Week 2 HW: Read, Write, Edit DNA
Contents
Part 1: Benchling and In-Silico Gel Art
Simulated lambda DNA digestions:
[grid]
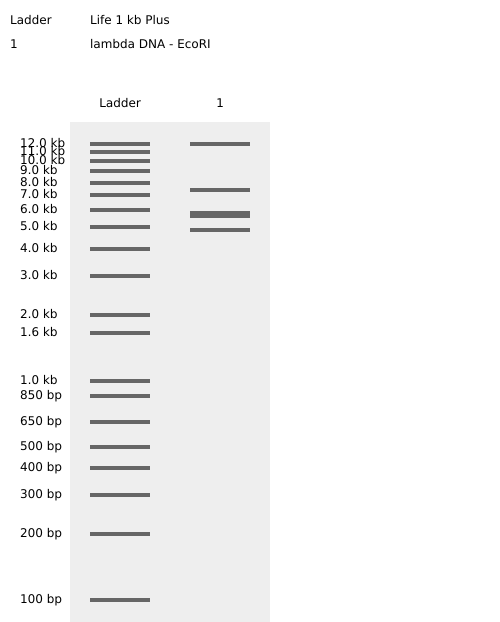
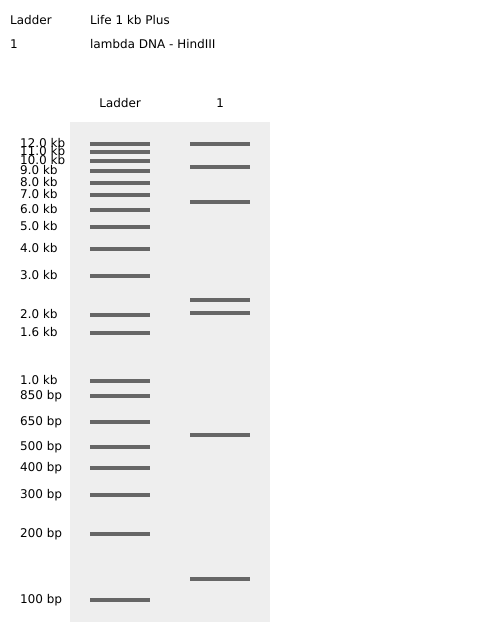
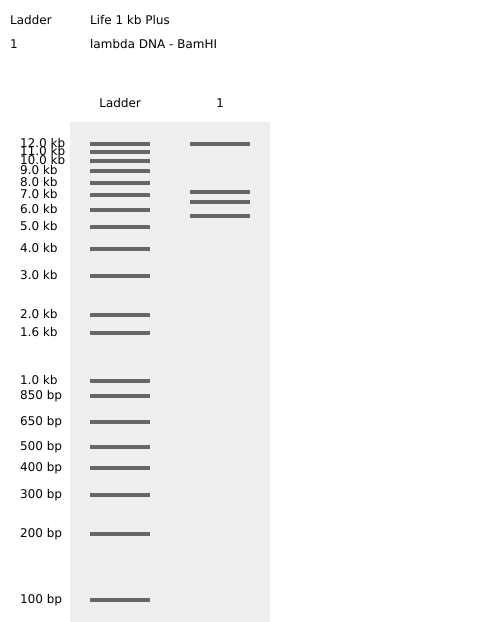
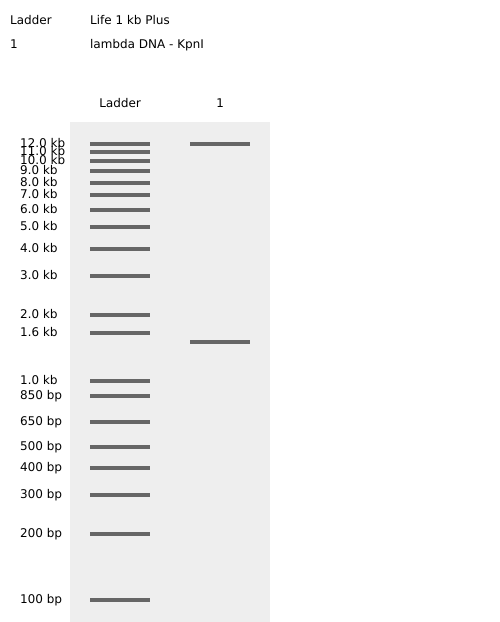
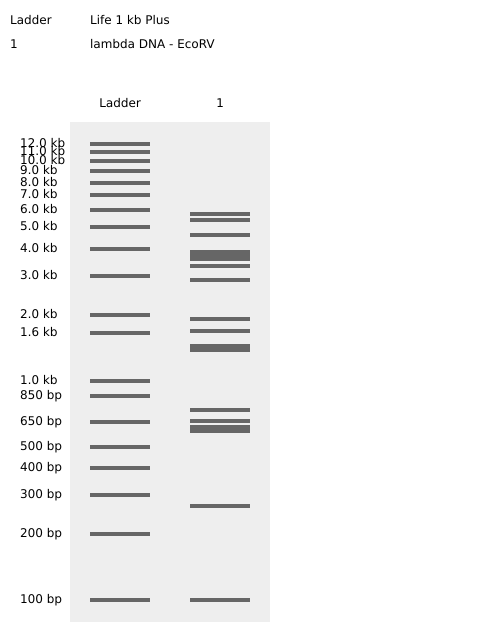
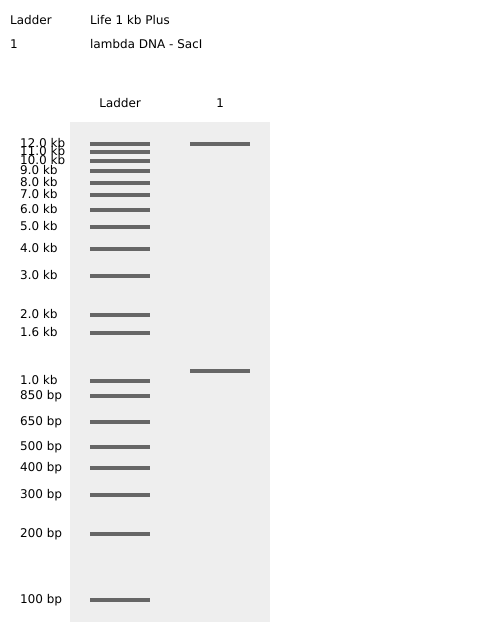
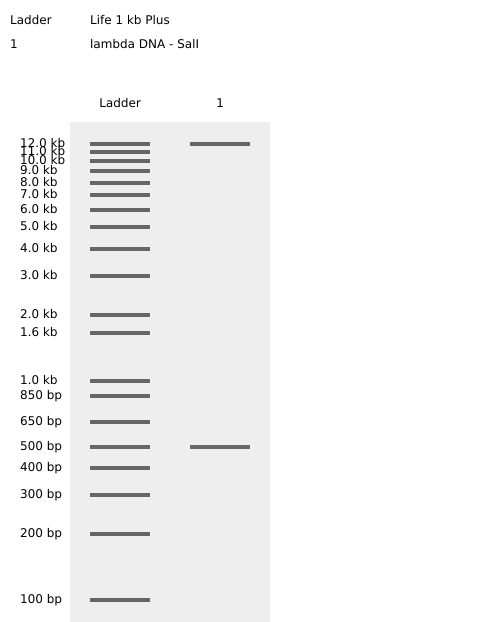 [/grid]"
[/grid]"
I couldn’t figure out how to use Ronan’s website other than the randomization button unfortunately. As a result, I went with a pretty simple smiley face design for my in-silico art.

Part 3: DNA Design Challenge
3.1 Protein
I chose PETase, a naturally occurring enzyme from Ideonella sakaiensis, which degrades poly(ethylene terephthalate) into monomers of mono-2-hydroxyethyl terephthalate.
sp|A0A0K8P6T7|PETH_PISS1 Poly(ethylene terephthalate) hydrolase OS=Piscinibacter sakaiensis OX=1547922 GN=ISF6_4831 PE=1 SV=1 MNFPRASRLMQAAVLGGLMAVSAAATAQTNPYARGPNPTAASLEASAGPFTVRSFTVSRP SGYGAGTVYYPTNAGGTVGAIAIVPGYTARQSSIKWWGPRLASHGFVVITIDTNSTLDQP SSRSSQQMAALRQVASLNGTSSSPIYGKVDTARMGVMGWSMGGGGSLISAANNPSLKAAA PQAPWDSSTNFSSVTVPTLIFACENDSIAPVNSSALPIYDSMSRNAKQFLEINGGSHSCA NSGNSNQALIGKKGVAWMKRFMDNDTRYSTFACENPNSTRVSDFRTANCS
3.2 Reverse translate
To reverse translate in Benchling, it asks what codon optimization scheme you want to use. For this initial DNA sequence, I just used Escherichia coli K12 as my organism, matching codon usage to the frequency found in the E. coli genome.
