024-02434-8
Personal notes/drafting
abstract formula:
1 sentence on the broad problem: Biologic therapeutics are critically important for a number of diseases, but require careful and specific conditions at all points on the supply chain to maintain efficacy.
1-2 sentences on the specific problem: How to transport cell therapies and biologics at room temperature, decentralizing medicine
1 sentence on the broad goal: We aim to express biologic compounds in extremophiles from the deep subsurface where energy and nutrients are limited.
2-3 sentences on methods: aerobic microbes from oxic abyssal marine sediment that proliferated at 10C with provision of nutrients and higher conc O2; might need to consider eukaryotic protein folding in prokaryotes; low O2 environment - maybe sealing the cells (post-therapeutic production, pre-shipping) into an airtight capsule would prevent metabolic activity including the breakdown of said therapeutics?
1 sentence on future work: maybe also try extremophiles found within old rock samples
1 sentence on conclusion/impact: expands access to biologics, especially to under-resourced communities
I couldn’t figure out how to use Ronan’s website other than the randomization button unfortunately. As a result, I went with a pretty simple smiley face design for my in-silico art.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I’m interested in PhaC, a PHA synthase. This is an enzyme involved in the synthesis of polyhydroxyalkanoates (PHAs), a class of biopolymer that is considered a potential non-petroleum-derived thermoplastic. PHAs are also of interest for possible medical uses as biodegradable polymers. PhaC is the enzyme that catalyzes the polymerization step, adding on monomers to the chain.
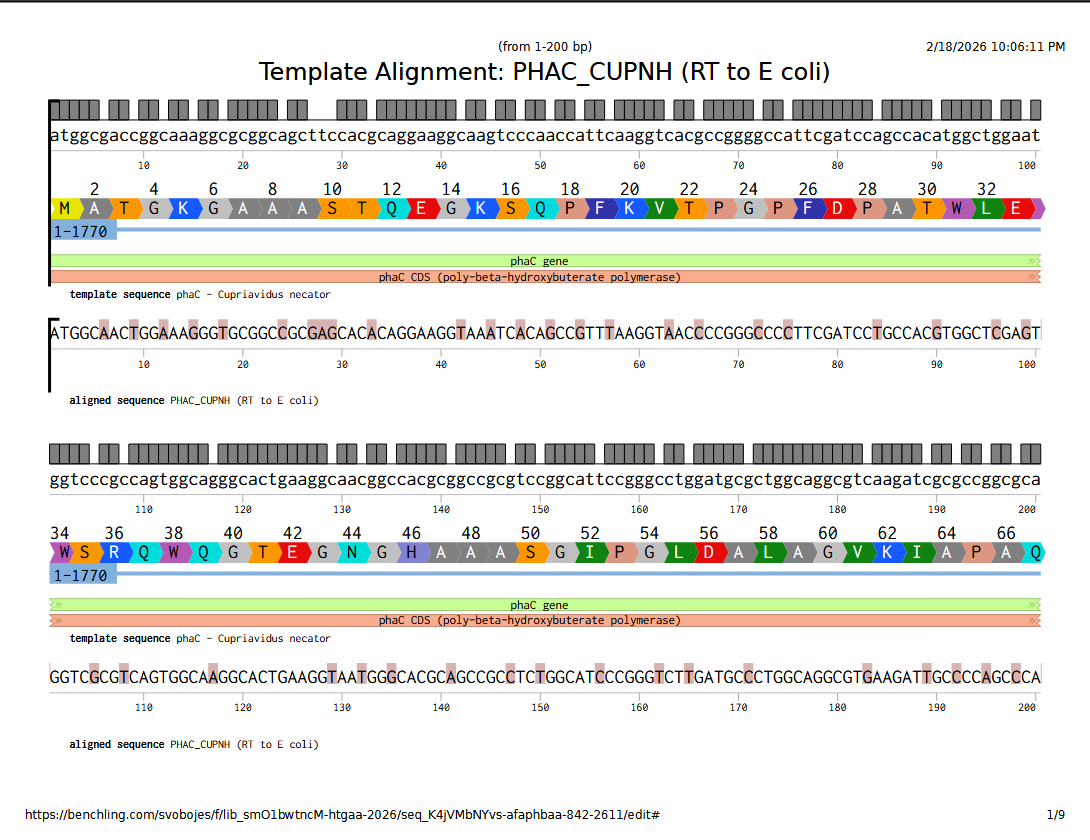
I selected PhaC from Cupriavidus necator H16 whose primary product is poly(3-hydroxybutyurate). From UniProt, the accession number is P23608 · PHAC_CUPNH.
I used the Benchling back-translate tool set to match Escherichia coli K-12 naturally occuring codon usage because it didn’t have the native host C. necator as an option. They are in the same phylum (Pseudomonadota), so maybe it will be similar.
They are not that similar, it turns out; although that may have less to do with codon usage frequency and more to do with when the reverse translate tool used which codons. Here’s the DNA sequence alignment comparing the genomic sequence from C. necator with the E. coli optimized reverse translation. This sequence alignment was performed in Benchling, using MAFFT with pre-set parameters.
Full alignment viewable here.
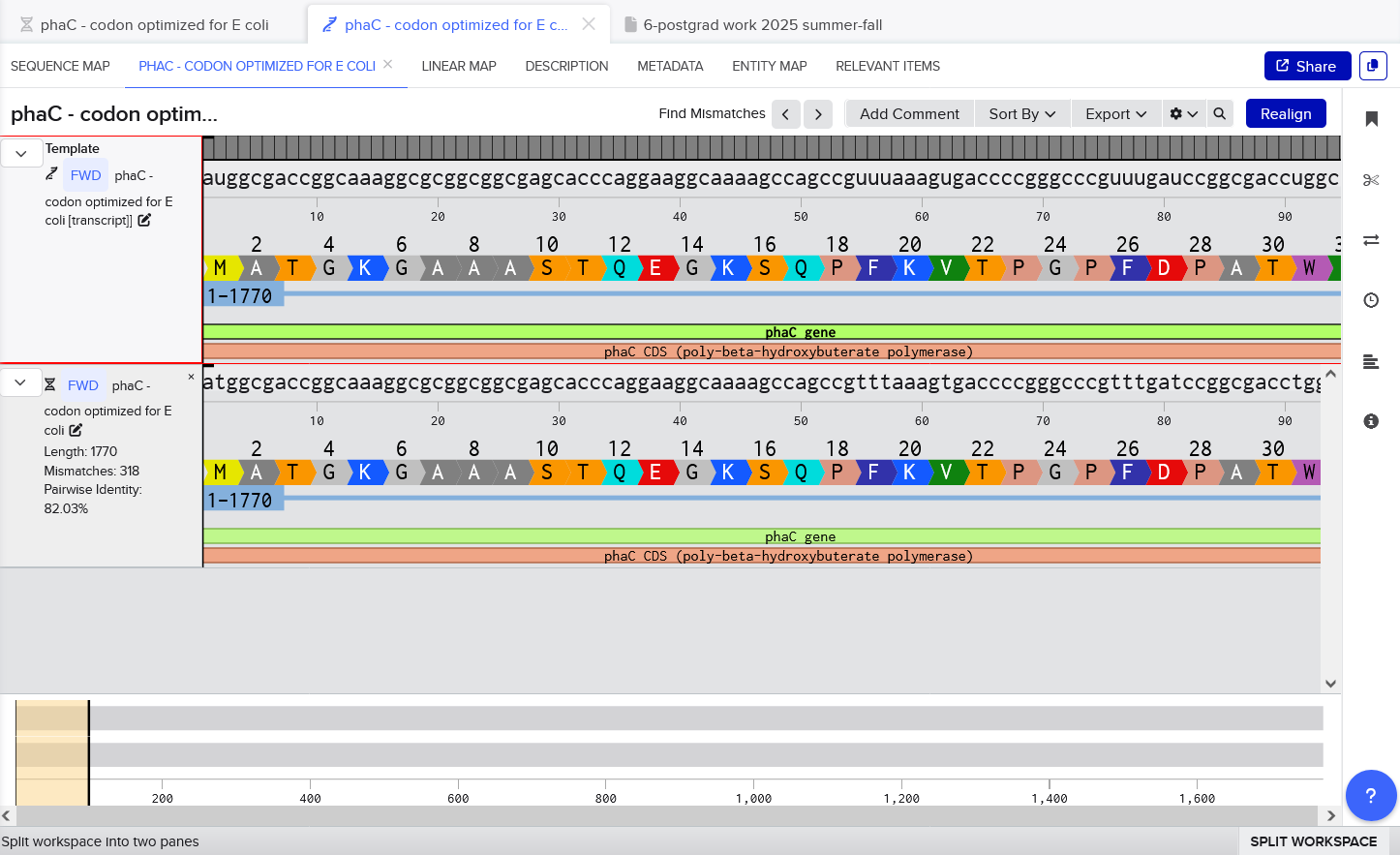
3.3 Codon optimize
I once again used the Benchling tool to codon optimize for E. coli K-12, but this time, I selected the Best Codon option in Benchling, and this was performed off the original C. necator phaC DNA sequence - although it should produce the same sequence if it was done as a reverse translate from the amino acid sequence too (since i confirmed that the phaC sequence does translate to the PhaC sequence with 100% identity).
This sequence could be used to express PhaC in E. coli. I would probably put the gene onto an expression plasmid, under a strong constitutive promoter, just to ensure it works. After transforming E. coli with the plasmid, I would test expression by looking at protein production with a Western blot, and looking at cells under a microscope to look for PHA granules. I need to do a little more literature searching on heterologous expression of PhaC in E. coli - I think maybe other enzymes are needed for PHB synthesis.
3.5 Optional - how does it work in natural biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level. Different reading frames on the same string of DNA bases gives different codons that are off-set by which base (1-3) starts it. In this way, genes for multiple proteins can overlap on the same sequence of DNA.
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein! I created the transcript by using Benchling to create a new RNA sequence off the reverse of my coodon-optimized sequence. I kept the annotations, so the translation should still be visible. Then I made a new alignment in Benchling using MAFFT with the automatic parameters. Again, the sequences match perfectly - although it’s not 100% identity because technically the T/U difference between DNA and RNA are considered mismatches, but we can see visually across the bottom of the screenshot that we don’t have any actual mismatches.
Part 4: Prepare a Twist DNA Synthesis Order
Following the instructions in the Week2 Homework, I added the J23106 promoter and an RBS at the beginning of my codon-optimized phaC sequence. My coding sequence already had a start and stop codon, so I didn’t need to add those. I inserted the 7x-His tag just before the stop codon, and then I put the terminator after the stop codon at the end.
I then set up the Twist order, as if I was going to order this cassette to be synthesized. Again, following the instructions for upload, I chose cloning vector pTwist Amp High Copy to make a full plasmid. My sequence was high complexity, so I went through the Twist codon optimization process to improve the sequence for easier synthesis. I chose E. coli as my host strain again, and selected the ORF that matched my gene. I chose the promoter and RBS, and terminator regions as regions to preserve during the codon optimization process so that it kept the sequences for the genetic parts that I chose. The optimized sequence was no longer high complexity as the regions of high GC% and repeats were changed.
What DNA would you want to sequence (e.g., read) and why? I’d like to sequence the genomes of all cyanobacterial strains known to produce PHAs or specifically PHB (some already are sequenced, I think). I want to align all the known cyanobacterial PHA-synthases, and then align with the assembled genomes of the cyanobacterial strains known to produce PHAs that maybe aren’t annotated yet to try to find the PHA-synthases and add those to my comparisons.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? I would use third-generation sequencing on an Oxford nanopore. By using long-read technology, I would get much longer contigs, to make genomic assembly easier.
5.2 Write
What DNA would you want to synthesize (e.g., write) and why? I’d like to get a CRISPR-Cas12a multiplexed gRNA cassette synthesized. This would allow multiple genomic edits to occur simultaneously, if the appropriate repair templates are included (one for gRNA target).
What technology or technologies would you use to perform this DNA synthesis and why? I would submit an order to Twist to get this synthesized because it has multiple internal repeats because of the CRISPR region, which means traditional DNA synthesis technologies would struggle with this sequence.
5.3 Edit
What DNA would you want to edit and why? I’d like to improve PHA-synthase expression in my cyanobacterial chassis strain of choice (specific strain yet to be determined). This could be accomplished through promoter replacement if we’re staying in the genome rather than adding a plasmid, but I’d also be interested in knocking out other biosynthetic pathways to improve carbon flux towards PHA synthesis. So I’d want to edit the genomic DNA of a cyanobacterial chassis.
What technology or technologies would you use to perform these DNA edits and why? I’d use a CRISPR-Cas12a vector because it allows for multiplexed targeting, so I could make multiple genomic edits. Cas12a both processes the CRISPR-gRNA cassette and makes the cuts, so it requires fewer components than Cas9. Additionally, there’s some evidence suggesting Cas12a shows less off-target effects than Cas9.
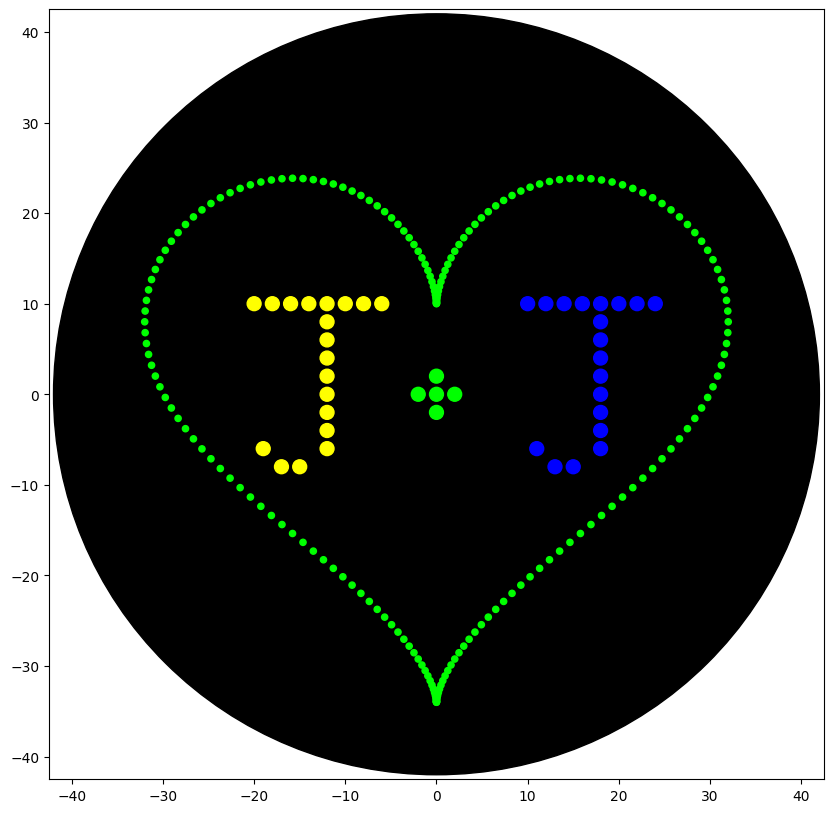
from opentrons import types
metadata = { # see https://docs.opentrons.com/v2/tutorial.html#tutorial-metadata
'author': 'JKS',
'protocolName': 'heartJ',
'description': 'writes the J+J inside a heart shape',
'source': 'HTGAA 2026 Opentrons Lab',
'apiLevel': '2.20'
}
##############################################################################
### Robot deck setup constants - don't change these
##############################################################################
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
well_colors = {
'A1' : 'Red',
'B1' : 'Yellow',
'C1' : 'Green',
'D1' : 'Cyan',
'E1' : 'Blue' # if in a 24-well plate, this needs to be moved to e.g. D2
}
def run(protocol):
##############################################################################
### Load labware, modules and pipettes
##############################################################################
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# Modules
temperature_module = protocol.load_module('temperature module gen2', COLORS_DECK_SLOT)
# Temperature Module Plate
temperature_plate = temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul',
'Cold Plate')
# Choose where to take the colors from
color_plate = temperature_plate
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate') ## TA MUST CALIBRATE EACH PLATE!
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
##############################################################################
### Patterning
##############################################################################
###
### Helper functions for this lab
###
# pass this e.g. 'Red' and get back a Location which can be passed to aspirate()
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)
def dispense_and_detach(pipette, volume, location):
"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""
assert(isinstance(volume, (int, float)))
above_location = location.move(types.Point(z=location.point.z + 5)) # 5mm above
pipette.move_to(above_location) # Go to 5mm above the dispensing location
pipette.dispense(volume, location) # Go straight downwards and dispense
pipette.move_to(above_location) # Go straight up to detach drop and stay high
###
### YOUR CODE HERE to create your design
###
### heart pattern taken from Selin Sahin (2023)
def heart_pattern(n, r, color_string, center_location):
# generate list of points forming the heart
scaling_factor = -2/r # calculate scaling factor to fit pattern within 40mm radius circle
angle_step = 2*math.pi/n
coords = []
for i in range(n):
angle = i * angle_step
x = scaling_factor*r*(16*math.sin(angle)**3)
y = scaling_factor*(-r*(13*math.cos(angle) - 5*math.cos(2*angle) - 2*math.cos(3*angle) - math.cos(4*angle)))
coords.append((x, y))
####PICK UP TIP HERE####
pipette_20ul.pick_up_tip()
print_every = 1 # 1=print every point; 2=print every other point; 3=print every third...
# now plot the points
for i, (x,y) in enumerate(coords):
#print(i,(x,y))
if i % (100*print_every) == 0: # 20uL/0.2uL = 100
# every 20th point we're printing starting with the first, aspirate 20uL total from Well 1
pipette_20ul.aspirate(min(20, math.ceil((len(coords)-i)/print_every)), location_of_color(color_string))
# print every other point we've calculated (was too dense otherwise)
if i % print_every == 0:
adjusted_location = center_location.move(types.Point(x, y))
dispense_and_detach(pipette_20ul, 0.2, adjusted_location)
####DROP TIP####
pipette_20ul.drop_tip()
##################################
#### DRAW PATTERN ####
##################################
heart_pattern(200, 50, 'Green', center_location)
###### write
# letter J1
pipette_20ul.pick_up_tip()
pipette_20ul.aspirate(8, location_of_color('Yellow'))
cursor = center_location.move(types.Point(x=-20, y = 12))
for i in range(8):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(y=-2)))
cursor = cursor.move(types.Point(x =2))
cursor = cursor.move(types.Point(x=-10, y=-4))
pipette_20ul.aspirate(8, location_of_color('Yellow'))
for i in range(8):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(x=2)))
cursor = cursor.move(types.Point(y =-2))
pipette_20ul.aspirate(3, location_of_color('Yellow'))
for i in range(2):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(x=-1)))
cursor = cursor.move(types.Point(x =-2))
cursor = cursor.move(types.Point(x=-1, y=2))
dispense_and_detach(pipette_20ul, 1, cursor)
pipette_20ul.drop_tip()
### +sign
pipette_20ul.pick_up_tip()
cursor = center_location.move(types.Point(x=-4))
pipette_20ul.aspirate(5, location_of_color('Green'))
for i in range(3):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(x=2)))
cursor = cursor.move(types.Point(x=2))
cursor = cursor.move(types.Point(x=-2, y=2))
dispense_and_detach(pipette_20ul, 1, cursor)
cursor = cursor.move(types.Point(y=-4))
dispense_and_detach(pipette_20ul, 1, cursor)
pipette_20ul.drop_tip()
# letter J2
pipette_20ul.pick_up_tip()
pipette_20ul.aspirate(8, location_of_color('Blue'))
cursor = center_location.move(types.Point(x=10, y = 12))
for i in range(8):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(y=-2)))
cursor = cursor.move(types.Point(x =2))
cursor = cursor.move(types.Point(x=-10, y=-4))
pipette_20ul.aspirate(8, location_of_color('Blue'))
for i in range(8):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(x=2)))
cursor = cursor.move(types.Point(y =-2))
pipette_20ul.aspirate(3, location_of_color('Blue'))
for i in range(2):
dispense_and_detach(pipette_20ul, 1, cursor.move(types.Point(x=-1)))
cursor = cursor.move(types.Point(x =-2))
cursor = cursor.move(types.Point(x=-1, y=2))
dispense_and_detach(pipette_20ul, 1, cursor)
pipette_20ul.drop_tip()
# Don't forget to end with a drop_tip()
Post-lab questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. A paper published this month in ACS Synthetic Biology details a new workflow for automating MoClo plasmid assembly and transformation, with a semi-automated colony PCR on an Opentrons OT-2 and Opentrons Flex. These workflows are designed to be user-friendly and output the Opentrons protocol from user-supplied CSV files, which provided README files describe how to produce.
Alternatively, the authors also developed a graphical user interface which requires no coding ability. This is a novel application because it is only the second automation of MoClo/Golden Gate cloning for Opentrons system (as opposed to advanced high-throughput liquid handling systems), and this new workflow does not require Python ability as the previously published AssemblyTron workflow.
These workflows were validated by assembling plasmids with the MoClo Yeast Toolkit and MoClo SubtiToolKit, and transforming these plasmids into Saccharomyces cerevisiae and sequentially Escherichia coli and Bacillus subtilis, respectively. With both toolkits, the automated procedure achieved efficiency comparable to the manual procedures (> 90% and 60%, respectively).
