Week 4 HW: Protein Design Part I
Contents
Part A: Conceptual Questions
Need to answer 9/11 questions; I skipped 7 and 11.
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
$$ 500g * \frac{1 mol AA}{100g} = 5 mol AA $$ $$ 5 mol * \frac{6.02*10^{23} molecules}{1 mol} = 3.01 E24 molecules $$ - Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We break down the proteins during digestion to the constituent amino acids. These amino acids are then used in our cells to build human proteins. - Why are there only 20 natural amino acids?
It’s been hypothesized that the 20 naturally occurring amino acids fairly effectively cover the “chemical space”, which would indicate that more complex or diverse amino acids are not needed for increasing function. This includes variation in chemical properties like molecular size, hydrophobicity, and charge, but also rotational conformations. These twenty sufficiently cover the space for effective function while also being relatively low in energy (easy to synthesize). Another paper hypothesizes that all twenty natural amino acids predate the RNA world, and in fact were naturally synthesized prebiotically with mineral catalysts - thus suggesting that the development of the three-base 64-codon alphabet actually was because a two-base 16-codon alphabet would restrict to sixteen instead of the existing 20 amino acids.- Doig, AJ. Frozen, but no accident – why the 20 standard amino acids were selected. 2017. FEBS J, 284: 1296-1305. doi: 10.1111/febs.13982
- Bywater RP. Why twenty amino acid residue types suffice(d) to support all living systems. 2018. PLoS One, 13(10):e0204883. doi: 10.1371/journal.pone.0204883
- Brazil, R. The alphabet soup of life: Why are there 20 amino acids? 2018. ChemistryWorld. https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- Can you make other non-natural amino acids? Design some new amino acids.
There are a new non-cannonical amino acids that people have designed and used, by changing the residue for an unnatural one. - Where did amino acids come from before enzymes that make them, and before life started?
In 2018, Bywater suggested that amino acids were synthesized prebiotically, with the simpler structures occurring through aqueous reactions, and more complex structures requiring mineral catalysts. Many amino acids have been identified on meteorites, suggesting that amino acids could have originated in outer space, but more likely that the conditions to synthesize the “simpler” amino acids exist in multiple places. Other researchers have suggested that the “complex” amino acids must have been biosynthesized by early proteins made up of “simple” amino acids, and in particular, that histidine, phenylalanine, cysteine, methionine, tryptophan and tyrosine had to come after molecular oxygen because they have redox functionality.
- Doig, AJ. Frozen, but no accident – why the 20 standard amino acids were selected. 2017. FEBS J, 284: 1296-1305. doi: 10.1111/febs.13982
- Bywater RP. Why twenty amino acid residue types suffice(d) to support all living systems. 2018. PLoS One, 13(10):e0204883. doi: 10.1371/journal.pone.0204883
- Brazil, R. The alphabet soup of life: Why are there 20 amino acids? 2018. ChemistryWorld. https://www.chemistryworld.com/features/why-are-there-20-amino-acids/3009378.article
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect D-amino acids would form a left-handed helix because L-amino acids form right-handed helices. Can you discover additional helices in proteins?- Why are most molecular helices right-handed?
In general, naturally occuring amino acids are L-enantiomers, which leads to right-handed helices because of steric hindrance requiring the side chains to point outwards. - Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Because beta sheets are flat, they can stack, and the large surface area means that the side-chains can have interactions (especially hydrophobic side-chains) between the sheets. - Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloids are ordered protein aggregates consisting of repeating beta sheet motif. Proteins that have an alternative folding structure with a lot of beta sheets become amyloids when they self-assemble into fibrils, and the alternative conformation with the beta sheets is energetically stable. Amyloid diseases usually are from a single amyloid-forming protein. Because of their tendency to self-assemble, I think you could use amyloid beta sheets as materials for DNA origami.- Riek R. The Three-Dimensional Structures of Amyloids. 2017. Cold Spring Harb Perspect Biol;9(2):a023572. doi: 10.1101/cshperspect.a023572.
- Ow SY, Dunstan DE. A brief overview of amyloids and Alzheimer’s disease. 2014. Protein Sci;23(10):1315-31. doi: 10.1002/pro.2524.
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I chose PhaC from Cupriavidus necator. PhaC is a polyhydroxyalkanoate-synthase, used in biopolymer production. I selected it because engineering PhaC is one of my potential final projects. The C-terminal domain is believed to be the catalystic domain, and it has a solved crystal structure. The N-terminal domain does not have a solved crystal structure, and is believed to potentially be involved in substrate specificity.Identify the amino acid sequence of your protein. \
5HZ2_1|Chain A|Poly-beta-hydroxybutyrate polymerase|Cupriavidus necator (381666) AFEVGRNVAVTEGAVVFENEYFQLLQYKPLTDKVHARPLLMVPPCINKYYILDLQPESSLVRHVVEQGHTVFLVSWRNPDASMAGSTWDDYIEHAAIRAIEVARDISGQDKINVLGFCVGGTIVSTALAVLAARGEHPAASVTLLTTLLDFADTGILDVFVDEGHVQLREATLGGGAGAPCALLRGLELANTFSFLRPNDLVWNYVVDNYLKGNTPVPFDLLFWNGDATNLPGPWYCWYLRHTYLQNELKVPGKLTVCGVPVDLASIDVPTYIYGSREDHIVPWTAAYASTALLANKLRFVLGASGHIAGVINPPAKNKRSHWTNDALPESPQQWLAGAIEHHGSWWPDWTAWLAGQAGAKRAAPANYGNARYRAIEPAPGRYVKAKALQHHHHHH
- How long is it? What is the most frequent amino acid?