ATGAATTTTCCGCGCGCAAGTCGTTTAATGCAAGCGGCGGTACTGGGCGGCTTGATGGCAGTGTCGGCAGCTGCGACGGCTCAGACCAATCCGTATGCACGCGGTCCGAATCCAACCGCGGCCAGCCTGGAAGCATCCGCGGGTCCTTTTACTGTTCGAAGCTTCACAGTGAGCCGGCCGTCGGGCTATGGCGCTGGGACCGTGTATTATCCAACTAACGCGGGAGGCACCGTAGGTGCGATTGCTATCGTACCCGGCTACACAGCGCGTCAGTCCTCAATTAAATGGTGGGGCCCCCGCTTAGCGTCGCACGGTTTTGTTGTCATTACCATTGATACGAATAGTACCCTAGACCAACCATCGTCTCGTTCGTCTCAGCAGATGGCCGCGCTGCGCCAGGTTGCCAGCCTCAACGGCACGAGCTCATCTCCGATCTACGGTAAAGTCGATACGGCACGCATGGGCGTGATGGGATGGTCAATGGGCGGCGGTGGTAGTCTGATTAGTGCGGCGAATAACCCGTCTTTGAAAGCCGCCGCCCCGCAGGCCCCGTGGGATAGTAGCACAAACTTTTCCTCAGTTACTGTCCCGACCCTTATCTTCGCCTGTGAGAACGACTCCATTGCGCCTGTGAATAGCTCAGCCCTGCCGATATACGATTCAATGAGCCGTAATGCCAAGCAGTTTCTTGAAATCAATGGCGGAAGCCATAGCTGCGCAAACAGTGGGAATAGCAACCAAGCCCTGATTGGTAAAAAGGGGGTGGCGTGGATGAAACGCTTCATGGATAACGACACCAGGTACTCGACCTTCGCATGTGAAAACCCTAACAGCACGCGCGTGAGCGATTTTCGTACCGCCAACTGCTCG
3.3 Codon optimize
To practice codon optimization, I stayed within the Benchling tool. This time, I chose my potential host organism: Pseudomonas putida, a soil bacteria that is a fairly common chassis. This time, I selected to only use the best (most frequently occuring) codons, to theoretically improve expression. I also avoided BsaI cut sites, in case I want to use Golden Gate cloning with this construct.
ATGAACTTCCCGCGCGCCAGCCGCCTGATGCAGGCCGCCGTGCTGGGCGGCCTGATGGCCGTGAGCGCCGCCGCCACCGCCCAGACCAACCCGTACGCCCGCGGCCCGAACCCGACCGCCGCCAGCCTGGAGGCCAGCGCCGGCCCGTTCACCGTGCGCAGCTTCACCGTGAGCCGCCCGAGCGGCTACGGCGCCGGCACCGTGTACTACCCGACCAACGCCGGCGGCACCGTGGGCGCCATCGCCATCGTGCCGGGCTACACCGCCCGCCAGAGCAGCATCAAGTGGTGGGGCCCGCGCCTGGCCAGCCACGGCTTCGTGGTGATCACCATCGACACCAACAGCACCCTGGACCAGCCGAGCAGCCGCAGCAGCCAGCAGATGGCCGCCCTGCGCCAGGTGGCCAGCCTGAACGGCACCAGCAGCAGCCCGATCTACGGCAAGGTGGACACCGCCCGCATGGGCGTGATGGGCTGGAGCATGGGCGGCGGCGGCAGCCTGATCAGCGCCGCCAACAACCCGAGCCTGAAGGCCGCCGCCCCGCAGGCCCCGTGGGACAGCAGCACCAACTTCAGCAGCGTGACCGTGCCGACCCTGATCTTCGCCTGCGAGAACGACAGCATCGCCCCGGTGAACAGCAGCGCCCTGCCGATCTACGACAGCATGAGCCGCAACGCCAAGCAGTTCCTGGAGATCAACGGCGGCAGCCACAGCTGCGCCAACAGCGGCAACAGCAACCAGGCCCTGATCGGCAAGAAGGGCGTGGCCTGGATGAAGCGCTTCATGGACAACGACACCCGCTACAGCACCTTCGCCTGCGAGAACCCGAACAGCACCCGCGTGAGCGACTTCCGCACCGCCAACTGCAGC