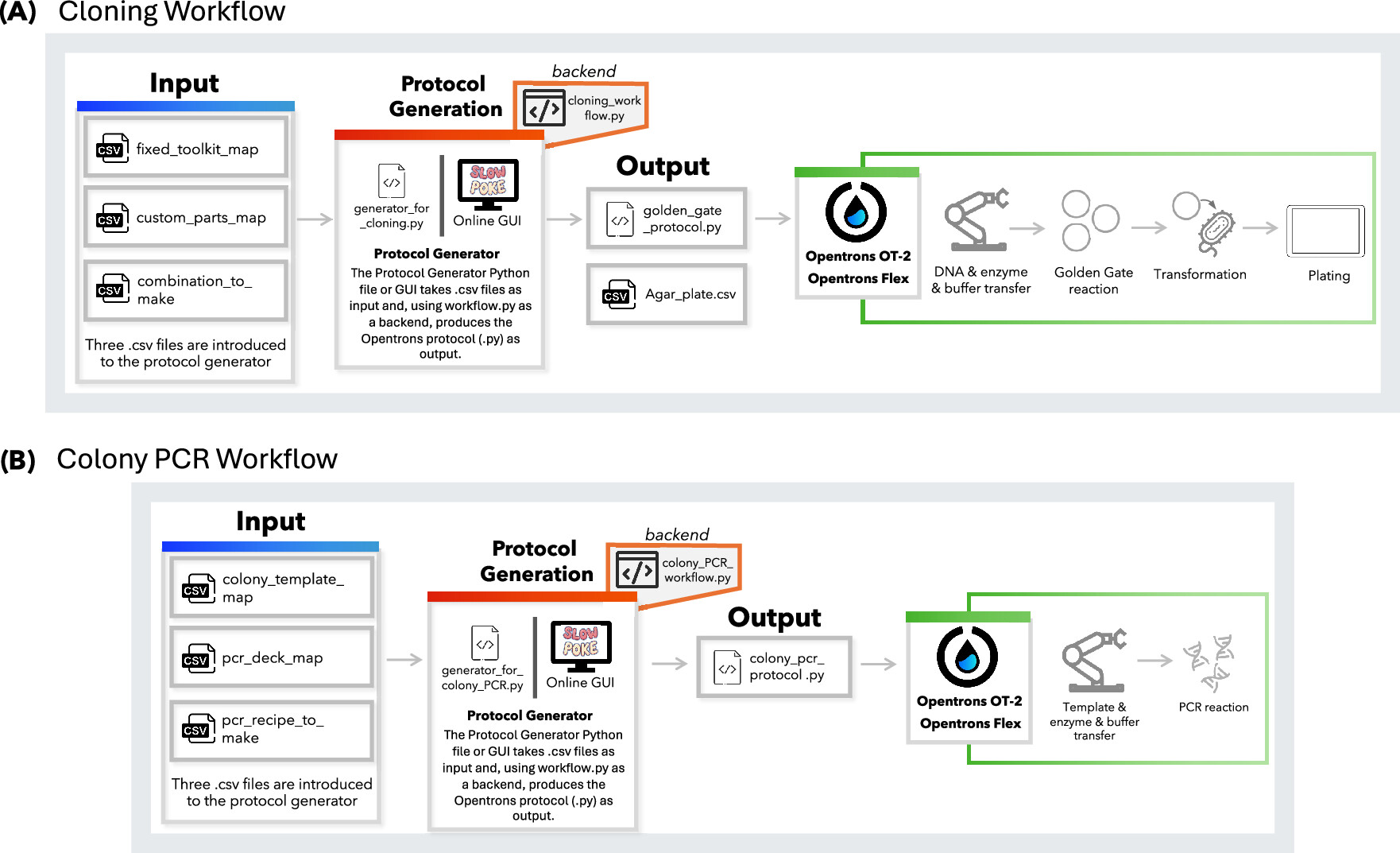
Figure 1: Schematic overview of the protocol design workflows developed for the Opentrons platform. Protocols can be generated using either the generator.py Python script via the command line or the online Slowpoke tool, which features a user-friendly GUI. Both tools run the workflow.py files in the backend. (A) Workflow for Golden Gate-based cloning, where users define genetic part layouts and assembly combinations. (B) Workflow for colony PCR, including colony selection, reagent layout, and reaction recipe input.
Malci, K; Meng, F; Galez, H; et al. Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex. 2026. ACS Synthetic Biology, 15(2): 511-521. DOI: 10.1021/acssynbio.5c00629
Write a description about what you intend to do with automation tools for your final project. I’d want to utilize the Opentrons set-up in the Victoria node to enable the possible execution of my medium-term aim with as little scientist benchtime as possible. I don’t know the exact make and model of all modules that the Victoria Opentrons has, but below is a series of possible steps that might be automatable (best use of automation would be medium or high throughput, depending on the number of designs we are able to test):
Gibson Assembly or MoClo plasmid assembly
Transfer reaction components into wells
Heat block for digestion/ligation/PCR steps
Transformation of expression plasmid
Transfer plasmids and competent cells into wells
Heat block for heat shock
Transfer media into wells
Heated shaker for recovery
Incubator for overnight growth
Stamp onto new plate or pick into multiple liquid cultures for culturing
Incubator or heated shaker for overnight growth
Readout
Transfer cells (and reagents) into wells
Plate reader for fluorescent or colorimetric output
Final project ideas
Brainstorming:
Identification of PhaC analog in Cyanobacterium aponium UTEX 3222 and overproducing or engineering for increased efficiency
BLAST/align with known PHA-synthases
Compare efficiency / mutations that improved turnover in other PhaC - test analogous mutations (aligned location, similar or different AAs). improved substrate specificity?
Site-specific saturation mutagenesis? Would be good use for automation
Quorum sensing based killswitch (i.e. cell dies if it escapes bioreactor)
Has to have some kind of inducible element or won’t grow after initial transformation
What’s good at quorum sensing already?
Something else??? Something in E coli that can be done on Opentron
Because it’s more convenient for a final project to be executed in Victoria remotely
Cyanobacterial expression plasmid across multiple cyano species
needs to include E coli machinery for manipulation and production (and conjugation, for relevant species)
Ideas:
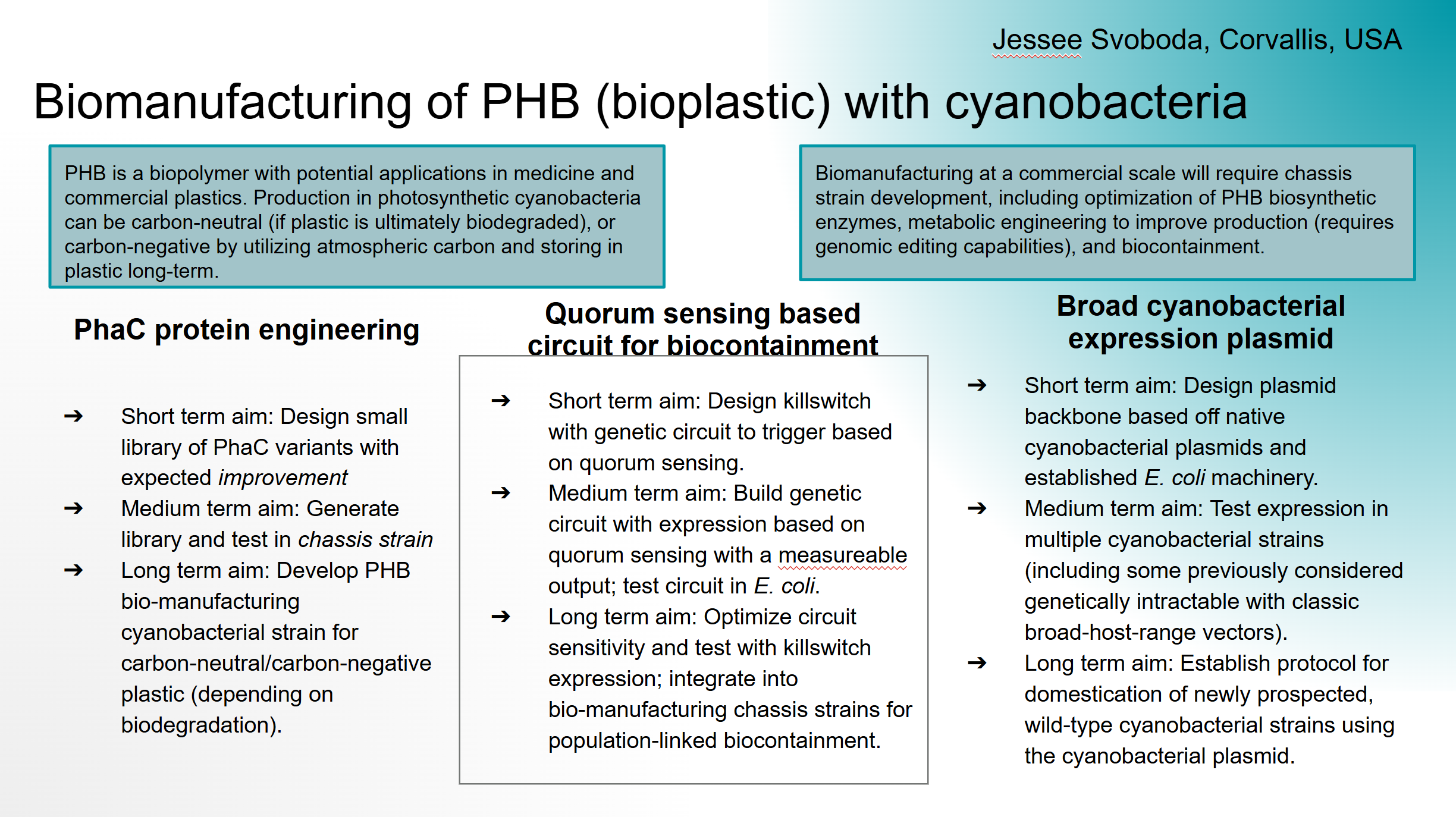
PhaC protein engineering
Short term aim: Design small library of PhaC variants with expected improvement
Medium term aim: Generate library and test in chassis strain
Long term aim: Develop PHB bio-manufacturing cyanobacterial strain for carbon-neutral/carbon-negative plastic (depending on biodegradation).
Quorum sensing based circuit for biocontainment
Short term aim: Design killswitch with genetic circuit to trigger based on quorum sensing.
Medium term aim: Build genetic circuit with expression based on quorum sensing with a measureable output; test circuit in E. coli.
Long term aim: Optimize circuit sensitivity and test with killswitch expression; integrate into bio-manufacturing chassis strains for population-linked biocontainment.
Broad cyanobacterial expression plasmid
Short term aim: Design plasmid backbone based off native cyanobacterial plasmids and established E. coli machinery.
Medium term aim: Test expression in multiple cyanobacterial strains (including some previously considered genetically intractable with classic broad-host-range vectors).
Long term aim: Establish protocol for domestication of newly prospected, wild-type cyanobacterial strains using the cyanobacterial plasmid.
Need to answer 9/11 questions; I skipped 7 and 11.
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) $$ 500g * \frac{1 mol AA}{100g} = 5 mol AA $$
$$ 5 mol * \frac{6.02*10^{23} molecules}{1 mol} = 3.01 E24 molecules $$
Why do humans eat beef but do not become a cow, eat fish but do not become fish? We break down the proteins during digestion to the constituent amino acids. These amino acids are then used in our cells to build human proteins.
Why are there only 20 natural amino acids? It’s been hypothesized that the 20 naturally occurring amino acids fairly effectively cover the “chemical space”, which would indicate that more complex or diverse amino acids are not needed for increasing function. This includes variation in chemical properties like molecular size, hydrophobicity, and charge, but also rotational conformations. These twenty sufficiently cover the space for effective function while also being relatively low in energy (easy to synthesize). Another paper hypothesizes that all twenty natural amino acids predate the RNA world, and in fact were naturally synthesized prebiotically with mineral catalysts - thus suggesting that the development of the three-base 64-codon alphabet actually was because a two-base 16-codon alphabet would restrict to sixteen instead of the existing 20 amino acids.
Doig, AJ. Frozen, but no accident – why the 20 standard amino acids were selected. 2017. FEBS J, 284: 1296-1305. doi: 10.1111/febs.13982
Bywater RP. Why twenty amino acid residue types suffice(d) to support all living systems. 2018. PLoS One, 13(10):e0204883. doi: 10.1371/journal.pone.0204883
Can you make other non-natural amino acids? Design some new amino acids. There are a new non-cannonical amino acids that people have designed and used, by changing the residue for an unnatural one.
Where did amino acids come from before enzymes that make them, and before life started?
In 2018, Bywater suggested that amino acids were synthesized prebiotically, with the simpler structures occurring through aqueous reactions, and more complex structures requiring mineral catalysts. Many amino acids have been identified on meteorites, suggesting that amino acids could have originated in outer space, but more likely that the conditions to synthesize the “simpler” amino acids exist in multiple places. Other researchers have suggested that the “complex” amino acids must have been biosynthesized by early proteins made up of “simple” amino acids, and in particular, that histidine, phenylalanine, cysteine, methionine, tryptophan and tyrosine had to come after molecular oxygen because they have redox functionality.
Doig, AJ. Frozen, but no accident – why the 20 standard amino acids were selected. 2017. FEBS J, 284: 1296-1305. doi: 10.1111/febs.13982
Bywater RP. Why twenty amino acid residue types suffice(d) to support all living systems. 2018. PLoS One, 13(10):e0204883. doi: 10.1371/journal.pone.0204883
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? I would expect D-amino acids would form a left-handed helix because L-amino acids form right-handed helices.
Can you discover additional helices in proteins?
Why are most molecular helices right-handed? In general, naturally occuring amino acids are L-enantiomers, which leads to right-handed helices because of steric hindrance requiring the side chains to point outwards.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? Because beta sheets are flat, they can stack, and the large surface area means that the side-chains can have interactions (especially hydrophobic side-chains) between the sheets.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Amyloids are ordered protein aggregates consisting of repeating beta sheet motif. Proteins that have an alternative folding structure with a lot of beta sheets become amyloids when they self-assemble into fibrils, and the alternative conformation with the beta sheets is energetically stable. Amyloid diseases usually are from a single amyloid-forming protein. Because of their tendency to self-assemble, I think you could use amyloid beta sheets as materials for DNA origami.
Riek R. The Three-Dimensional Structures of Amyloids. 2017. Cold Spring Harb Perspect Biol;9(2):a023572. doi: 10.1101/cshperspect.a023572.
Ow SY, Dunstan DE. A brief overview of amyloids and Alzheimer’s disease. 2014. Protein Sci;23(10):1315-31. doi: 10.1002/pro.2524.
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
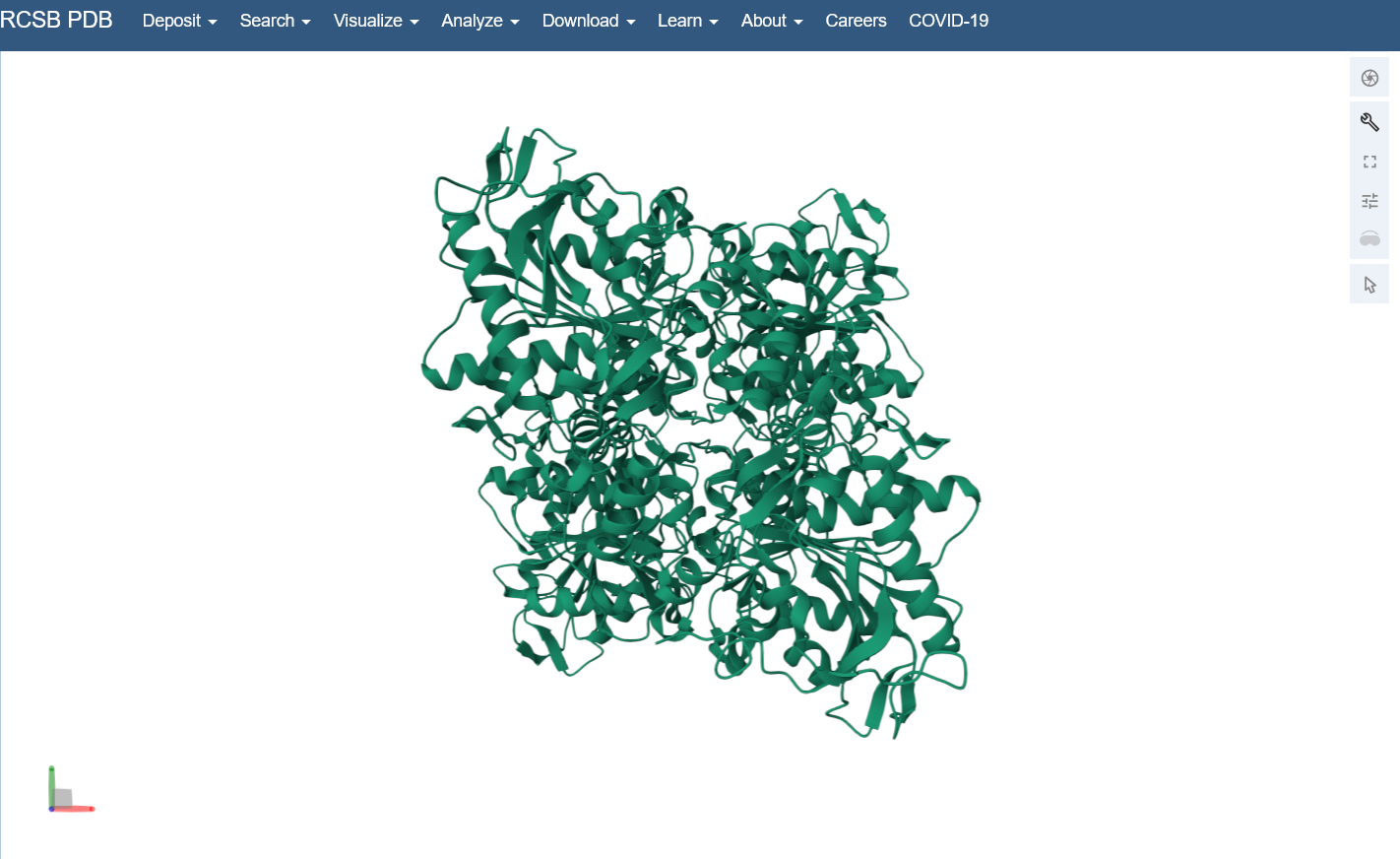
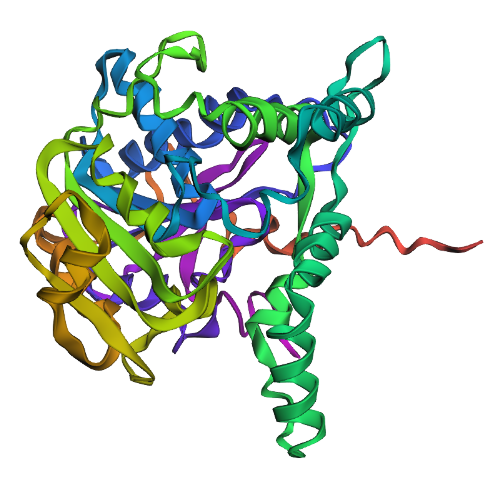
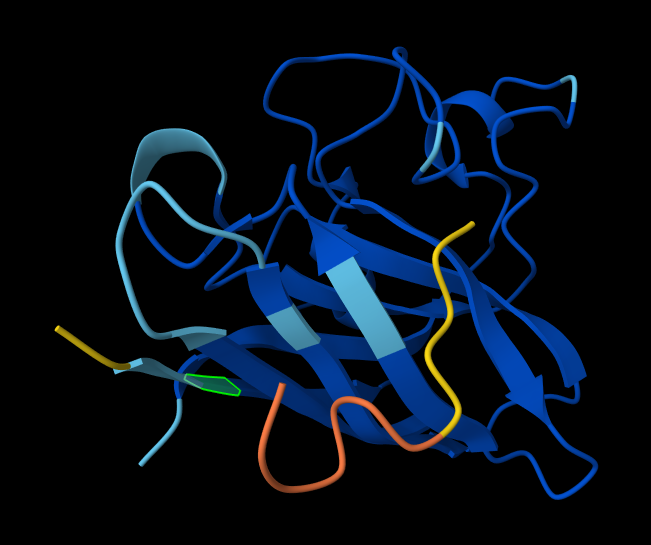
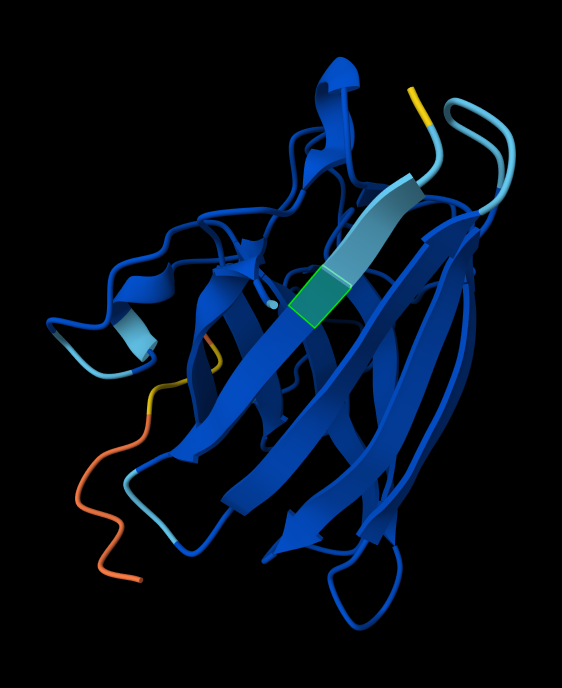
Briefly describe the protein you selected and why you selected it. I chose PhaC from Cupriavidus necator. PhaC is a polyhydroxyalkanoate-synthase, used in biopolymer production. I selected it because engineering PhaC is one of my potential final projects. The C-terminal domain is believed to be the catalystic domain, and it has a solved crystal structure. The N-terminal domain does not have a solved crystal structure, and is believed to potentially be involved in substrate specificity.
Identify the amino acid sequence of your protein. \
How long is it? What is the most frequent amino acid? 390 amino acids (when i removed the His-tag at the end). Most frequent amino acid is A (alanine).
How many protein sequence homologs are there for your protein? BLAST found 250 sequence homologs - mostly belonging to other bacteria that biosynthesize PHAs.
Does your protein belong to any protein family? It’s classified as a transferase.
Identify the structure page of your protein in RCSB C. necator PhaC (C-terminal domain) has been uploaded to RCSB PDB here.
When was the structure solved? Is it a good quality structure? The structure was solved in 2016 by two different and unrelated groups, which is a good sign for repeatability (PDB 5HZ2 and 5T6O). It has a resolution of 1.8Å, which is a good quality structure.
Are there any other molecules in the solved structure apart from protein? Yes, there is a sulfate ion and a glycerol molecule.
Does your protein belong to any structure classification family? Nothing that I could find on SCOP.
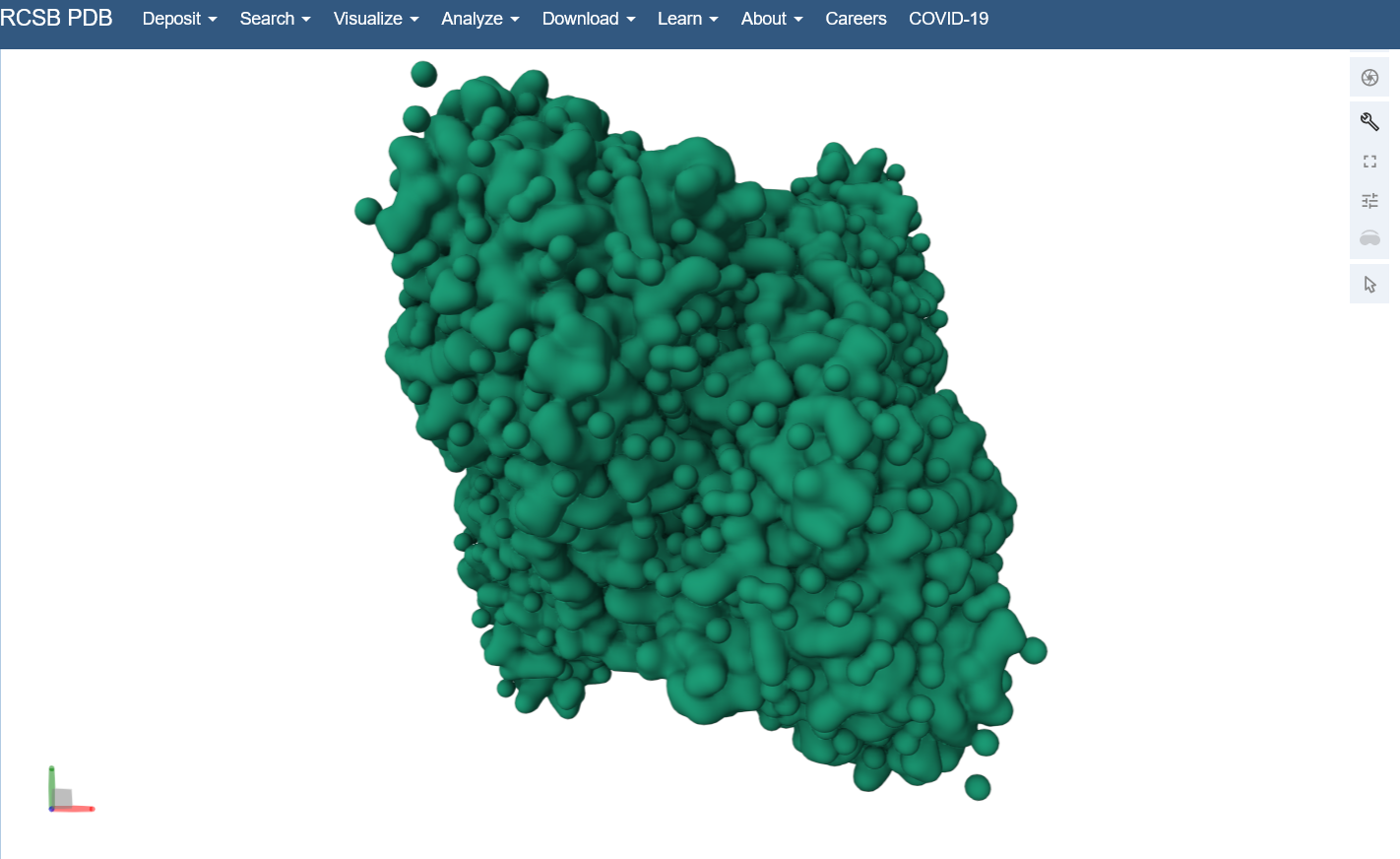
Open the structure of your protein in any 3D molecule visualization software: I used the structure viewer on the PDB website because I wasn’t able to download PyMol on my laptop (not enough memory space).
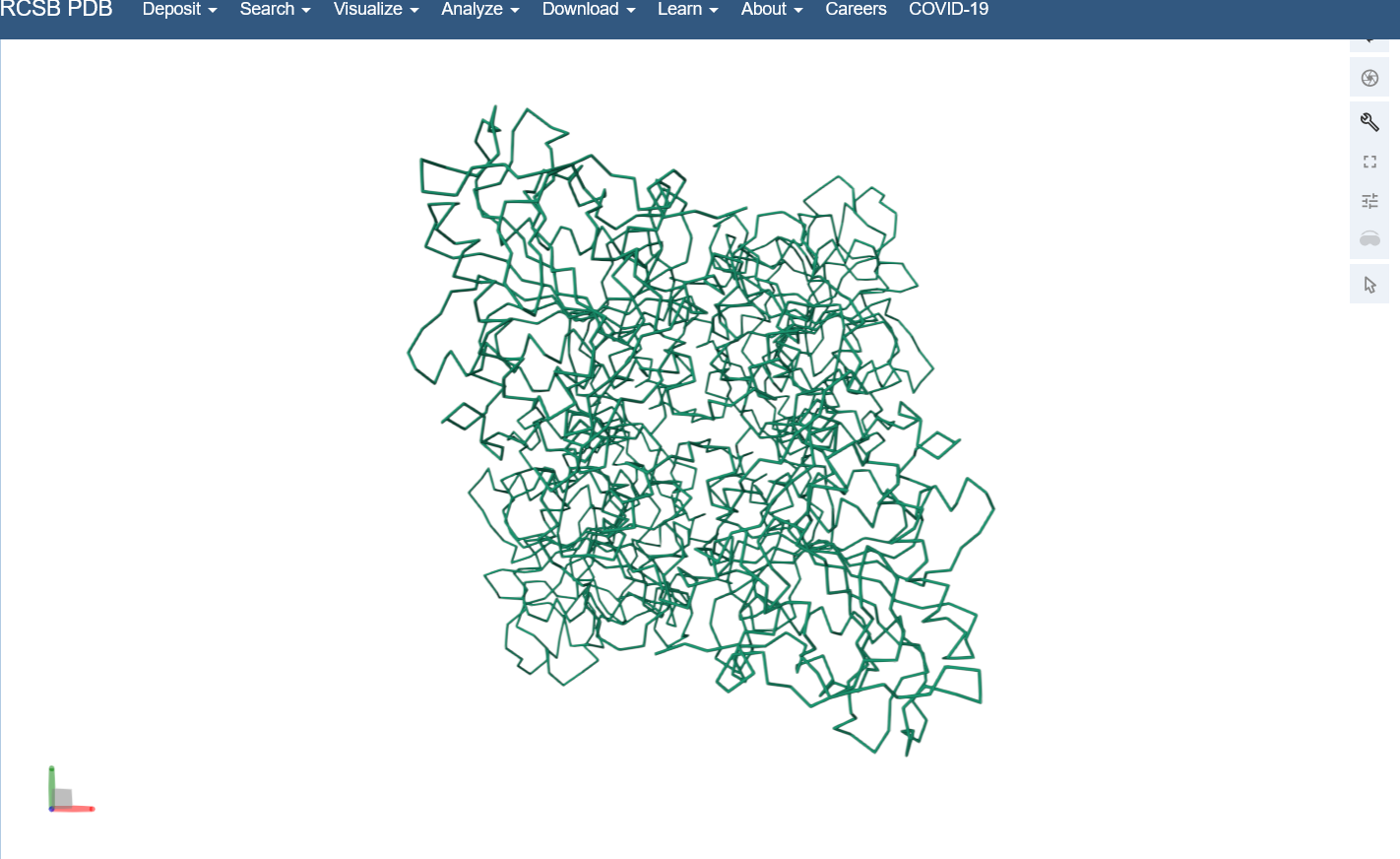
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
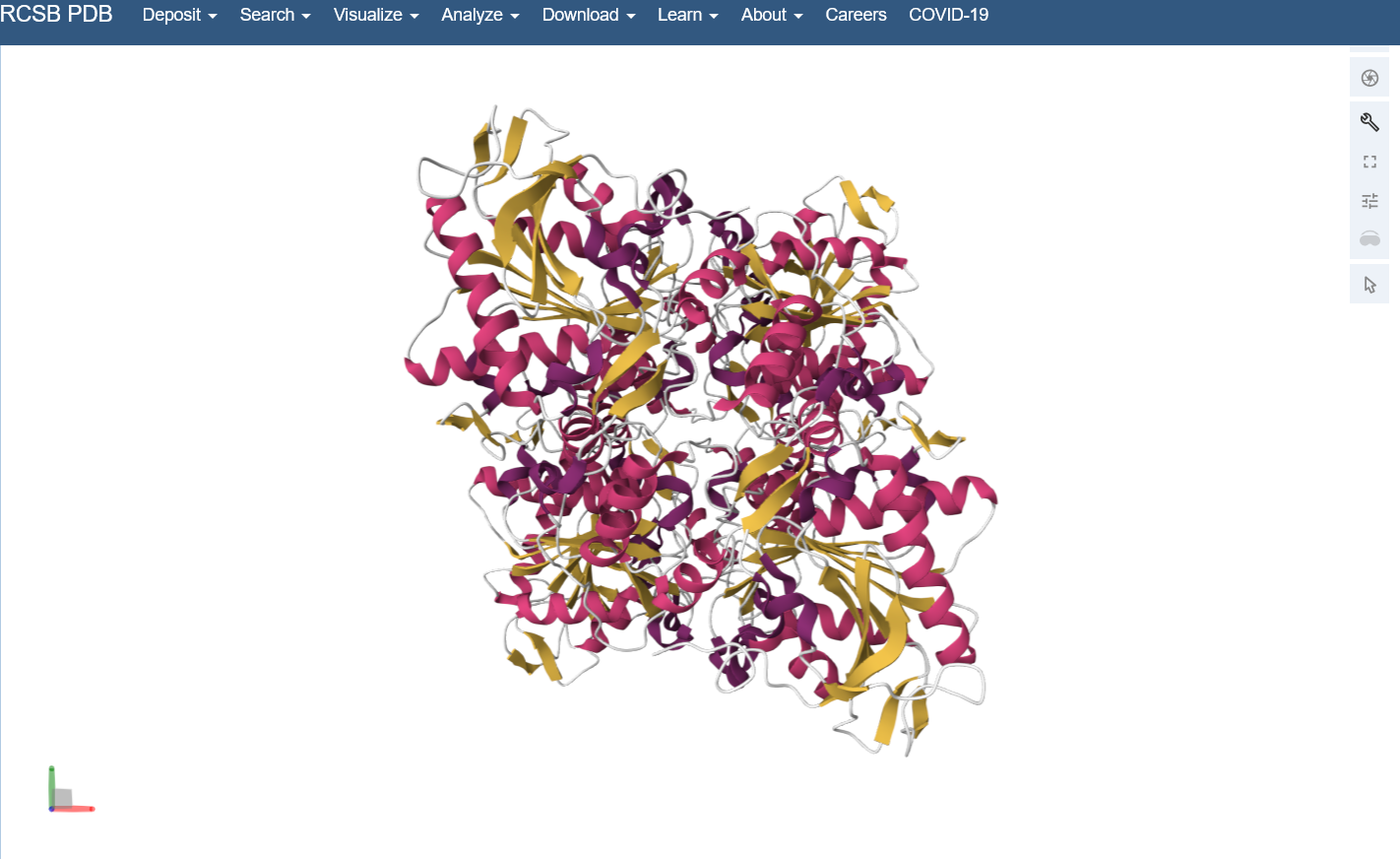
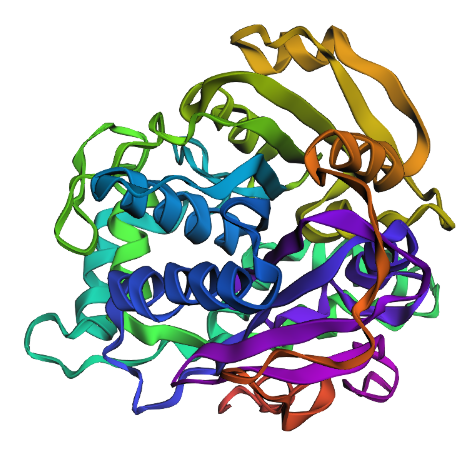
Color the protein by secondary structure. Does it have more helices or sheets? I think it looks like it has more helices.
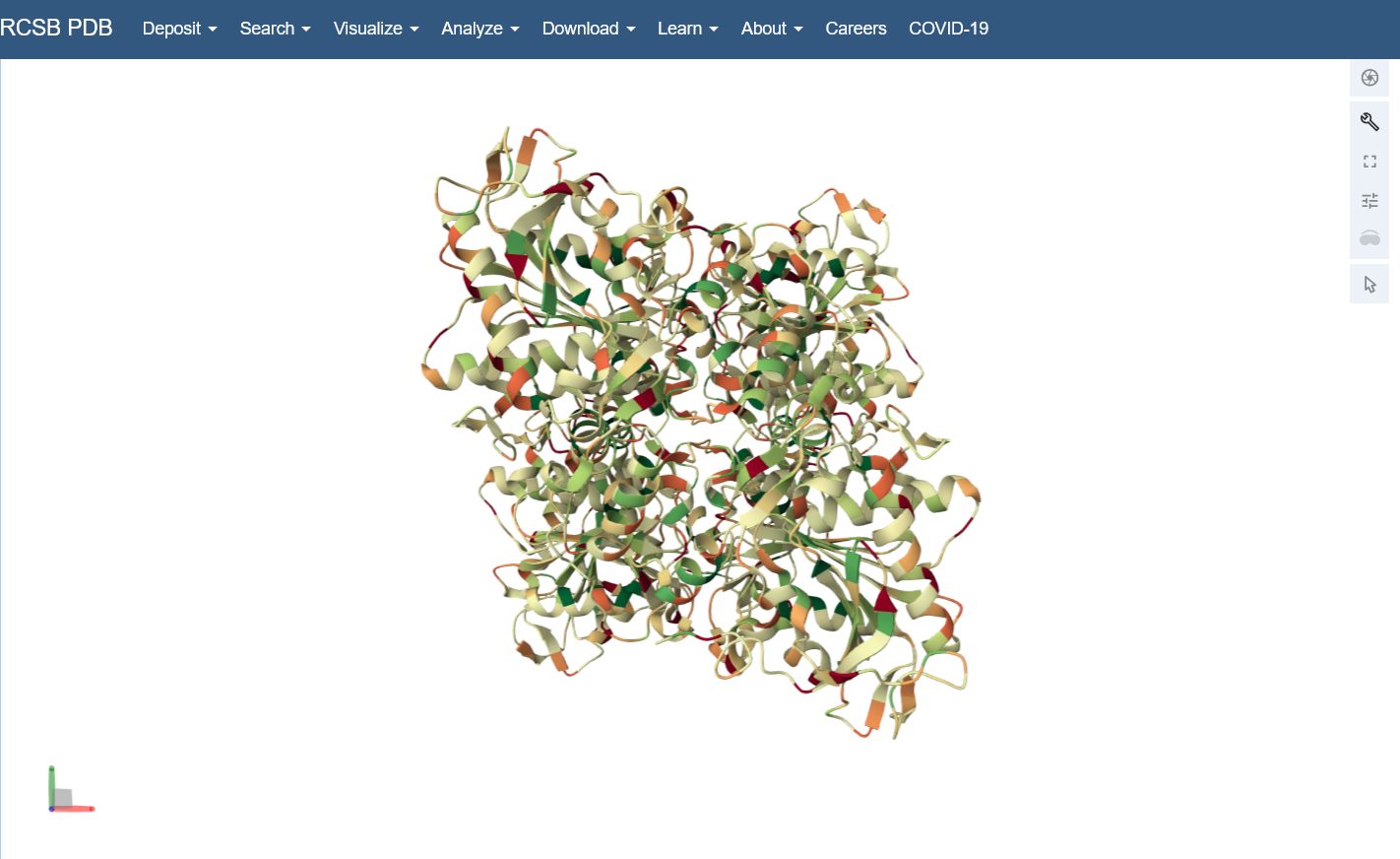
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? I colored by hydrophobicity of residue in the PDB structure viewer, because it was all one color when I selected color by residue molecule type. Not sure what was up with that, but I figured hydrophobicity would let me look at the hydrophobic vs hydrophilic residues. The hydrophobic residues are more clustered towards the insides of the structure.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? Yes, you can kind of see the indentation in the center of the screenshot below.
Part C: Using ML-based Protein Design Tools
I’m continuing with the C-terminal domain of PhaC, 5HZ2 in PDB. Colab notebook.
C1. Protein Language Modeling
Deep Mutational Scans
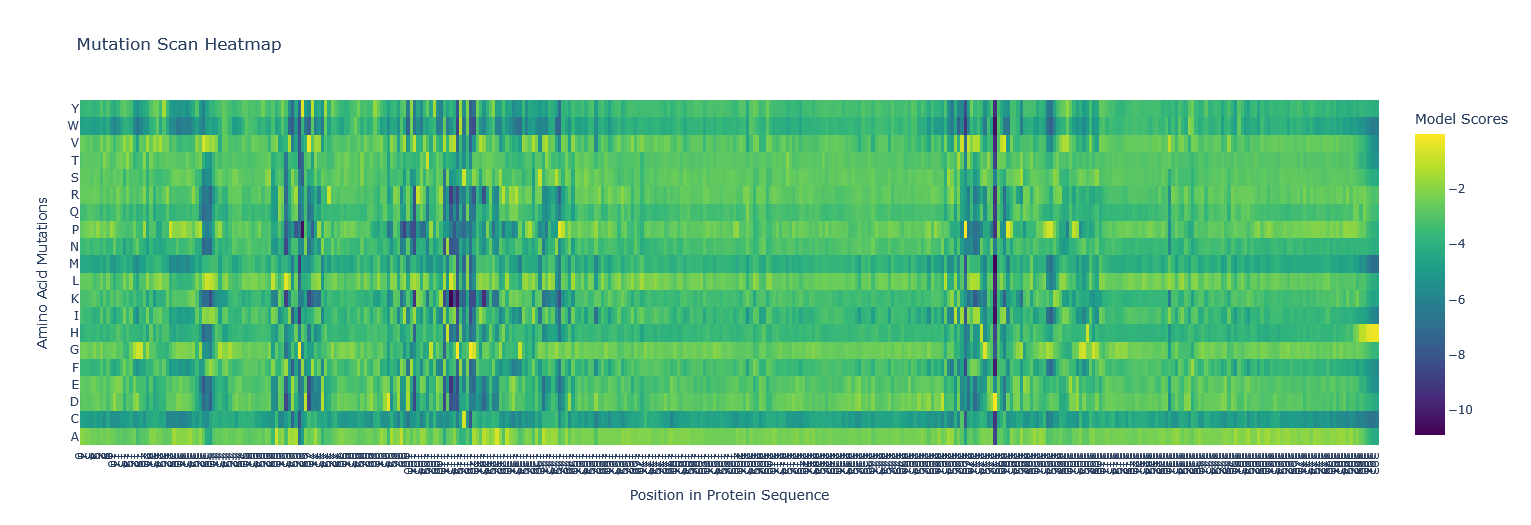
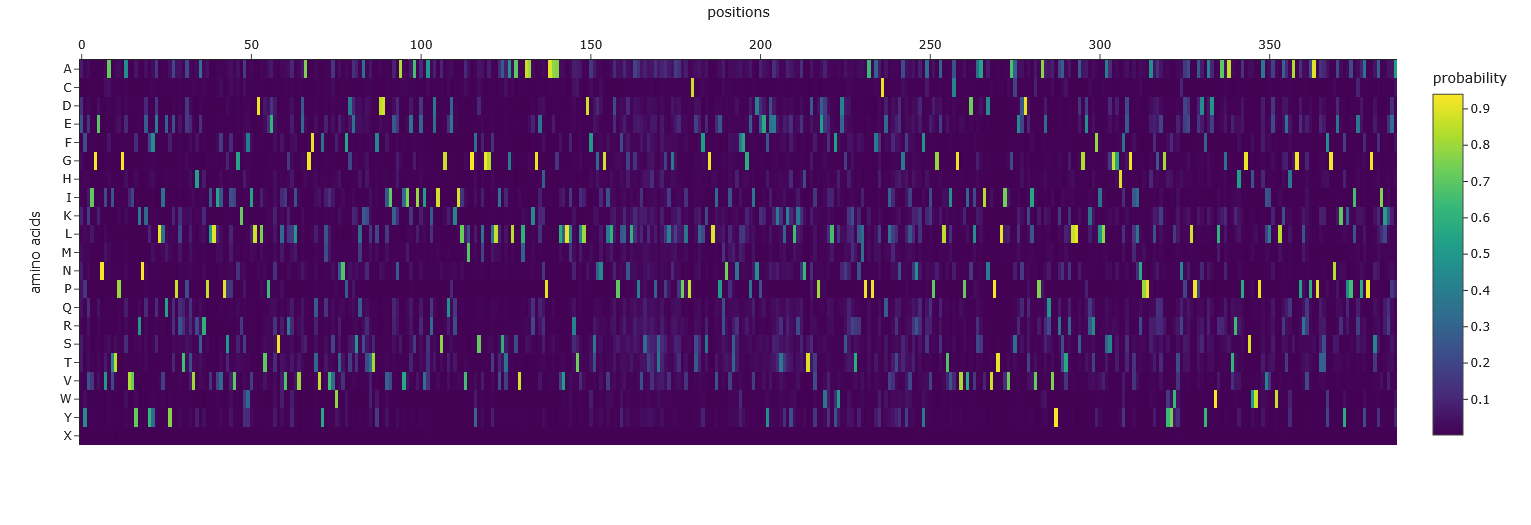
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. I copied the FASTA protein sequence from PDB into the first line of cell3 of the Colab notebook replacing the string labeled “protein_sequence”.