3.4 Now what?
I optimized the codons for P. putida, so I would choose to express this in P. putida. I would probably put the gene onto an expression plasmid first, under a strong constitutive promoter, just to ensure it works. After transforming P. putida with the plasmid, I would test expression by looking at protein production with a Western blot, and also culturing with a sample of PET plastic, to check for degradation. Ultimately, I would want to integrate this gene into the genome of the bacteria, possibly under an inducible promoter for use in open-release plastic pollution bioremediation.
Part 4: Prepare a Twist DNA Synthesis Order
There is actually a way to use Twist’s expression vectors, so I wouldn’t have to design the whole expression cassette. For example, if I wanted to express my E. coli codon-optimized PETase gene in E. coli, I could select one of Twist’s pET expression vectors; in this case, I chose pET-blank(Kan). It has a T7 promoter and RBS already included in it, and lacO for inducible expression. I believe the host strain would need a T7 polymerase, but my PETase gene should be expressed in the presence of IPTG.
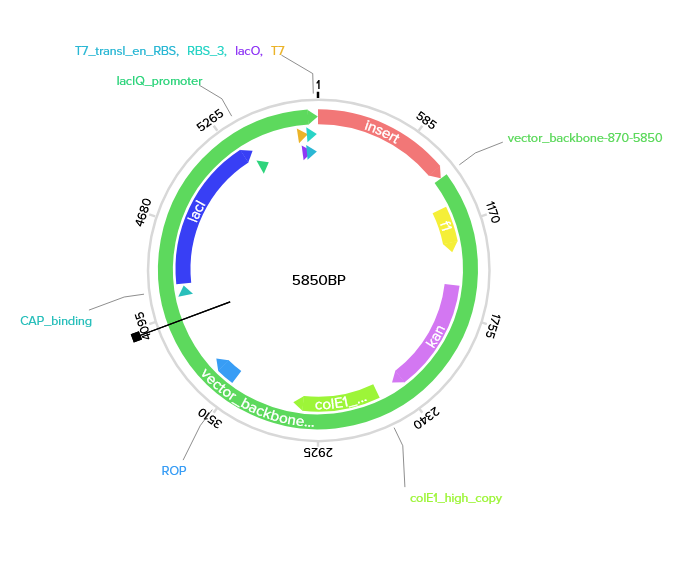
Part 5: DNA Read/Write/Edit
5.1 Read
- What DNA would you want to sequence (e.g., read) and why? I would want to sequence metagenomic 16S sequences from soil samples. This gives me a baseline for bacterial community structure (prior to engineered strain addition).
- In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? This would be Illumina sequencing probably, so second generation. This requires PCR amplification of the 16S variable region, adapter ligation, and then library pooling. The output would be many, many sequences that I would be able to compare to published 16S sequences to identify bacterial species present. I’d choose this method because it multiplexes better than Sanger sequencing, and it doesn’t need to be long-read like nanopore.
5.2 Write
- What DNA would you want to synthesize (e.g., write) and why? I would want to synthesize the CRISPR cassette for my kill-switch because it’s somewhat difficult and time-intensive to stitch together out of oligos.
- What technology or technologies would you use to perform this DNA synthesis and why? I’d order it from Twist because it has multiple internal repeats, and they’re one of the few companies with the technology to accomplish that.
5.3 Edit
- What DNA would you want to edit and why? I’d like to edit the P. putida genome to include PETase and MHETase genes, as well as a killswitch circuit for biocontainment to prevent unintended ecological effects during application.
- What technology or technologies would you use to perform these DNA edits and why? I’d like to use CRISPR-Cas9 because it is the most flexible when it comes to genomic integration location. I could identify a few good neutral sites for integration and design sgRNAs to target these locations. Then I could insert repair templates (including homology arms) onto vectors and transform those sequentially with the CRISPR-Cas9 plasmid (repair template should be first). The repair templates could either be wholly synthesized, or assembled through overlap PCR or Gibson assembly (primer design for homology arms and overlaps for assembly). I might need a antibiotic, fluorescent, or other marker to scan for initial transformation and also genomic integration post-plasmid loss - in that case, I would also need to consider a step to remove the marker.