Can you explain any particular pattern? (choose a residue and a mutation that stands out) Position 277 seems important - Aspartic acid is the only yellow/high score. Everything else is mostly dark blue, so very negative, which I think means not likely to be able to mutate. So likely, this is either important structurally or catalytically. Asp is one of the few charged amino acids, so that makes me think it might be catalytic.
Latent Space Analysis
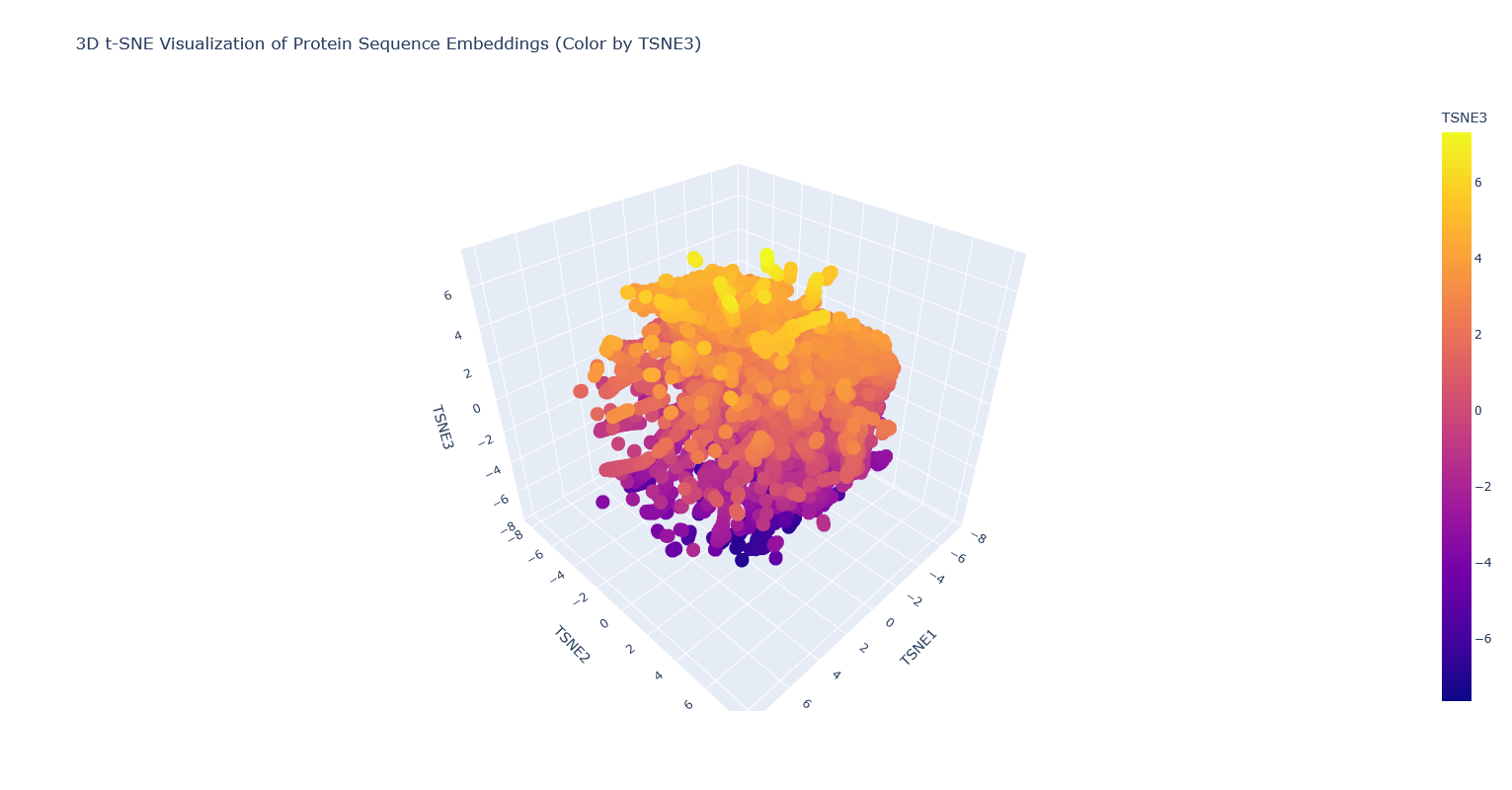
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins? I think they probably mostly do, but it’s kind of hard to tell, because there are so many proteins that it’s hard to visually see which are clustered vs overlapping clusters, and also many of the proteins are just labeled “automated matches” which isn’t really helpful for identification.
Place your protein in the resulting map and explain its position and similarity to its neighbors. It’s nearest to a lipase, a few esterases/thioesterases, and some acetyl-transferases. These are all also from bacteria. I think this makes sense, because these are all kind of involved in biosynthesis of (sometimes long) carbon-containing molecules.
Note: PhaC is the partially covered black dot surrounded by orange-yellow dots.
Code for visualization:
New cell after cell53 of the Colab. i wrote the following code based off existing Python knowledge, and mostly looking at the prior couple cells.
# add my protein sequence to the sequences array
#make list collection to match the first thing in sequences that was printed above
record = SeqRecord(seq=Seq(protein_sequence), id='5hz2', name='PhaC', description='PhaC - polyhydroxyalkanoate synthase (Cupriavidus necator)', dbxrefs=[])
#print the original length of sequences array to compare
print(len(sequences))
#append my new entry to the sequences array
sequences.append(record)
#print new length of sequences array to compare to the old (should be one greater here)
print(len(sequences))
#print the final item of the sequences array (should be my new one)
sequences[len(sequences)-1]
Then ran former cell 54 (currently cell 55 since i added a new one) as usual. Separated out the visualization generation code into a separate cell. Ran the initial dataframe creation. Made a new cell to confirm what my sequence descriptor was:
protein_sequence_annotations[15177]
Then visualized with the following code in a single cell. The chunk that was added is after the fig_3d.update_layout and before fig_3d.show(). This chunk was adapted from the bit that was posted by Noureldin Rihan on the Discourse forum.
# Visualize with Plotly 3D scatter plot, coloring by TSNE3
fig_3d = px.scatter_3d(
tsne_df_3d,
x='TSNE1',
y='TSNE2',
z='TSNE3',
color='TSNE3', # Color points based on the third t-SNE component
title='3D t-SNE Visualization of Protein Sequence Embeddings (Color by TSNE3)',
hover_name=protein_sequence_annotations[:len(embeddings_array)] # You can replace this with sequence IDs if available
)
fig_3d.update_layout(
height=800 # Increase the height of the plot
)
#change color and size of my protein so it is easier to find in the huge latent space
#code adapted from Noureldin Rihan on Discourse forum https://forum.htgaa.org/t/issues-with-latent-space-analysis/382
# get the protein's index
my_point = tsne_df_3d.iloc[protein_sequence_annotations.index("PhaC - polyhydroxyalkanoate synthase (Cupriavidus necator)")]
# color it differently
fig_3d.add_scatter3d(
x=[my_point["TSNE1"]],
y=[my_point["TSNE2"]],
z=[my_point["TSNE3"]],
marker=dict(
size=10, # Choose the dot size
color="Black" # Choose a color
),
text=["PhaC - polyhydroxyalkanoate synthase (Cupriavidus necator)"],
hovertemplate="<b>%{text}</b><br>TSNE1: %{x:.2f}<br>TSNE2: %{y:.2f}<br>TSNE3: %{z:.2f}<extra></extra>"
)
fig_3d.show()
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? This looks like a smaller and less intricate structure than the solved structure. I’m not sure what’s up with that.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations? I replaced all the Es with Ds and removed the His-tag at the end of the sequence. This yielded the following structure:
I think it looks similar. So at least with the small mutations it’s resilient. larger mutations probably not.
C3. Protein Generation
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. The output from the third cell after the Inverse Folding with MPN heading:
Based on the heatmap, it has far less flexibility in sequence than the original.
Then after the heatmap cell, there was the last cell that gave a different output that also looked like a predicted sequence, so I’m unclear which one we should look at:
Input this sequence into ESMFold and compare the predicted structure to your original. Replacing the original 5HZ2 protein sequence with the new sequence from the last cell into ESMFold (cell 54) gives us this predicted structure below. Which I guess looks kind of similar to the original predicted structure, but still to me does not look like the PDB structure. The new sequence doesn’t have a His-tag at the end, but it does kind of look like it has a linear tail like a His-tag, which is neat.
Part D: Group Brainstorm on Bacteriophage Engineering
What do we know:
E. coli DnaJ binds to denatured proteins to prevent/disassemble aggregates (native function in heat-shock).
DnaJ binds to the hydrophilic tail of MS2-L protein.
point mutation of highly conserved proline in DnaJ results in no lysis (so maybe no more binding of MS2-L tail?)
removal of MS2-L tail recovers lysis function (meaning DnaJ is only necessary when tail exists)
suggests hydrophilic tail aggregates in some way that prevents lysis except in presence of DnaJ to stop aggregation
so stability should be improved if we can figure out how the tail is interacting with the tail of other MS2-L molecules, and then mutating that away so there is no aggregation and dependence on DnaJ
graph TB;
A[sequence and structure of MS2-L] -->|if geometry and chemical interactions are known| B[view interactions between MS2-L copies]
A -->|if geometry and interactions are not known| C[model interactions with AlphaFold or something that can do protein interactions]
B -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions]
C -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions]
D -->|use knowledge of hydrophobicity/charge/etc. OR use ESM2 mutational scan and select ones that it finds unlikely| E[Select dissimilar AAs to substitute in interacting residues]
E -->|AlphaFold or similar| F[model protein folding in new AA sequence with selected mutations]
F -->|something that can model protein interactions| G[model interactions between mutant MS2-L copies]
G -->|select mutations that have similar hydrophilicity as original tail but less interaction with each other and maybe also with DnaJ| H[test mutations in lab]
Potential problems:
don’t know what can model protein-protein interactions
we might have covered this in class but i don’t remember. i can rewatch the lectures
what if modeling doesn’t show interactions between the tails? we know there probably has to be one…
might have to simplify by only modeling the tail section, but that is probably known already (will have to model folding and interactions with full protein sequence in later steps probably)
could start with DnaJ, what in MS2-L binds with the essential proline in DnaJ, and assume that it’s spatially close to that. then test various mutations of nearby residues
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. Imported from Uniprot into Benchling. Manually changed A at residue 5 to V (because this sequence includes the starting M which is not traditionally counted, I assume). Screenshot shows the mutation by aligning with the original sequence.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:Colab
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
These binders are from couple different runs because each run gives me one or more binders that contain amino acid single letter code X, which AlphaFold can’t handle because it’s non-standard.
KRVYVVAVEHWE
WLVPAVVLEWKK
WRYYVAGLRWKE
WRYYAAGARHGE
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
index
Binder
Pseudo Perplexity
1
KRVYVVAVEHWE
31.639343
2
WLVPAVVLEWKK
14.543342
3
WRYYVAGLRWKE
20.310199
4
WRYYAAGARHGE
9.566312
5
FLYRWLPSRRGG
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
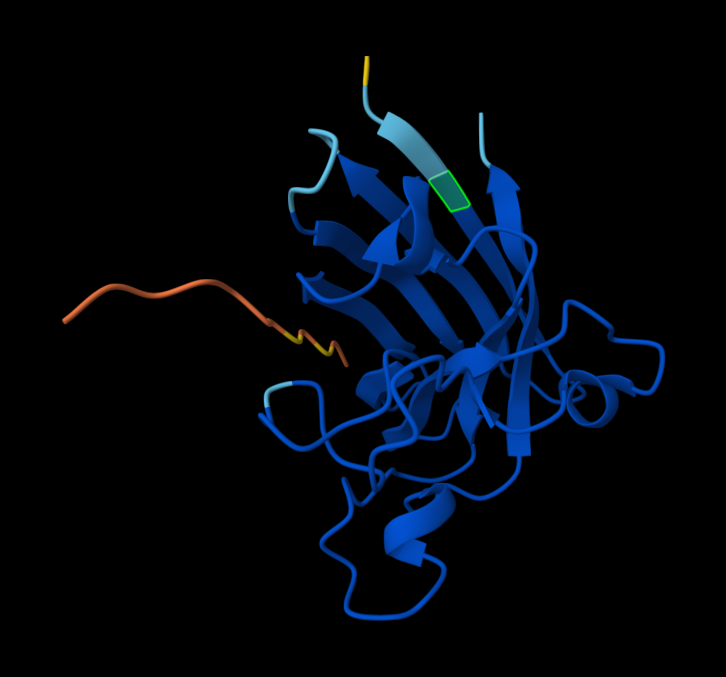
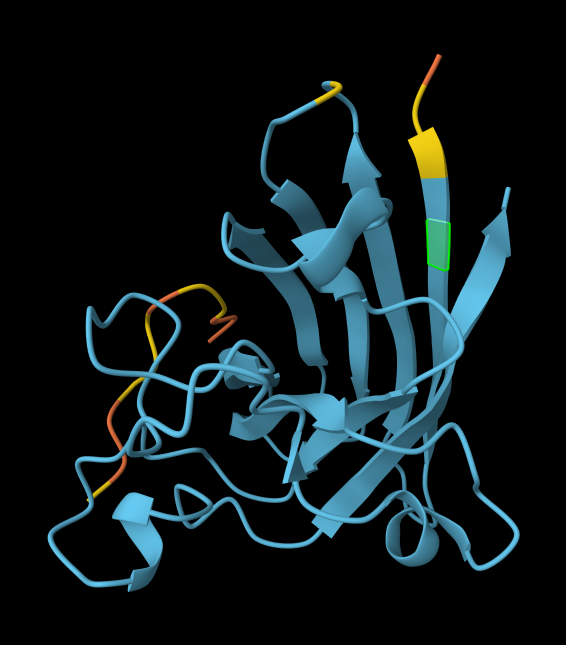
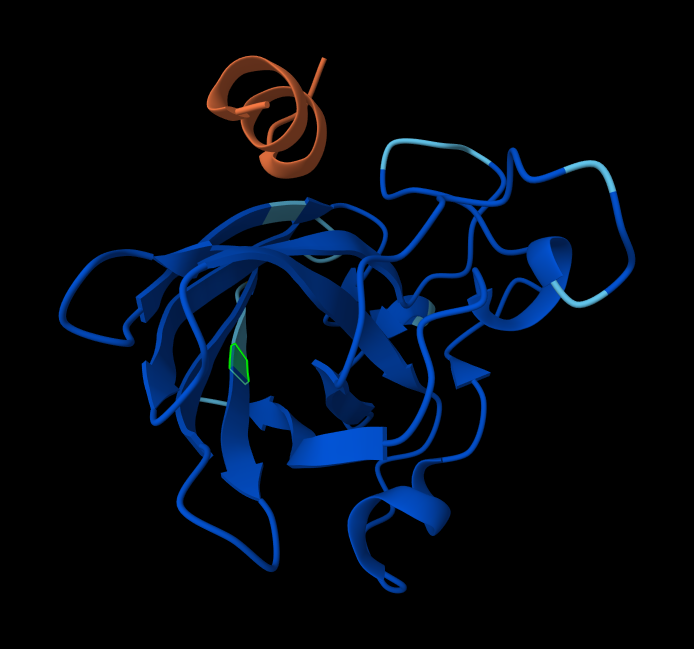
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
index
Binder
Pseudo Perplexity
ipTM score
Localization
1
KRVYVVAVEHWE
31.639343
0.31
Within the beta-barrel, but not near the N-terminus.
2
WLVPAVVLEWKK
14.543342
0.34
Partially within the beta-barrel, partially within the more disordered region. Not near the N-terminus. More on the surface of the barrel, but a little buried within the disordered region.
3
WRYYVAGLRWKE
20.310199
0.29
Adjacent to the beta-barrel, but not near the N-termins. On the surface, possibly sterically interfering with the barrel because it’s an alpha-helix rather than linear.
4
WRYYAAGARHGE
9.566312
0.42
On top of beta-barrel, with one end somewhat near the N-terminus. On the surface of the barrel.
5
FLYRWLPSRRGG
0.30
In disordered region, not near the N-terminus or the beta-barrel. On the surface.
AlphaFold peptide 1, highlighted residue is A4V.
AlphaFold peptide 2, highlighted residue is A4V.
AlphaFold peptide 3, highlighted residue is A4V.
AlphaFold peptide 4, highlighted residue is A4V.
AlphaFold known peptide, highlighted residue is A4V.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder. Three of my 4 peptides have ipTM values above the known binder. Even my one peptide that has a lower value is almost the same (0.29 vs 0.3). Three of my peptides have very similar values, but one standout is much higher (0.42 vs 0.3). This would suggest that at least that peptide, if not all of them, is worth pursuing further.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes: Predicted binding affinity; Solubility; Hemolysis probability; Net charge (pH 7); Molecular weightCompare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
index
Binder
Pseudo Perplexity
ipTM score
Binding affinity
1
KRVYVVAVEHWE
31.639343
0.31
6.739
2
WLVPAVVLEWKK
14.543342
0.34
6.450
3
WRYYVAGLRWKE
20.310199
0.29
6.637
4
WRYYAAGARHGE
9.566312
0.42
6.401
5
FLYRWLPSRRGG
0.30
6.361
Actually for my peptides, the higher ipTM scores tend to have lower binding affinities predicted by PeptiVerse. The highest ipTM score was 0.42 from peptide 4 - it had the lowest pseudo perpexity score and one of the lower binding affinities. The second lowest ipTM score was 0.31 from peptide 1 - it had the highest psueo perplexity score and the highest binding affinity. The known peptide had a similar binding affinity as the rest of my peptides: 6.361. It’s actually lower than two of them and pretty close but slightly lower than the other two.
Peptide 1:
Property
Prediction
Value
Unit
Solubility
Soluble
0.549
Probability
Hemolysis
Non-hemolytic
0.099
Probability
Binding affinity
Weak binding
6.739
pKd/pKi
Net charge (pH 7)
-0.14
Peptide 2:
Property
Prediction
Value
Unit
Solubility
Soluble
0.904
Probability
Hemolysis
Non-hemolytic
0.091
Probability
Binding affinity
Weak binding
6.450
pKd/pKi
Net charge (pH 7)
0.76
Peptide 3:
Property
Prediction
Value
Unit
Solubility
Soluble
0.598
Probability
Hemolysis
Non-hemolytic
0.052
Probability
Binding affinity
Weak binding
6.637
pKd/pKi
Net charge (pH 7)
1.77
Peptide 4:
Property
Prediction
Value
Unit
Solubility
Soluble
0.982
Probability
Hemolysis
Non-hemolytic
0.023
Probability
Binding affinity
Weak binding
6.401
pKd/pKi
Net charge (pH 7)
1.85
Known peptide:
Property
Prediction
Value
Unit
Solubility
Soluble
0.608
Probability
Hemolysis
Non-hemolytic
0.047
Probability
Binding affinity
Weak binding
6.361
pKd/pKi
Net charge (pH 7)
2.76
Choose one peptide you would advance and justify your decision briefly. I’d probably choose either peptide 1 or peptide 4.
Peptide 1: has the highest pseudo complexity score. It has a similar ipTM as the known peptide, and a higher binding affinity. It also has good solubility, hemolysis, and charge predictions. AlphaFold predicted it to be within the beta-barrel.
Peptide 4: has the lowest pseudo perplexity score. It has a higher ipTM than the known peptide, and a similar binding affinity. It also has good solubility, hemolysis, and charge predictions. AlphaFold predicted it to be near the N-terminus.
I’d move forward with peptide 4 because of it has similar properties as the known peptide, but has possible binding location near the A4V mutation.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card.Colab
Make a copy and switch to a GPU runtime: T4 GPU runtime
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). I chose the first 10 residues, roughly centered around the A4V mutation.
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
index
Binder
Binding affinity
Hemolysis
Solubility
1
CTRDYPVCRACR
7.1381
0.0499
1.0000
2
ACRGRRFAFFRV
6.8598
0.0189
1.0000
3
GSRRWWVYWHWR
7.5707
0.0225
1.0000
4
VWAAIWRREYGK
6.4160
0.0222
1.0000
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies? These peptides are different from the PepMLM peptides. I’d go through the same process I did with the PepMLM peptides to evaluate these peptides: modeling with AlphaFold and and evaluate with PeptiVerse.
Part C: Final Project: L-Protein Mutants
We didn’t get to this part of the project unfortunately. But we did have some planning discussion.
My assumption was that DnaJ stabilizes the L-protein by preventing aggregation that would otherwise occur with the long tail.
Peter suggested:
Sooo, the phage genome is very tightly regulated, I decided to take a look on how this regulation work, and it’s mainly based on RNA secondary structures
How the lysis protein is regulated:
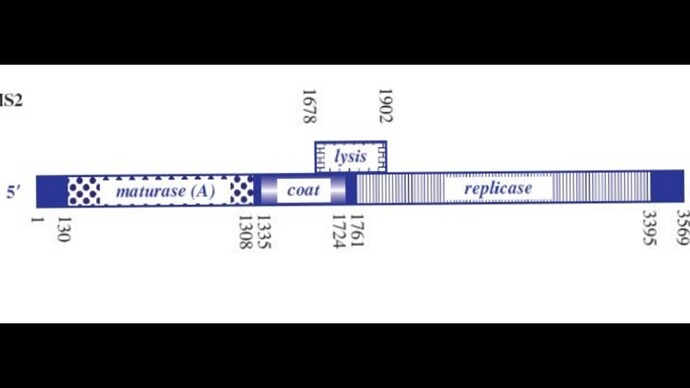
The start codon and the shine-Dalgarno sequence are buried in an RNA hairpin, rendering virtually inaccessible to the ribosome, only when a ribosome slips during Coat protein’s translation termination does it get get translated, this has a very rare 5% chance of occuring
How the replicase protein is regulated:
There’s a 19 nt hair called the operator or TR (translation repression) located upstream of the replicase protein, as the CP is translated, dimers form, that binds the TR hairpin, repressing replicase translation and signaling the beginning of the capsid assembly
One of the things I noticed, the TR hairpin overlaps with the lysis protein too, so in theory, it does repress it too
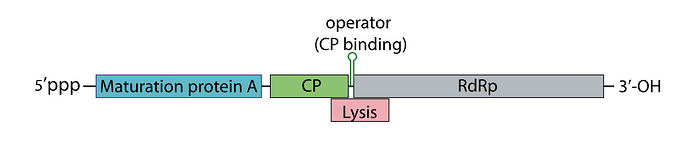
I’ve attached a linear map of the MS2 genome to follow along, here is its source too: Emesvirus ~ ViralZone
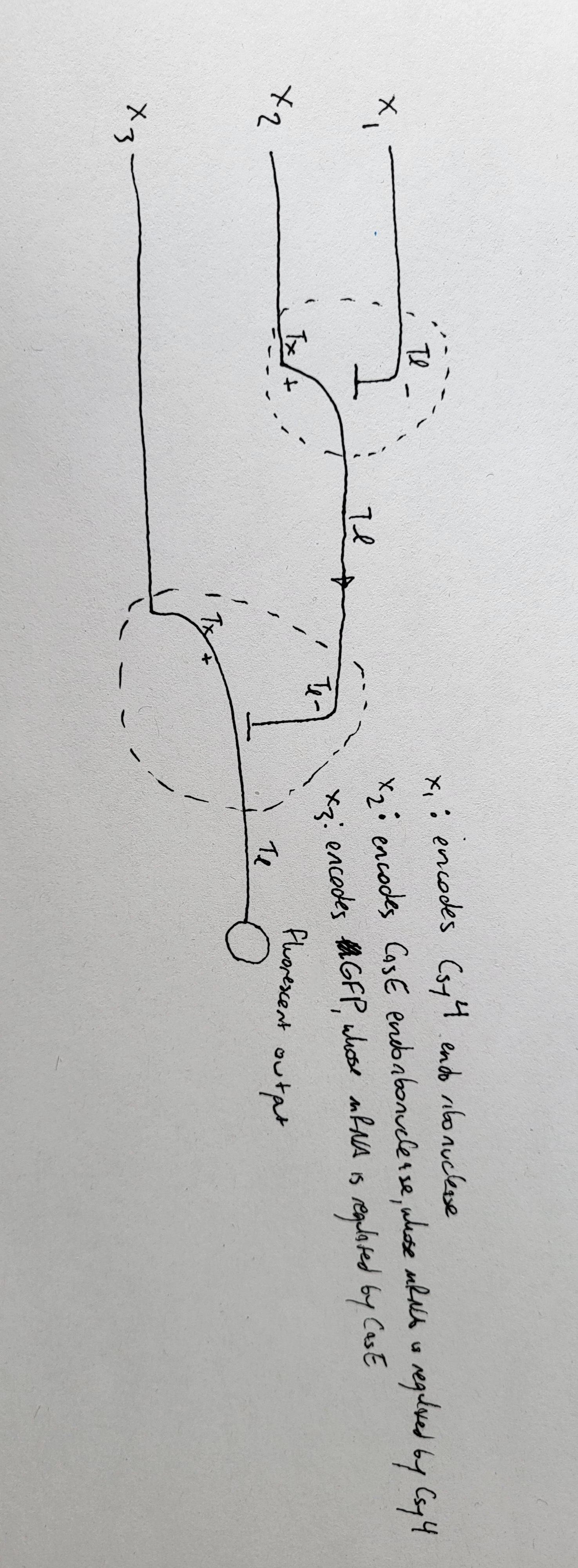
Here’s the genome engineering idea I arrived at: the first 40 amino acids of the L protein seem to be dispensable, and they’re the ones that cause it to interact with the chaperone DnaJ. What if we shift the start codon from its original position at 1678 to 1795? This would produce an L protein without the troublesome soluble N-terminus.
There are several problems though:
We need to model the MS2 gRNA. Most models can only handle short sequences, while the MS2 genome is 3569 nt long, which is pretty large for current tools. One model that might work is RNAPro, but I couldn’t find a web server or a Colab notebook to run it. The source code is on Hugging Face, but I don’t have much coding experience so I couldn’t get it running.
If the start codon is shifted to this position, the L protein will compete with the replicase for translation, so we’d need to ensure there’s a strong SD sequence for the new L start site.
The translation regulation would basically be lost, since L translation would no longer be coupled to CP. That creates a risk of premature lysis, where L protein is translated at lethal levels before new virions are assembled.
I was wondering if there’s a way to bury the SD sequence for the 1795 L site so that it’s only accessible when the CP dimer binds to the TR hairpin. That might help mitigate the premature lysis problem. I’m not sure though whether the L region would stay accessible long enough to induce lysis. I also couldn’t find a paper on the assembly kinetics.
Another idea I had was increasing the CP dimer affinity to the TR hairpin so that the L region can stay accessible for long enough before assembly proceeds.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA polymerase - a high fidelity DNA polymerase, which means that it is an enzyme that adds single nucleotides to extend a DNA chain along a template with some sort of proof-reading ability. It is used for PCR, which means it has to be thermostable.
dNTPs - single nucleotide bases to be used by the polymerase to make DNA
buffer - buffer is used primarily for controlling the pH of the PCR reaction, but it also includes MgCl2 which is a required co-factor for the DNA polymerase.
What are some factors that determine primer annealing temperature during PCR? Primer annealing temperature is affected by the length of the primer and the GC content primarily.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR is a method to produce many copies of a DNA sequence for which you already have a template. It requires a thermocycler, and PCR mix (thermostable DNA polymerase, dNTPs, appropriate buffer). To use it, you need to have template DNA and primers designed to bookend the sequence of interest. Restriction digests can linearize circular DNA or trim DNA sequences. It requires a heat block or incubator, the relevant restriction enzymes, and appropriate buffer. To use it, you need to have (typically a medium or high concentration amount) DNA that contains your sequence of interest already bookended by restriciton enzyme cutsites. Restriction digests can produce sticky ends or blunt ends; PCR will always produce blunt ends. Both methods will typically require some sort of purification step before further use (DNA cleaning and concentrating; gel extraction). PCR is useful when you need more of a particular sequence of DNA, when you want to make point mutations within a sequence (multi-step process), to add short sequences to the ends of the DNA sequence (such as restriction enzyme cutsites, adaptors, or overlaps). Restriction digestion is useful when you need to remove an insert from a plasmid backbone, to linearize a vector for electrophoresis or other analysis, and for restriction-digest cloning (including ensuring insert and vector have appropriate sticky ends for directional insertion).
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? Ideally you would design and test in silico to ensure overlaps are appropriate. My first couple times trying Gibson assembly, i wrote it out by hand to convince myself i had done it correctly, but many molecular biology software options can now assist with this as well. You can exactly confirm your purified DNA fragments prior to Gibson assembly by sequencing them, but you can also just get a good idea of their size (which would at least tell you if you PCR’d a very different or non-specific products) by running them on a gel.
How does the plasmid DNA enter the E. coli cells during transformation? During a heat shock transformation, you shock the E. coli cells with an abrupt temperature change from on ice at 0°C (or sometimes room temperature around 20°C) to 42°C. This opens pores within the cell membrane that allow DNA to enter the cells, due to prior treatment with CaCl2 to neutralize the negative charge of the DNA.
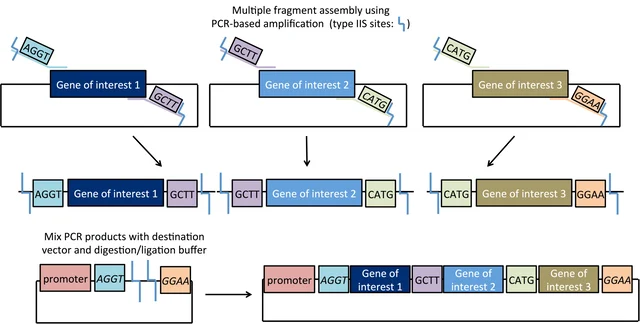
Describe another assembly method in detail (such as Golden Gate Assembly).
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly can be conceptualized as a cross between restriction digest cloning and Gibson Assembly. Like restriction digest cloning, restriction enzymes are used to digest both the insert and the vector to create compatible sticky ends for directional insertion. However, it uses Type IIS restriction enzymes (such as AarI) that cut outside their recognition site. Therefore with correct design, the recognition sites are removed in assembly. This allows for plasmid construction similar to Gibson assembly: design your insertion fragments and vector backbone to have compatible overhangs/overlaps with the adjacent sequences (often added during primer design in PCR), then add all fragments to the reaction mix which includes both a nuclease and a ligase for assembly. In Golden Gate assembly, the Type IIS restriction enzyme(s) find their recognition sites, cut nearby (at a pre-identified base), resulting in the designed 4-base overhangs. These overhangs can connect with matching overhangs from either the original construct or the intended adjacent fragment, which will be ligated into a closed dsDNA molecule (if the original construct is re-ligated, then the Type IIS enzyme again finds the recognition site and cuts again, thereby improving the efficiency).
Figure from Addgene’s Golden Gate Cloning page.
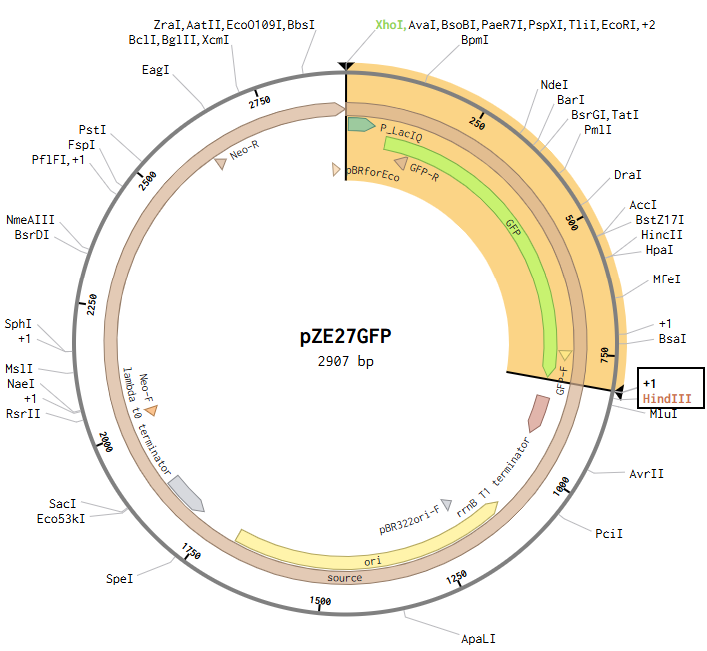
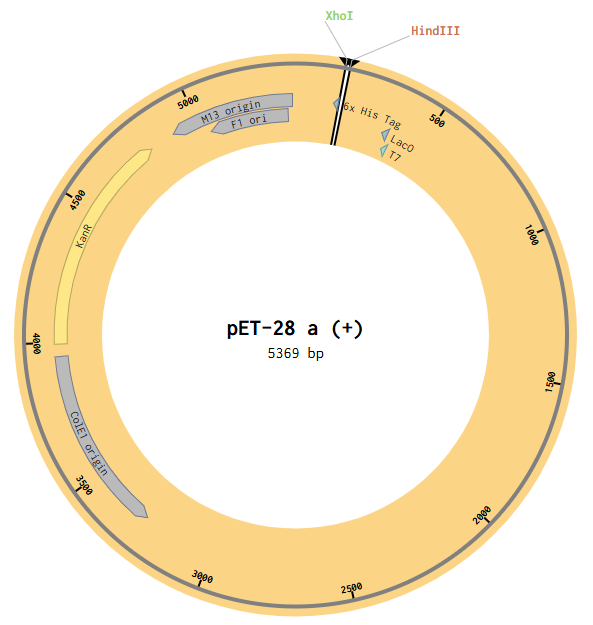
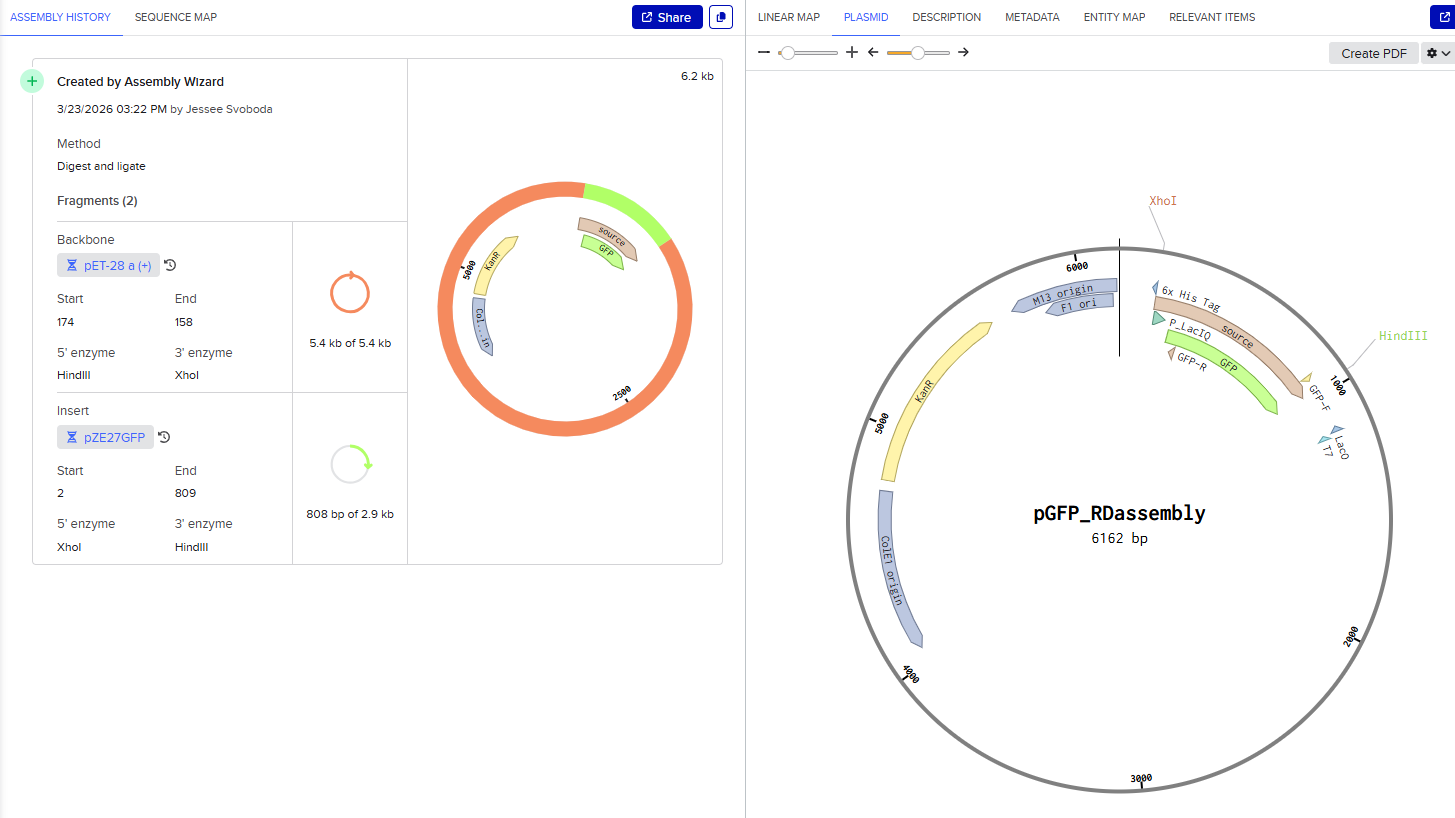
Model this assembly method with Benchling or Asimov Kernel! To compare assembly methods, I used Benchling’s Assembly Wizard tool to simulate the same plasmid construction using restriction digest, Gibson assembly, and Golden Gate assembly. My target plasmid is called “pGFP”, with a pET28a(+) backbone and an insert containing the gene for green fluorescent protein (GFP) under constitutive promoter P_LacIQ from plasmid pZE27GFP. I started by importing both pET28a(+) and pZE27GFP into Benchling from Addgene. I used Benchling’s auto-annotation tool on pET28a(+) for annotations. pZE27GFP was already annotated, but was missing the annotation for P_LacIQ, so I added an annotation from that by downloading the Genbank file from the Addgene site and using CTRL-F on the sequence to identify it in the original file. I wanted these annotations so that I knew the locations of the relevant sequences in my files for easier visual identification during the cloning simulation. Note that the GFP translation in the pZE27GFP file didn’t include the stop codon, but the stop codon was present, just not included in that translation annotation, and I was too lazy to fix this, so I just remembered that my sequence of interest included the three bases past the end of the translation annotation.
Restriction Digest
Opening the pZE27GFP file to the plasmid map view, I selected the Digests tool to show all single cutters on the map, and identified ones that were near the ends of goal insertion sequence (outside P_LacIQ and GFP): XhoI and HindIII.
Then I opened the pET28a(+) file to the plasmid map view, and selected the Digest option to only show the selected enzymes, and found these two enzymes cut in the insertion locus on the plasmid (between the T7 promoter and the His-tag).
Since both enzymes were present on both starting constructs, I used the Assembly Wizard tool for Restriction Digest cloning, and selected the backbone and insert by highlighting the above sequences with the selected enzymes.
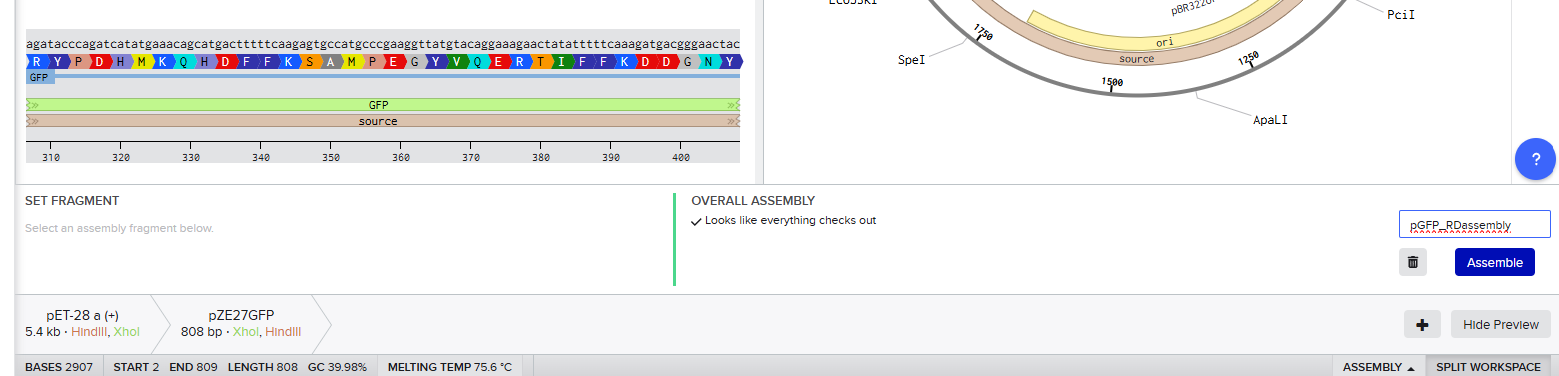
This resulted in a final assembly of pGFP_RDassembly. Note that both the XhoI and HindIII recognition sites are preserved in the final construct. While sticky-ended enzymes allow for directional insertion, this insert does not require directional insertion because it contains both the promoter and the gene. This is important because technically the insert is backwards for the vector as intended (for the T7 promoter and His-Tag on the backbone).
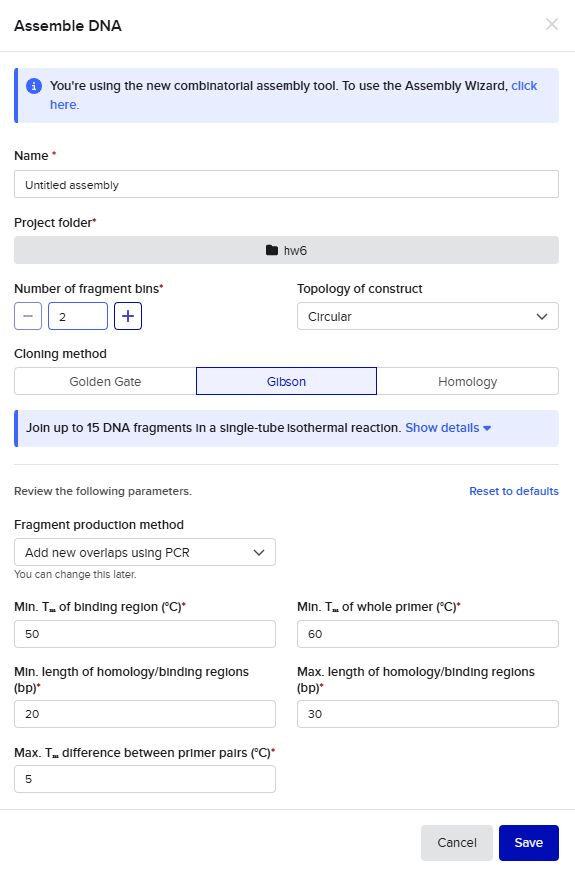
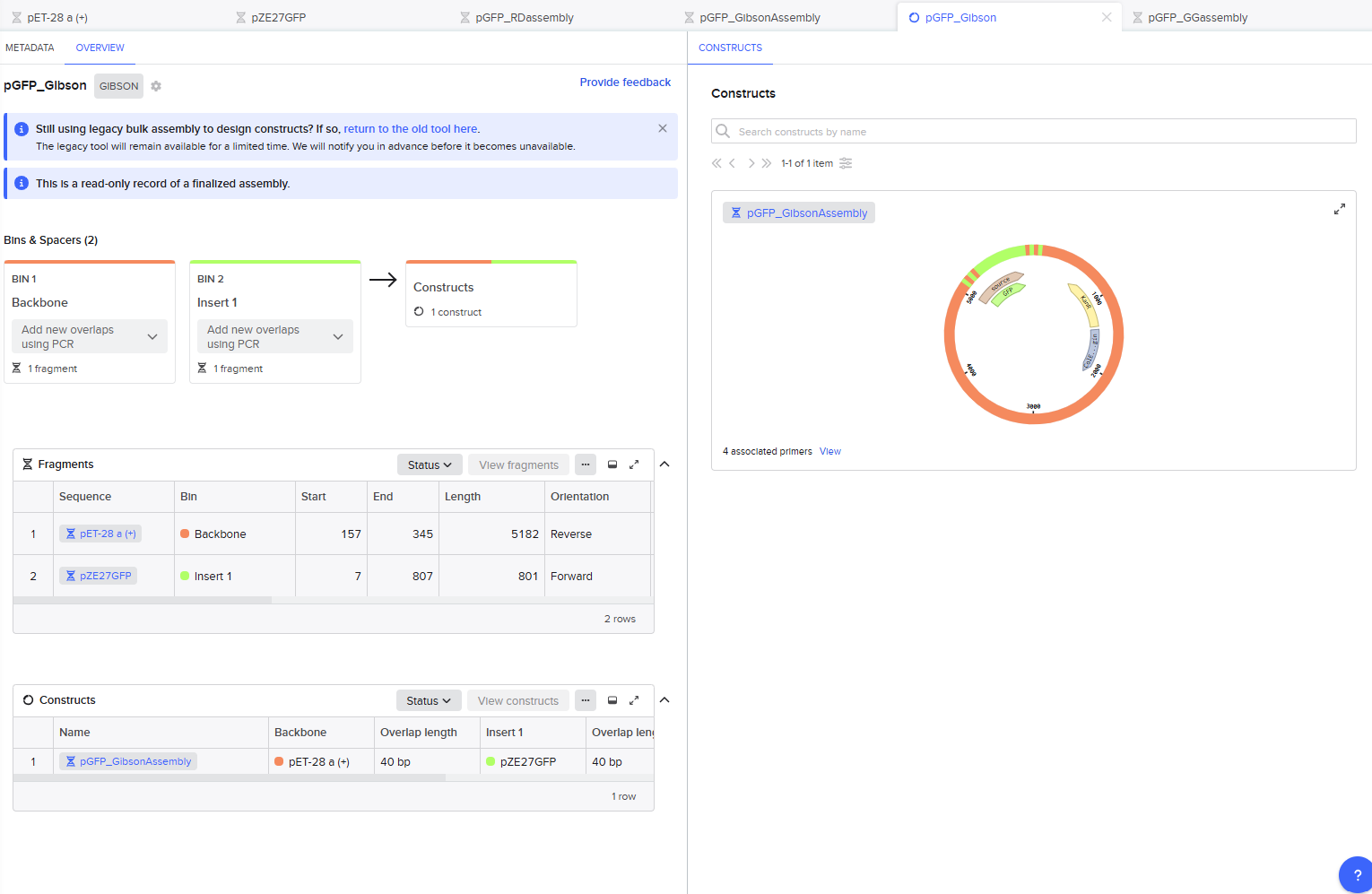
Gibson Assembly
For the Gibson assembly method, I started by opening the Assembly Wizard, and selecting the Gibson option, and opting to try the new combinatorial assembly tool instead. I retained all the default options.
This resulted in a final assembly of pGFP_GibsonAssembly. The primers were auto-generated by the tool, and are visible in the Benchling files for pET28a(+) and pZE27GFP the the naming convention following “pET28a-GA_forward”. The PCR products used in the final assembly are here (insert) and here (backbone).
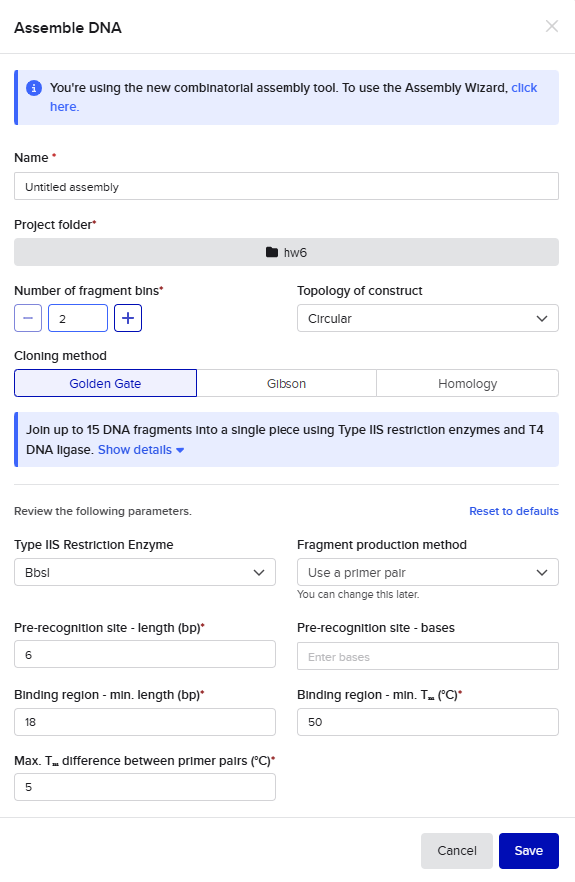
Golden Gate Assembly
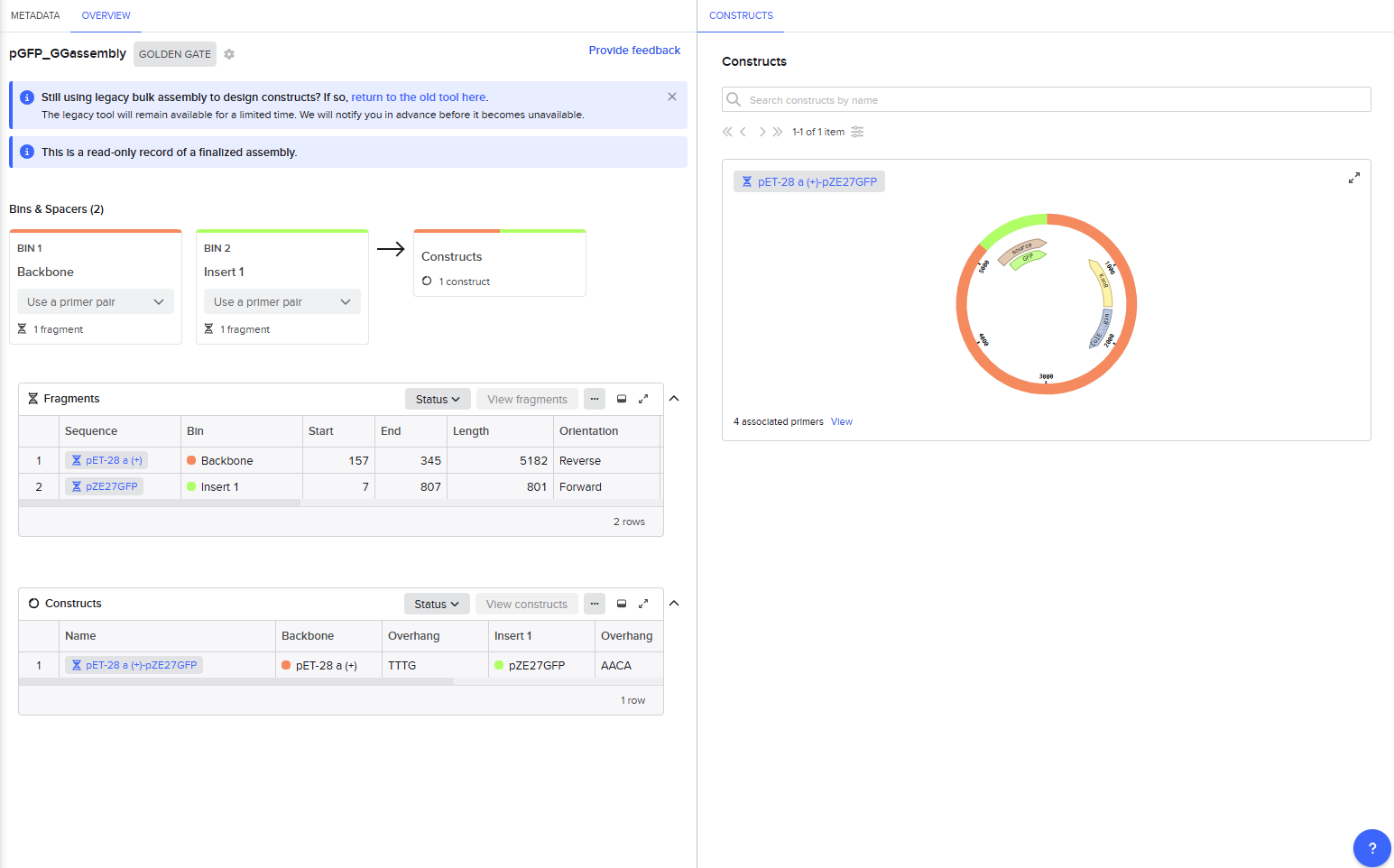
For the Golden Gate assembly method, I similarly started by opening the Assembly Wizard, used the new combinatorial assembly tool for Golden Gate. I retained all the default options. I selected “Use a primer pair” as the option under “Fragment production method”, and then retained the default options that auto-populated. Upon selecting my insert and backbone sequences, the tool threw a warning for a recognition site for the Type IIS enzyme within one of those sequences, so I went into the tool settings to instead select AarI as my enzyme. AarI was chosen somewhat arbitrarily because I’ve used it before; if it had also thrown an error, I would have simply gone down the list until I found a compatible enzyme that wouldn’t cut inside my sequences.
This resulted in a final assembly of GFP_GGassembly. The primers were auto-generated by the tool, and are visible in the Benchling files for pET28a(+) and pZE27GFP the the naming convention following “pET28a-GG_forward”. The PCR products used in the final assembly are here (insert) and here (backbone). Note that both fragments contain AarI recognition sites, but the final construct does not.
Asimov Kernel
See repository JKS_hw6 in Asimov Kernel.
Repressilator:
Repressilator reconstruction:
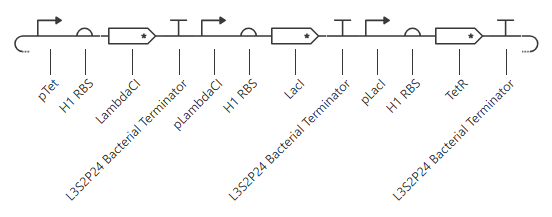
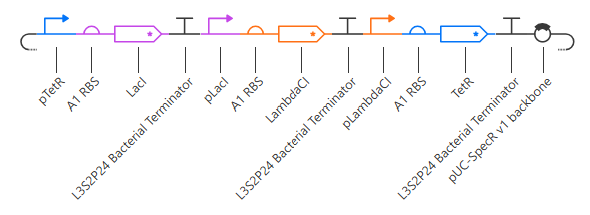
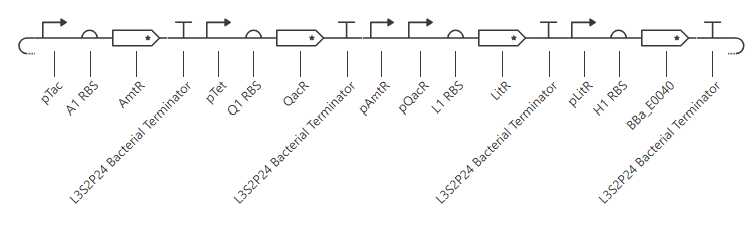
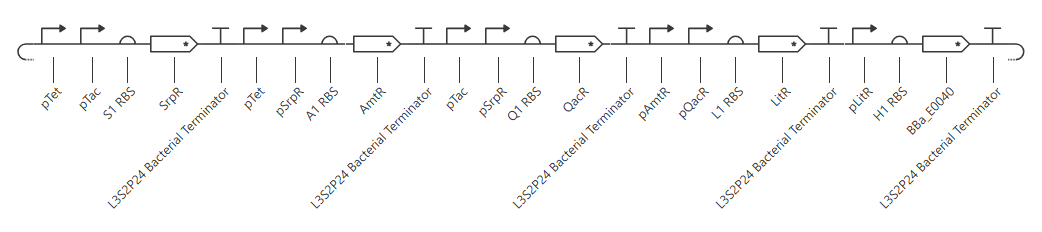
My initial attempt looks like
The Terminator chosen (L3S2P24 Bacterial Terminator) is the only one available in the Characterized Bacterial Parts repo. The H1 terminator was chosen arbitrarily as the shortest RBS; I just wanted the same RBS for each promoter-gene combo.
I wanted to add a backbone, but there’s no backbone available in the Characterized Bacterial Parts repo. Because the homework instructions said to use only the parts in this repo, I figured I’d try this first without the backbone.
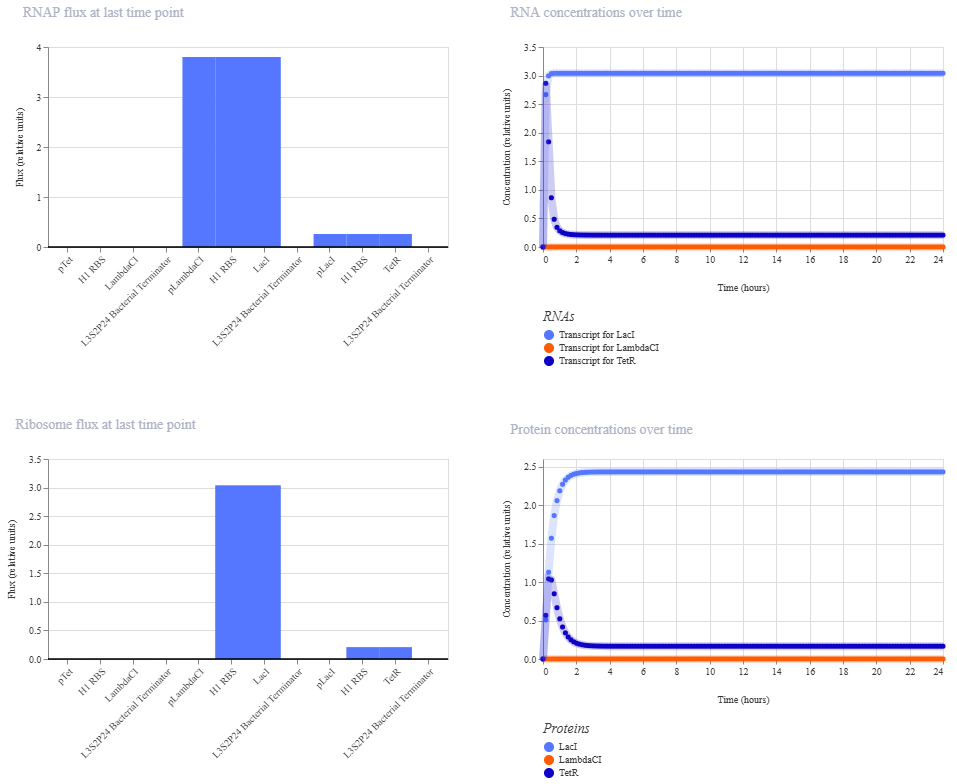
Unfortunately, this didn’t work. The outcome of my first simulation (E coli, 24h, 30min, no ligands) is below. Notice the lack of oscillations in the transcript and protein concentrations over time.
I have two potential solutions for this that I can think of before I check the pre-made Repressilator: first, I don’t have a backbone, which I do think I need, but I did still get a simulation without it, so maybe I don’t. Second, I don’t have a reporter protein. My recollection of the Repressilator paper includes a fluorescent output, so I’ll try adding a reporter gene next.
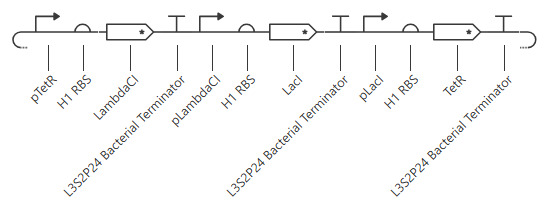
Second attempt:
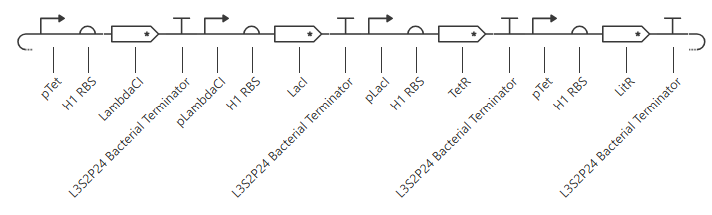
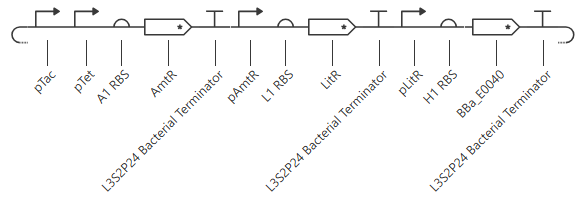
pTet was chosen arbitrarily - it could have been any of the three promoters used prior. H1 RBS was used again for consistency. LitR was chosen arbitrarily as a reporter gene because I couldn’t find a fluorescent protein within the Characterized Bacterial Parts repo.
Unfortunately, this gave more or less the same kind of output with no oscillations. I’ll try adding in a backbone from outside the Characterized Bacterial Parts repo, but if that doesn’t work then I’ll have to go back and reference the demonstration repressilator. Adding pUC-SpecR-v1 backbone, but it didn’t change the output.
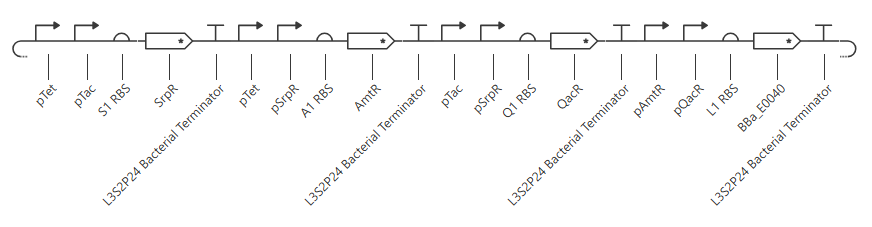
Checking the repressilator in the Bacterial Demos repo, I’m honestly not totally sure why mine didn’t work. It looks really similar:
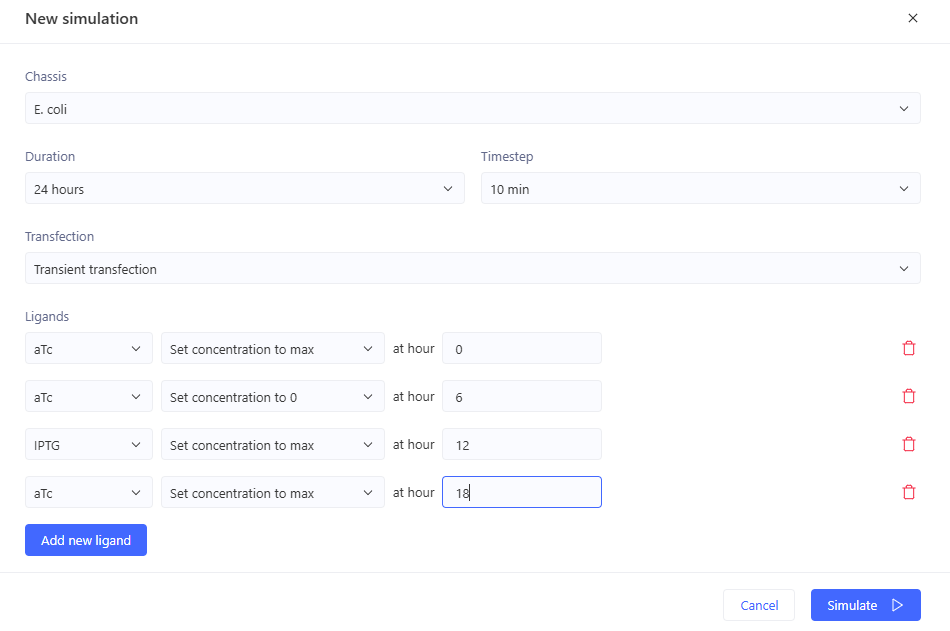
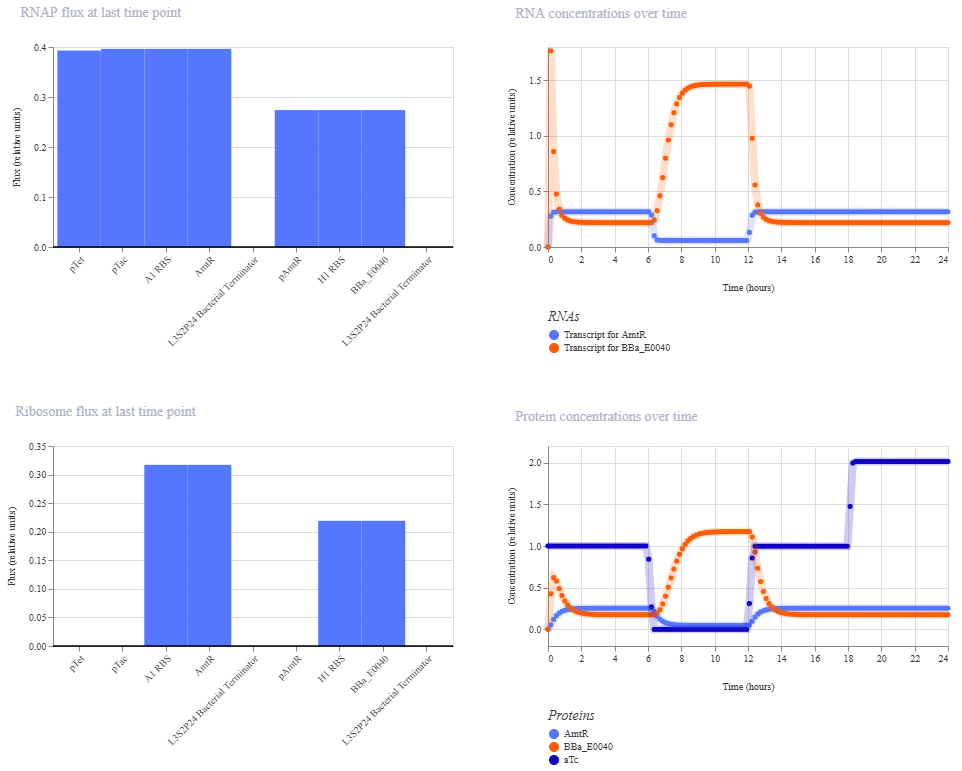
The terminator and backbone used are the same ones as I used. It has LacI/LambdaCI swapped from my original construct, but it should still work. Oh! I see - I accidentally grabbed pTet not pTetR originally. I went back and removed my pTet-LitR section, to return to my original construct, and then I replaced the pTet with pTetR.
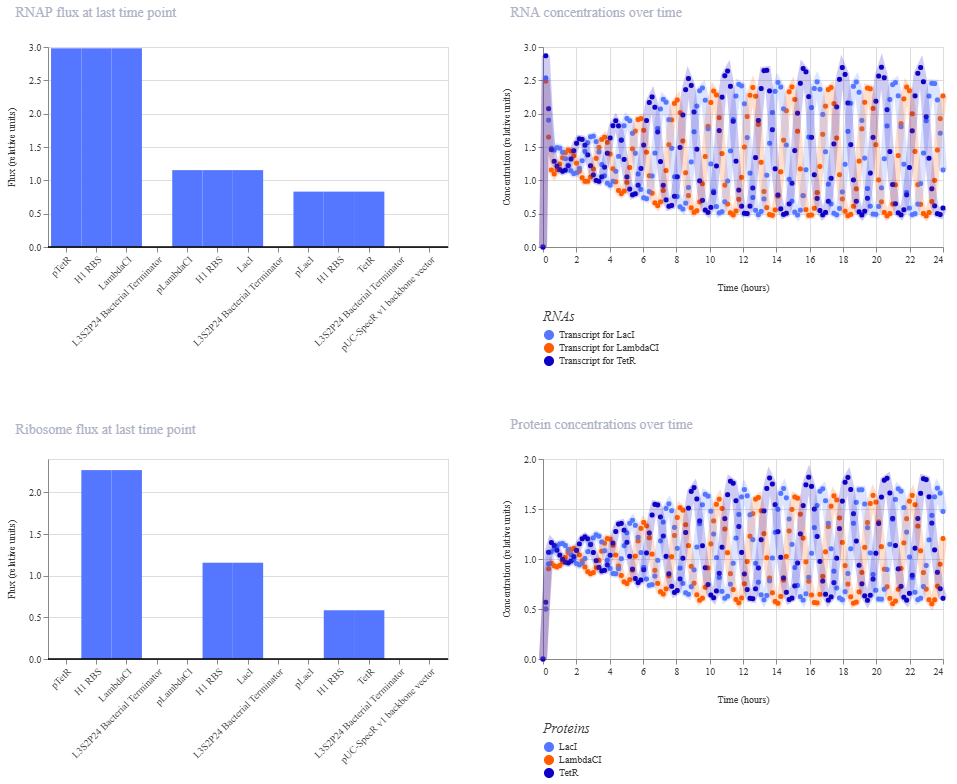
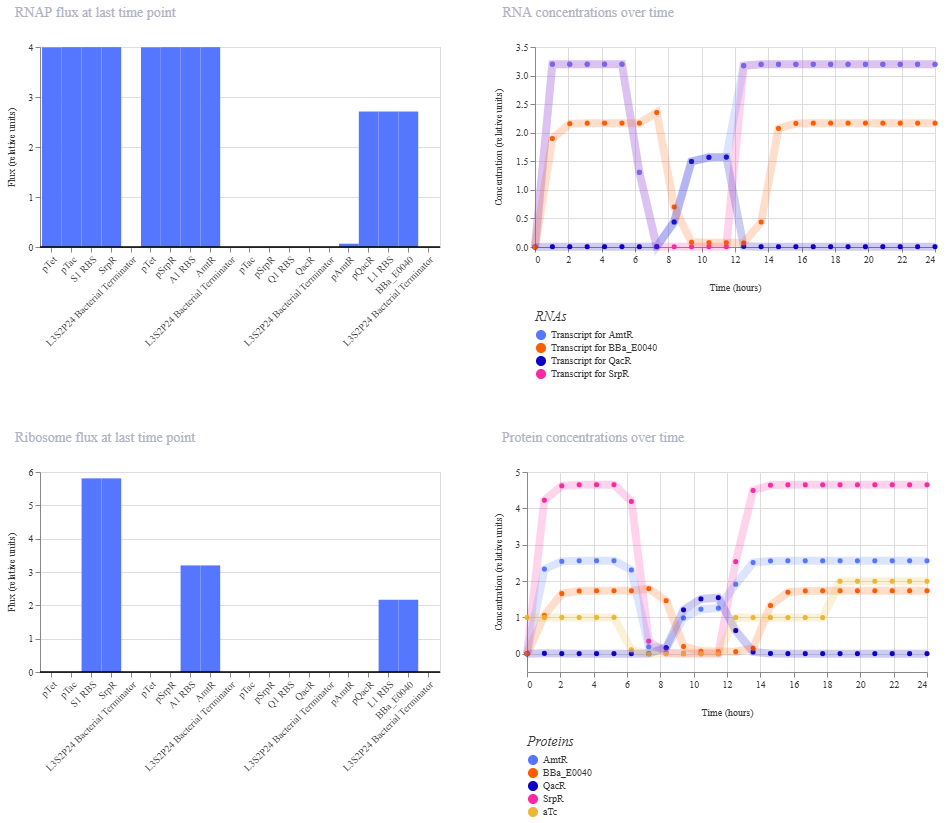
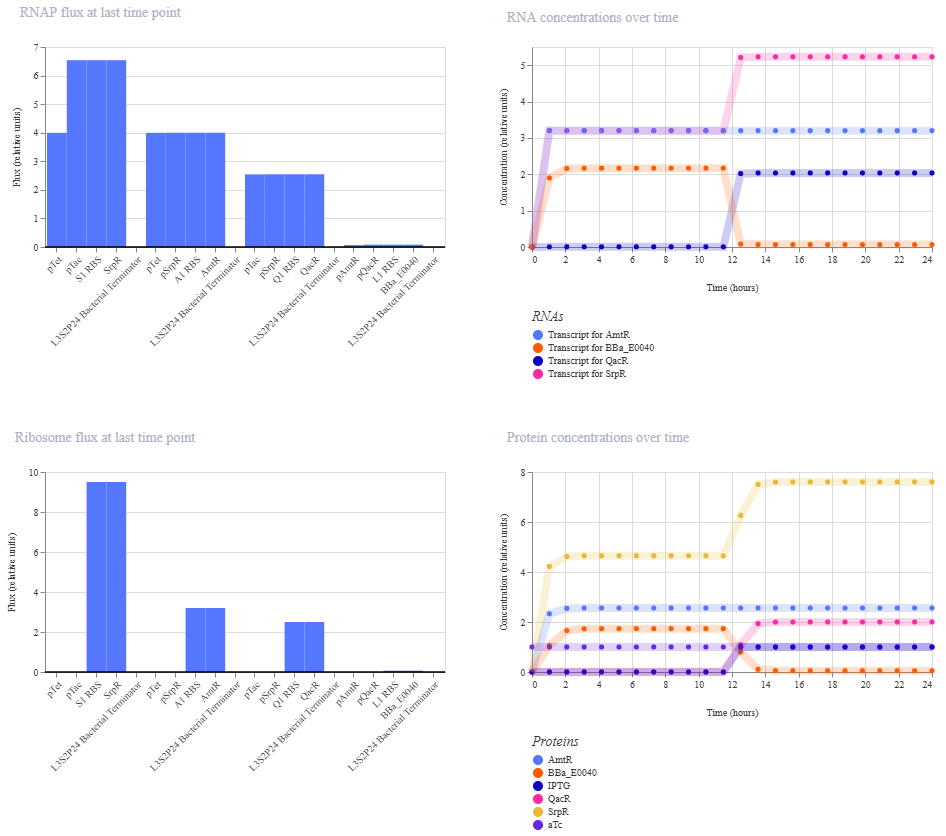
This worked! Here’s my new output:
And here’s the oscillations that I wanted to see. Awesome!
Construct1: OR gate
Construct 1: OR gate
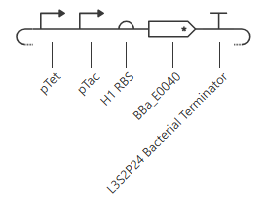
Initial construct
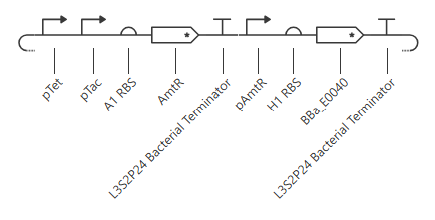
pTet is activated by aTc, pTac is activated by IPTG. BBa_E0040 is from the iGEM registry; encodes for GFP.
If aTc or IPTG is present, then GFP will be expressed.
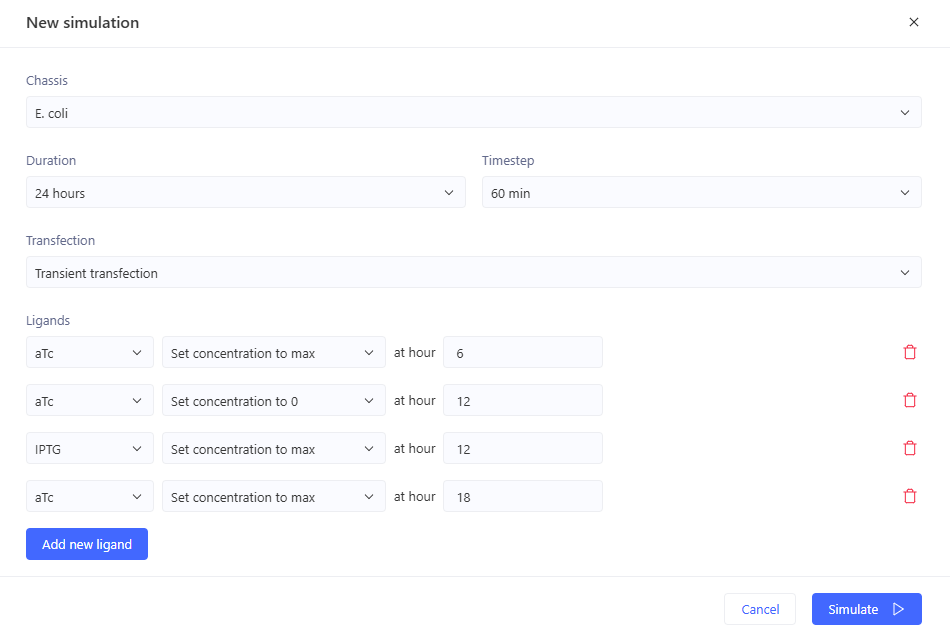
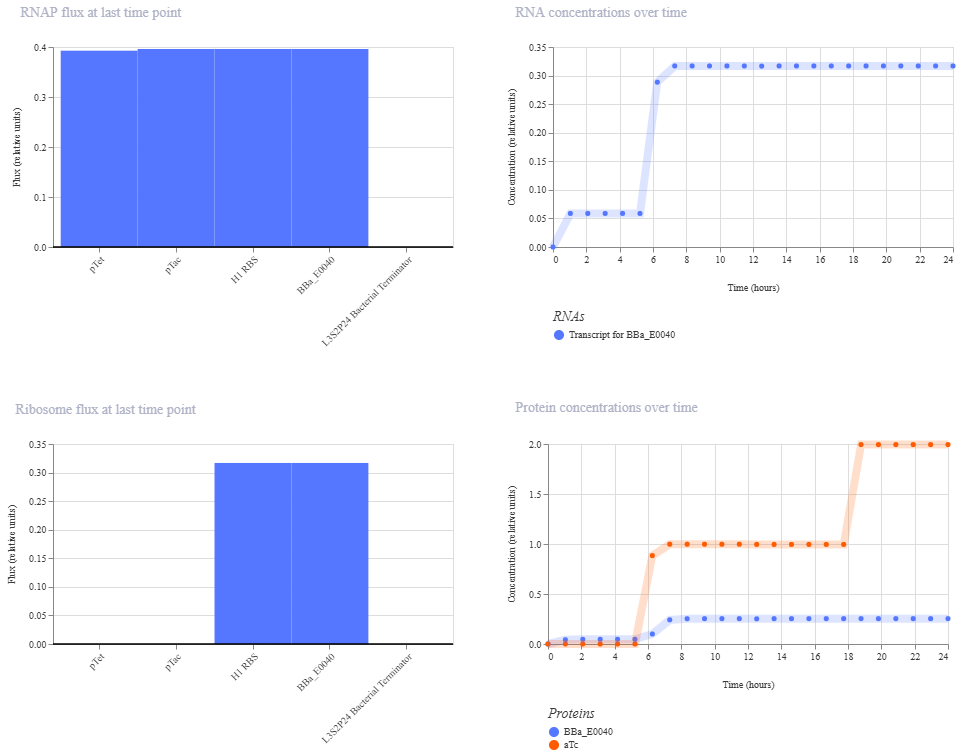
I’m a little surprised that there was as much of a difference between aTc and IPTG alone, but considering we are just looking at expression or not (rather than how much expression), i think this still worked. I am curious if I flip the order of pTet and pTac if that changes it at all. Kept the ligand amounts and times the same.
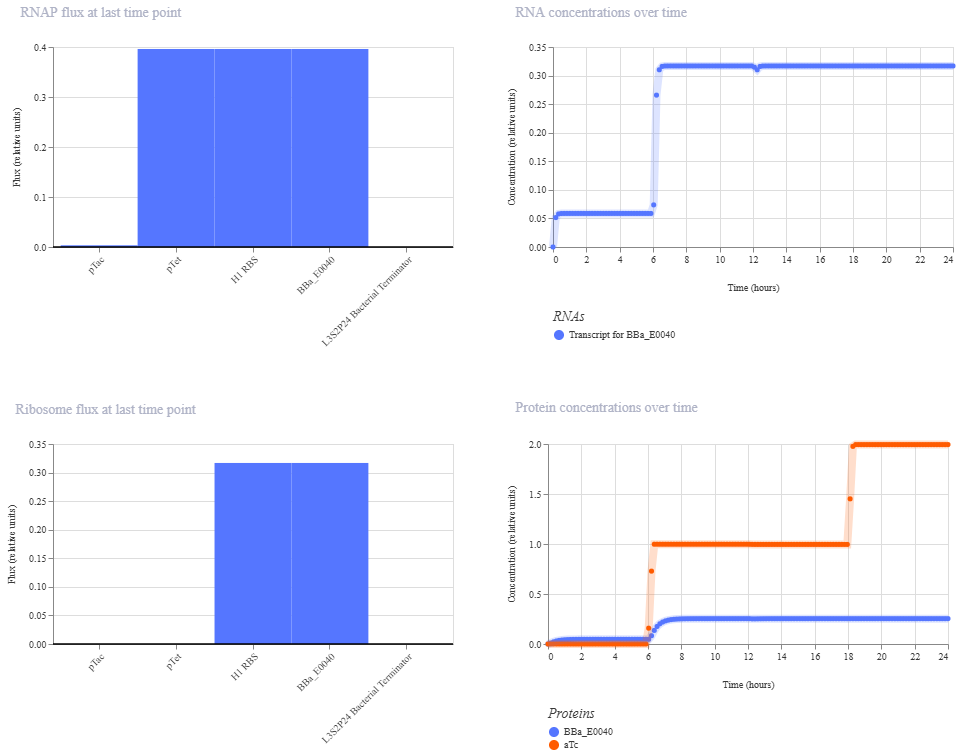
Just about the same. This makes me think that maybe setting the aTc concentration to 0 at time 12hr is maybe not working well, or maybe pTac is just that much stronger of a promoter than pTet.
Construct2: NOR gate
Construct2: NOR gate
Initial construction:
pTet is induced by aTc, pTac is induced by IPTG. BBa_E0040 encodes GFP.
If neither aTc nor IPTG are present, then GFP will be expressed.
Expected output:
aTc
IPTG
Output
0
0
1
1
0
0
0
1
0
1
1
0
Simulation:
0-6hrs: aTc => no output 6-12hrs: no ligands => GFP 12-18hrs: IPTG => no output 18-24hrs: aTc+IPTG => no output \
Expected outcome achieved.
Construct3: XOR gate
Construct3: NOR gate
I wanted to try to see if i could independently come up with a XOR gate without directly copying the one in the Bacterial Demos repo. Looking at my OR gate and NOR gate, I thought I’d be able to, but when I started to try to sketch it out, I kept getting stuck. Originally, I was thinking an OR gate minus an AND gate, and I had designs for both of those.
OR gate
Expected output:
aTc
IPTG
Output
0
0
0
1
0
1
0
1
1
1
1
1
AND gate
Expected output:
aTc
IPTG
Output
0
0
0
1
0
0
0
1
0
1
1
1
However, I couldn’t figure out how to combine these in a way that made sense. After drawing out probably a couple dozen circuits, I ended up consulting the XOR gate in the Bacterial Demos repo. Looking over it briefly (but not trying to track out the outcomes directly), I figured out a tiered method to design the circuit.
Line1: start with the output: GFP, under a repressible promoter. Line2: then below that draw that promoter’s transcription factor. add in a repressible promoter (but leave room for more if needed). Line3: then below that, draw the new promoter’s transcription factor. add in one of the two inducible promoters (leave room for more promoters if needed). But we have two inputs, so we need two inducible promoters. They can’t be on the same protein, because that wouldn’t give an OR gate. So add another promoter on line2. Line2: add another repressible promoter to the transcription factor for the GFP promoter. Line3: Below that, draw in the new promoter’s transcription factor, under the control of the other inducible promoter (leave room for more promoters if needed). But the inducible promoters need to be able to cancel each other out. Line3: So add the same repressible promoter to each transcription factor on this line. Line4: Below that, draw in that new promoter’s transcription factor, under the control of BOTH inducible promoters.
This yields the following circuit:
Expected outcome:
aTc
IPTG
SrpR
AmtR
QacR
LitR
Output
0
0
0
1
1
0
1
1
0
1
1
0
1
0
0
1
1
0
1
1
0
1
1
1
1
1
0
1
This is the opposite of an XOR gate (yielding output at Neither input or Both inputs, rather than yielding output at Either of only one input), so i just need to add one more layer of repressible promoter to get what I’m hoping for I think. Or I can replace the LitR with GFP and remove the section with GFP under pLitR.
New circuit for XOR gate:
Expected outcome:
aTc
IPTG
SrpR
AmtR
QacR
Output
0
0
0
1
1
0
1
0
1
1
0
1
0
1
1
0
1
1
1
1
1
1
1
0
Simulation:
0-6 hrs: aTc only 6-12 hrs: nothing 12-18 hrs: IPTG only 18-24 hrs: aTc and IPTG
This did not give the expected outcome. GFP doesn’t fall again at the end like it should.
I think there was just something with the simulation; either i didn’t set up the ligands properly, or it wasn’t enough time to equilibrate or something. Because when I run the different ligand combinations individually, or just one change over 24 hours it works like expected.
Here is the outcome for aTc high the entire time, and adding high IPTG at 12 hours. So it does work as expected.
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs do analog computing instead of digital. So functions are additive (positive or negative) rather than just present/absent. This means that they can respond to an input that’s beyond (over or under) a certain threshold, instead of just is the input present or not. Non-digital dosage. IANNs can also stack with multiple layers for multiple inputs as well.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. IANNs can be used to identify cell types, such as cancer cells by differentiating them from the surrounding healthy cells. The cancer cells might not have a single unique signal to use as an identifier, but it might have a few different metabolites (or other signals) present in different amounts from the healthy cells. So an IANN can be used to recognize multiple inputs, and how much of those inputs are present (is it more/less than the baseline amount present in the healthy cells). The output might be fluorescence to tag tumor locations for a surgeon to excise, or maybe the output could be a medication for specific targeted release.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts? Some existing fungal materials include fungal leather and fabrics for clothing, primarily of mycelium or cellulose; biocement, which uses bacteria or fungi to produce calcium carbonate around gravel; and fungal composite materials, which uses a fungal mycelium around an organic or agricultural substrate. Fungal composite materials can be leather-like fabrics, packaging, acoustic insulation, thermal insulation, and hard particle board or brick-like building materials for furniture or architecture.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria? Engineered fungi might form mycelium materials that can produce different colors; contain biosensors; have different material properties like hardness/flexibility; or be able to actively bioremediate the location that the mycelium-made object is placed in. Fungi are eukaryotic instead of prokaryotic like bacteria, which means there is more diversity both within the cell (organelles) and on a cell-to-cell level (cell differentiation). This complexity both increases the difficulty of synthetic biology in fungi over bacteria, but also allows for engineering that complexity (such as only having the bioremediation turned on in the fruiting bodies).
First DNA Twist Order
Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website. My first sequence is the wild-type Cupriavidus necator PhaC. For cell-free synthesis, it will be transcribed by T7 polymerase, so it needs to have those components. I designed it in Kernel, using the parts from the iGEM repository and the PhaC_Cnecator gene from the Uniprot repository.
The promoter, Bba_Z0251 is the T7 promoter with the consensus sequence. The RBS, Bba_Z0261 is a wild-type T7 RBS that has been characterized as a strong RBS by an iGEM team. The terminator, Bba_K731721 is a wild-type T7 terminator that has been characterized by an iGEM team. The Uniprot PhaC_Cnecator part has no DNA sequnce in Kernel, so I remade this circuit in Benchling, using the PhaC_Cnecator sequence that I had previously codon optimized for E. coli expression in homework2; and copied the regulatory elements from Kernel.
This will be synthesized into a Twist cloning vector. Ronan suggested a chloramphenicol marker for constructs at the Ginkgo Nebula facility, so I’ll use pTwist-Chlor-HighCopy.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis avoids the requirements of a cold chain for shipping or storage, and it also can simplify complex living systems by instead adding in specific and known amounts of reagents (enzymes, nucleotides, amino acids, etc.). It is more beneficial than cell production in situations like biosensing in remote environments (infectious disease detection in remote or under-resourced locations) and biomanufacturing of toxic products (like some pharmaceuticals) because production won’t stop due to cell death.
Describe the main components of a cell-free expression system and explain the role of each component.
template nucleic acid: DNA or RNA encoding the gene of interest for protein
cell lysate (collection of active components, including the following - or the purified components could be added individually)
tRNA: recognizes RNA codons and adds new amino acids onto a protein chain during translation
polymerase: makes nucleic acids (DNA or RNA)
nucleotides: used by polymerase to make nucleic acids
buffer: maintains reaction pH to optimal level for enzyme function
other enzymes and cofactors, depending on the goal of the system (sometimes these are included through a cell lysate)
amino acids and ribosomes, if protein production is the goal
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment. Cell-free systems are essentially a series of chemical reactions (biological in nature, but still chemistry), which means that activation energy is required for some reactions. Energy provision regeneration is critical to ensure that the reactions continue to happen instead of stalling out early. Specifically, this is important in protein expression because translation is energetically expensive (requires ATP to attach amino acids to tRNAs). Cells generate ATP through a collection of metabolic processes; a cell-free system needs to be designed to ensure it has a way to generate ATP. One potential method is adding NAD and CoA to generate ATP from pyruvate without needing any additional enzymes.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why. Prokaryotic systems are simpler than eukaryotic systems. Eukaryotic systems might have more components, especially for production of functional proteins, for chaperones or post-translational modifications. A prokaryotic system might be good to produce antimicrobial peptides because you don’t need to worry about the product killing the host. A eukaryotic system might be better at producing functional antibodies because antibodies are eukaryotic proteins and therefore might be more functional in a eukaryotic system.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup. A membrane protein is difficult to produce in a cell-free system because it likely has a hydrophobic area and a hydrophilic area because it is natively located within a membrane. This means that it is unlikely to be folded into the correct structure without a hydrophilic space for the hydrophilic component of the protein. To optimize the expression of a membrane protein in a cell-free experiment, you would need to stabilize it, for example, by providing liposomes or membrane vesicles in which the membrane proteins could localize for correct folding.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each. Three possible reasons for a low protein yield in a cell-free system is insufficient transcription, insufficient translation, or a badly designed DNA template. Insufficient transcription could be due to not adding enough nucleotides into the reaction. This could be tested by adding an mRNA template into the reaction to see if this solves it. Insufficient translation could be due inactive tRNA, inactive ribosomes, or not enough amino acids. This could be tested by spiking more of those individual, purified components (or fresh cell lysate) into the reaction - it’s possible one of those has been degraded. A badly designed DNA template might have a promoter that isn’t recognized by the polymerase provided in the cell-free system; this could be tested with a control reaction that includes a DNA template known to work in this established system.
Reference
Hunt, AC; Rasor, BJ; Seki, K; et al. Cell-Free Gene Expression: Methods and Applications. 2024. ACS Chemical Reviews 125(1): 91-149. DOI: 10.1021/acs.chemrev.4c00116
Homework questions from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output? The SMC would produce PHB (bioplastic) using atmospheric carbon dioxide as a carbon source (effectively, photosynthesis producing PHB as the carbon storage molecule). The input is CO2 and sunlight. The output is PHB (and oxygen).
Could this function be realized by cell-free Tx/Tl alone, without encapsulation? Maybe, it would likely be at a low yield. The value of encapsulation here is to keep the intermediates in close spatial proximity to the biosynthetic enzymes for efficient biosynthesis of the final product. I’m also unsure if a thylakoid could exist without encapsulation. I’m not sure why it wouldn’t be able to; I just don’t think I’ve ever read of a cell-free thylakoid.
Could this function be realized by genetically modified natural cell? No. A cell, even a genetically modified one, would have to devote some carbon flux towards biomass and cell replication. Ideally, the synthetic cell wouldn’t have to, and all the carbon (consumed from atmospheric carbon dioxide) would go exclusively towards PHB production.
Describe the desired outcome of your synthetic cell operation. The synthetic cell would produce PHB from atmospheric carbon dioxide, with all carbon flux going towards PHB.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of? The membrane would be made up of lipids and cholesterol for flexibility. It also needs to include a thylakoid for light-harvesting.
What would you encapsulate inside? Enzymes, small molecules. Inside the SMC, I’d want the enzymes for PHB synthesis. This includes PhaC (the PHA synthase), and also all the enzymes required to build the precursor monomers. We’d maybe need a couple of Calvin Cycle enzymes, but it’s hard to say without drawing out all possible pathways of carbon flux - the idea would be for PHB to be the “energy storage” product. This might be easiest by using a cyanobacterial cell lysate, but ideally, we’d want to get to something simpler than that.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian) It would be bacterial. Especially at first, it would have to come from a cyanobacterium, likely Synechocystis sp. PCC 6803 because it’s well-studied. It would be ideal to understand the system to the extent that we could use any bacterial system (such as E. coli), and simply include whatever cyanobacteria-specific proteins or metabolites are needed.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?) The SMC would have to export the PHB. So some kind of membrane channel would need to be included.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.) Membrane: lipid, cholesterol, thylakoid membrane, chlorophyll, membrane channel Enzymes: bacterial Tx/Tl, PHB biosynthetic enzymes
How will you measure the function of your system? The system’s function would be measured by the PHB output, which could be BODIPY staining if PHB is not exported, or mass spectrometry if PHB is exported.
Homework questions from Peter Nguyen
Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept. A chlorophyll-based paint for self-healing concrete can improve air quality in buildings suffering degradation.
How will the idea work, in more detail? Write 3-4 sentences or more. Self-healing bioconcrete is either live cells, or a cell-free system, integrated into concrete that produces calcium carbonate from atmospheric CO2 when cracks are exposed to water (which then fills in the cracks). My idea is to create freeze-dry a cell-free system expressing chlorophyll to turn into a paint to go on the outside of this building material. The chlorophyll provides an energy regeneration capacity for the calcium-carbonate cell-free system, while also producing oxygen, thereby improving the local air quality; effectively, photosynthesis that generates calcium carbonate from CO2 and light instead of generating glucose. This would mean that any cracks or chips seen on the inside of the building could be sprayed with water and lit with a plant light, and the combination of the two cell-free systems would repair the crack. The chlorophyll paint would have the further benefit of being visibly green when activated, so the repair process could be visually tracked.
What societal challenge or market need will this address? How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)? This improves the self-healing concrete concept, which addresses the high CO2 emissions cost of traditional concrete manufacturing, as well as decreasing the amount of human work needed to repair broken concrete. The biggest limitation here is that it is one time use, but i think that making the chlorophyll into a paint addresses this because once the repair is completed, the new concrete could be painted over again.
Reference
Smirnova, M; Nething, C; Stolz, A; et al. High strength bio-concrete for the production of building components. 2023. NPJ Materials Sustainability, 1(4): s44296-023-00004-6. DOI: 10.1038/s44296-023-00004-6
Homework questions from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. Ionizing radiation is a safety and health concern for space exploration because of how damaging it is to living organisms. Ionizing radiation is more harmful than non-ionizing radiation because it is higher energy and can pass through more materials (thereby making it harder to shield from). While in low Earth orbit, where the ISS is, most of the radiation is protected against by Earth’s magnetic field, but the astronauts aboard the ISS still experience more radiation than people on Earth. Any space exploration beyond low Earth orbit has to deal with higher amounts of ionizing radiation.
Name the molecular or genetic target that you propose to study. Melanin from Cryptococcus neoformans, biosynthesized by Lac1 with phenolic substrate such as dopamine; and control pigment chlorophyll, biosynthesized by ChlP with substrate geranylgeranyl-chlorophyll a
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. C. neoformans is a fungus that utilizes the energy in radiation via radiosynthesis, analogous to plants utilizing the energy in sunlight via photosynthesis. A similarly radiotrophic fungus was grown on the ISS to investigate its potential as a shielding mechanism against the ionizing radiation in space. It’s known that the pigment melanin provides some protective effect against radiation, and it’s hypothesized that melanin plays an analogous role as chlorophyll in radiosynthesis and photosynthesis, respectively.
Clearly state your hypothesis or research goal and explain the reasoning behind it. Hypothesis: melanin will provide a greater protective effect against the radiation in space than chlorophyll a. The DNA in tubes with lac1 will have a lower mutation or fragmentation than tubes with chlP. The tubes containing lac1 will have a higher number of control (mRFP1) transcripts than tubes containing chlP. This difference in transcript counts might be attributable either to the higher DNA integrity due to melanin’s protection or to increased energy availability from radiosynthesis over photosynthesis (more radiation than sunlight in the test conditions).
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. All tubes contain BioBits cell-free expression system, the control gene for red fluorescent protein (mRFP1), and the substrates for both Lac1 and ChlP (dopamine and geranylgeranyl-chlorophyll a).
Negative control: no additional DNA
Condition 1: DNA encoding lac1 gene
Condition 2: DNA encoding chlP gene While these tubes could be visualized with the Molecular Fluorescence Viewer for red fluorescence, I believe visual analysis would be hampered by the pigment production. Better data would be obtained from purified nucleic acids. The DNA should be sequenced with long-reads to identify any fragmentation. The RNA should be used in RT-qPCR to quantify transcript counts.
Casadevall, A; Cordero, RJB; Bryan, R; et al. Melanin, radiation, and energy transduction in fungi. 2017. ASM Microbiology Spectrum, 5(2): 10.1128/microbiolspec.funk-0037-2016. DOI: 10.1128/microbiolspec.funk-0037-2016
Averesch, NJH; Shunk, GK; Kern, C. Cultivation of the dematiaceous fungus Cladosporium sphaeropermum aboard the International Space Station and effects of ionizing radiation. 2022. Frontiers in Microbiology, 13: 877625. DOI: 10.3389/fmicb.2022.877625
Williamson, PR; Wakamatsu, K; Ito, S. Melanin biosynthesis in Cryptococcus neoformans. 1998. ASM Journal of Bacteriology, 180(6): 1570-1572. DOI: 10.1128/jb.180.6.1570-1572.1998
Chen, GE; Canniffe, DP; Barnett, SFH; et al. Complete enzyme set for chlorophyll biosynthesis in Escherichia coli. 2018. Science Advances, 4(1): eaaq1407. DOI: 10.1126/sciadv.aaq1407
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. I’d like to measure the mass of produced PHB.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. How to measure the mass:
Centrifuge cell-free reaction to pellet insoluble PHB.
Aspirate off supernatant into waste.
Wash with water 2-3x, again pour off supernatant to waste.
Dissolve remaining PHB pellet in chloroform.
Weigh clean microtube.
Transfer chloroform solution into the weighed microtube.
Add methanol to precipitate out the PHB. Centrifuge to pellet.
Aspirate off supernatant (methanol and chloroform) to waste.
Leave tube open under fume hood to fully evaporate supernatant.
Weigh again; amount of PHB produced = [final tube weight]-[starting tube weight]
Once the PHB mass is measured, I could re-dissolve in chloroform for molecular weight and polydispersity measurements using gel permeation chromatography. I could also confirm that it is PHB on GC-MS.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? I’d use a mass balance, gel permeation chromatography, and gas chromatography-mass spectrometry.
References
Jossek, R; Steinbuchel, A. In vitro synthesis of poly(3-hydroxybutyric acid) by using an enzymatic coenzyme A recycling system. FEMS Microbiology Letters 1998, 168: 319-324. https://doi.org/10.1111/j.1574-6968.1998.tb13290.x
Satoh, Y; Tajima, K; Tannai, H; et al. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis from acetate with CoA recycling and NADPH regeneration in Vitro. Journal of Bioscience and Bioengineering 2002, 95(4): 335-341. https://doi.org/10.1016/S1389-1723(03)80064-6
Waters Part I: Molecular Weight
Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight? You can use an online calculator. Using the online calculator: 28006.60 Da. However, GFP’s self-cyclization into the active fluorophore results in a loss of around 20 Da, according to this week’s lab. So the better theoretical molecular weight should be 28006.60-20 = 27986.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and: m/z: Charge state n is 903.7148; charge state n+1 is 875.4421
Determine z for each adjacent pair of peaks.
$$ z = \frac{\frac{m}{z_{n+1}}}{\frac{m}{z_n} - \frac{m}{z_{n+1}}} $$
$$ z = \frac{875.4421}{903.7148 - 875.4421} = \frac{875.4421}{28.2727} $$
$$ z = 30.9642 = 31 $$
Determine the MW of the protein.
$$ MW = z*\frac{m}{z_n}-z = z(\frac{m}{z_n}-1) $$
$$ MW = 31*(903.7148-1) = 31*902.7148 $$
$$ MW = 27,984.1588 $$
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1.
$$ accuracy = \frac{|MW_{experiment} - MW_{theory}|}{MW_{theory}} $$
$$ accuracy = \frac{|27,984.1588 - 27986.60|}{27986.60} = \frac{2.4412}{27986.60} = 8.7227e-5 $$
$$ accuracy * 1,000,000 = 87.2275 ppm $$
This is >50ppm but it’s close, so this might be the right protein.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not? The picture is pretty blurry, so honestly i am having a hard time reading the numbers. But i think we can see isotope peaks labeled: 1473.7429, 1473.7950, [unreadable], 1474.0045, 1474.0481, 1474.1006. These all yield spacings around 0.05. This would indicate a charge state around 20.
Waters Part II: Secondary/Tertiary Structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)? Native protein conformation is the shape the protein is folded into when it is made by the cell, this is usually the active state for enzymes. Denatured protein conformation is when the protein is unfolded, and essentially a linear amino acid sequence. On mass spectometry, the denatured state exposes all possible sites for adding a charge for the clean z+1 peaks, whereas the native conformation has more limited (and frequently unknown) how many charges can and are added in different peaks. In a mass spec, the more linear/unfolded proteins add more charges, so the m/z peaks tend to be lower than those of a native protein (more peaks to the right).
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell? Once again, the low resolution of the screenshot is making it hard to read the numbers. A stretch that i’m decently confident about reads peaks at: 2545.1304, 2545.2222, 2545.3140, 2545.4058, 2545.4973. These all yield a spacing around 0.09. This would indicate a charge state around 11.
Waters Part III: Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
How many peptides will be generated from tryptic digestion of eGFP? 26, by my hand count. Using the online tool, 19. i think the difference is in not counting the very short peptides (< 5 amino acids, plus a couple of 4 AA peptides, likely because they have heavier side chains since it has a 500 Da cutoff).
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance. I saw 23 peaks, but only 21 are labeled, so I’m guessing maybe only the labeled ones are >10% abundance?
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer? No, there are more peaks in the chromatogram.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z. The m/z of the peptide at the most abundant charge state is 525.76712. The z of the most abundant charge state is 2 (because the highest peak has isotope peaks that are 0.5 m/z apart).
$$ \frac{MW+2H}{2} = 525.76712 $$
$$ MW + 2(1.00727) = 1051.53424 $$
$$ MW + 2.01454 = 1051.53424 $$
$$ MW = 1049.5197 $$
$$ [M+H]+ = MW+1H = 1049.5197 + 1.00727 $$
$$ [M+H]+ = 1050.52697 $$
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. The peptide is FEGDTLVNR.
$$ accuracy = \frac{|MW_{experiment} - MW_{theory}|}{MW_{theory}} $$
$$ accuracy = \frac{|1050.52697 - 1050.5214|}{1050.5214} = \frac{0.00557}{1050.5214} $$
$$ accuracy = 5.30212 e-6 * 1,000,000 = 5.3 ppm $$
This is <10 ppm, so it is probably the correct peptide.
What is the percentage of the sequence that is confirmed by peptide mapping? 88%
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? FEGDTLVNR; mono; +1; B, Y. Mostly matches up. D peak at 717 is very small and unlabeled, but it looks like there’s a peak approximately there. There’s no N peak at 289, nor an R peak at 175. Also the three smallest peaks don’t match up with anything in the in-silico fragmentation (56, 122, 214).
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Mostly - the peptides that are not covered in the peptide mapping are either too large (>20 AA) or too small (<5 AA) for confident identification according to the informaiton provided in the lab.
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
27986.60
27,984.16
87.2
This error is close to 50 ppm, so it might be GFP. Especially with the pretty good peptide mapping, I think this is likely GFP, though I am not as confident as I would like to be.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I made most of the big bulls-eye target in the upper right quadrant that occurred fairly early on in the editing time period. I made it largely during the recitation just by having it open in another window from the lecture and clicking another pixel whenever my timer ran out. I didn’t really contribute after that, but it was fun to see how people incorporated the target into one of the scissors handles, and then it ultimately disappeared. For future iterations, I’d really recommend publishing the viewable history link somewhere because I lost it after like a day and so then I wasn’t able to keep watching the changes and how they compared to previous versions.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction. \
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Lysate includes enzymes, nutrients, and cofactors; specifically, it includes the T7 RNA polymerase for rapid transcription of genes under T7 promoter.
Salts/Buffer
Potassium Glutamate: Potassium glutamate is a potassium source; potassium is an essential enzyme co-factor.
HEPES-KOH pH 7.5: HEPES buffer maintains pH at the optimal pH for enzyme efficiency; transcription and translation usually occurs at neutral pHs which the inside of a cell is. KOH is the hydroxide to adjust buffer to pH 7.5 to because potassium phosphate is used.
Magnesium Glutamate: Magnesium glutamate is a magnesium source; magnesium is an essential enzyme co-factor.
Potassium phosphate monobasic: Potassium phosphate is both a phosphate (energy) source and a potassium (enzyme co-factor) source; I’m unsure why both the monobasic and dibasic are included here. Monobasic is mildly acidic in comparison.
Potassium phosphate dibasic: Potassium phosphate is both a phosphate (energy) source and a potassium (enzyme co-factor) source; I’m unsure why both the monobasic and dibasic are included here. Dibasic is mildly basic in comparison.
Energy / Nucleotide System
Ribose: Ribose is a sugar molecule that is an essential component of nucleic acids and ATP. It’s used in nucleotide production (GMP from guanine) and possibly also energy regernation.
Glucose: Glucose is a sugar molecule that is used in ATP regeneration.
AMP: Adenosine monophosphate is a nucleotide used in transcription. It gets additional phosphate groups to become ATP, which is essential for energy, and so it is probably also used in ATP regeneration.
CMP: Cytidine monophosphate is a nucleotide used in transcription.
GMP: Guanosine monophosphate is a nucleotide used in transcription.
UMP: Uridine monophosphate is a nucleotide used in transcription.
Guanine: Guanine is the nucleoside base for GMP; it can be used to make GMP with ribose and phosphate probably?
Translation Mix (Amino Acids)
17 Amino Acid Mix: Amino acids are needed for translation because they are what proteins are made up of. I’m not sure why this is only 17 instead of 20.
Tyrosine: This is an amino acid needed for translation. I’m unsure why additional tyrosine would need to be added beyond the mix, maybe it’s not one of the 17?
Cysteine: This is an amino acid needed for translation. I’m unsure why additional cysteine would need to be added beyond the mix, maybe it’s not one of the 17?
Additives
Nicotinamide: Nicotinamide is part of NAD+/NADP+, and so is needed for energy regeneration in redox reactions.
Backfill
Nuclease Free Water: It’s an aqueous solution, so water fills out the rest of the reaction volume; nuclease-free water doesn’t contain active restriction enzymes to cleave DNA or RNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. The main difference appears to be in energy regeneration and cheapter components, because it uses nucleotide monophosphates instead of triphosphates. It also uses PEP-Mono (phosphoenol pyruvate, monosodium salt) for energy instead of nucleotides and sugars for energy generation through enzymatic pathways like glycolysis. PEP-Mono is a high energy phosphate-containing compound that can easily transfer phosphate groups for energy.
Bonus question: How can transcription occur if GMP is not included but Guanine is? GMP is produced with guanine, ribose, and phosphate that is provided separately in the cell-free mixture. not sure which specific enzyme(s) are involved
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. FPbase.org is not currently working, so i’m doing the best i can based off of skimming papers because i don’t have time to do a close reading of a whole bunch of them unfortunately. honestly, i’ll probably just try again later.
sfGFP: sfGFP was developed to be faster at folding into the active, fluorescent shape than wild-type GFP, resulting in a more robust and stable fluorescent protein.
mRFP1: Needs to bind to calcium?
mKO2
mTurquoise2
mScarlet_I
Electra2
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect. Because mRFP1 is considered to be “somewhat slowly-maturing”, I predict that changing the co-factor concentrations could improve maturation time for brighter fluorescence sooner. So I want to increase the potassium and magnesium concentrations.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. I wanted to test both the magnesium and the potassium concentrations for mRFP1. I chose wells Q2-G17 through Q2-G24 which are designated for mRFP1. Wells Q2-G17, G18, G19: I increased the potassium glutamate concentration by approximately 20% (specifically 19.9%). Wells G20, G21, G22: I increased the magnesium glutamate concentration by approximately 20% (specifically 17.9%). For the final two wells (G23 and G24), I increased the concentration of both salts to test the cumulative effect (potassium glutamate +19.9% and magnesium glutamate +17.9%).