390 amino acids (when i removed the His-tag at the end). Most frequent amino acid is A (alanine). - How many protein sequence homologs are there for your protein?
BLAST found 250 sequence homologs - mostly belonging to other bacteria that biosynthesize PHAs. - Does your protein belong to any protein family?
It’s classified as a transferase.
Identify the structure page of your protein in RCSB
C. necator PhaC (C-terminal domain) has been uploaded to RCSB PDB here.- When was the structure solved? Is it a good quality structure?
The structure was solved in 2016 by two different and unrelated groups, which is a good sign for repeatability (PDB 5HZ2 and 5T6O). It has a resolution of 1.8Å, which is a good quality structure. - Are there any other molecules in the solved structure apart from protein?
Yes, there is a sulfate ion and a glycerol molecule. - Does your protein belong to any structure classification family?
Nothing that I could find on SCOP.
- When was the structure solved? Is it a good quality structure?
Open the structure of your protein in any 3D molecule visualization software:
I used the structure viewer on the PDB website because I wasn’t able to download PyMol on my laptop (not enough memory space).Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
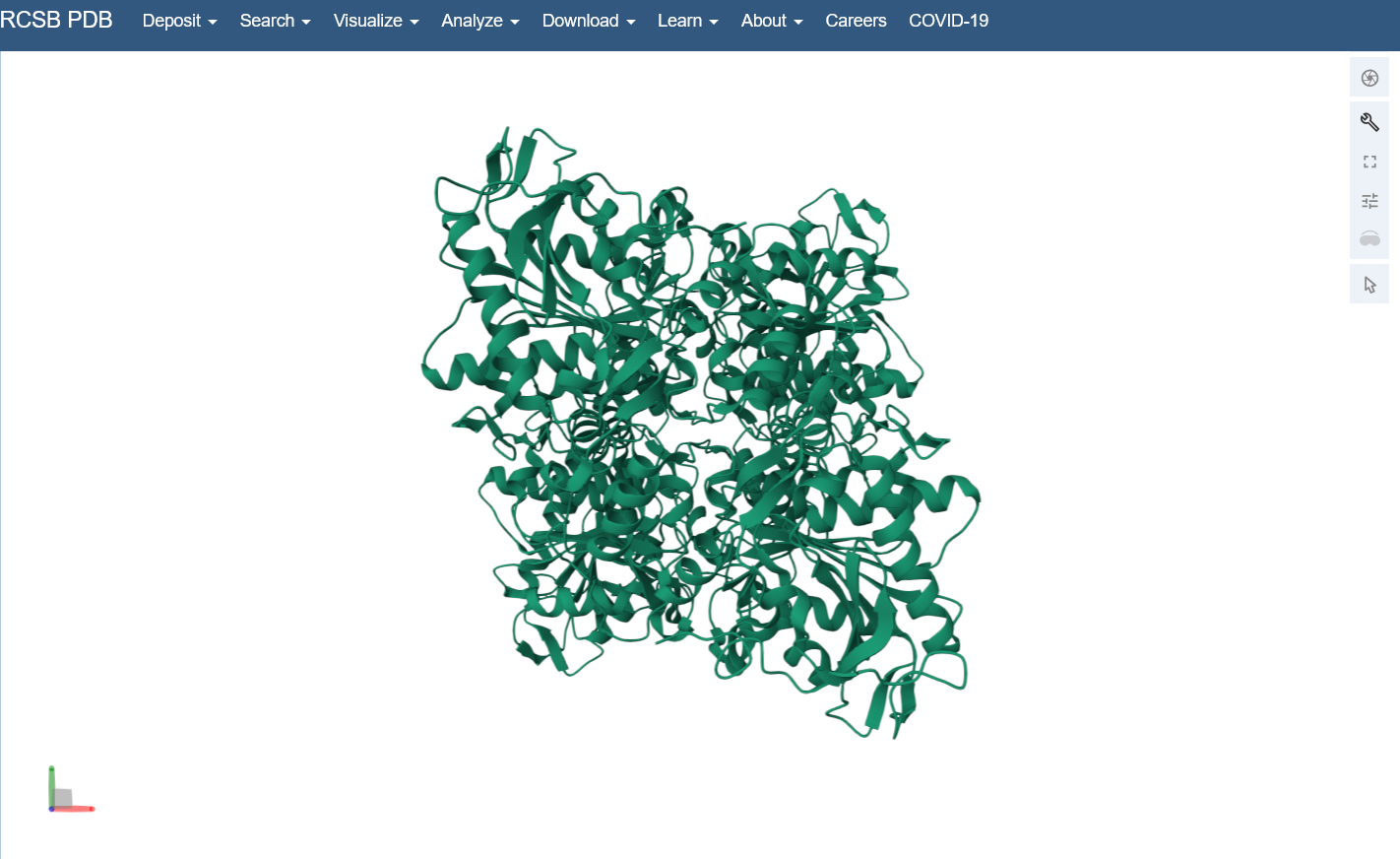
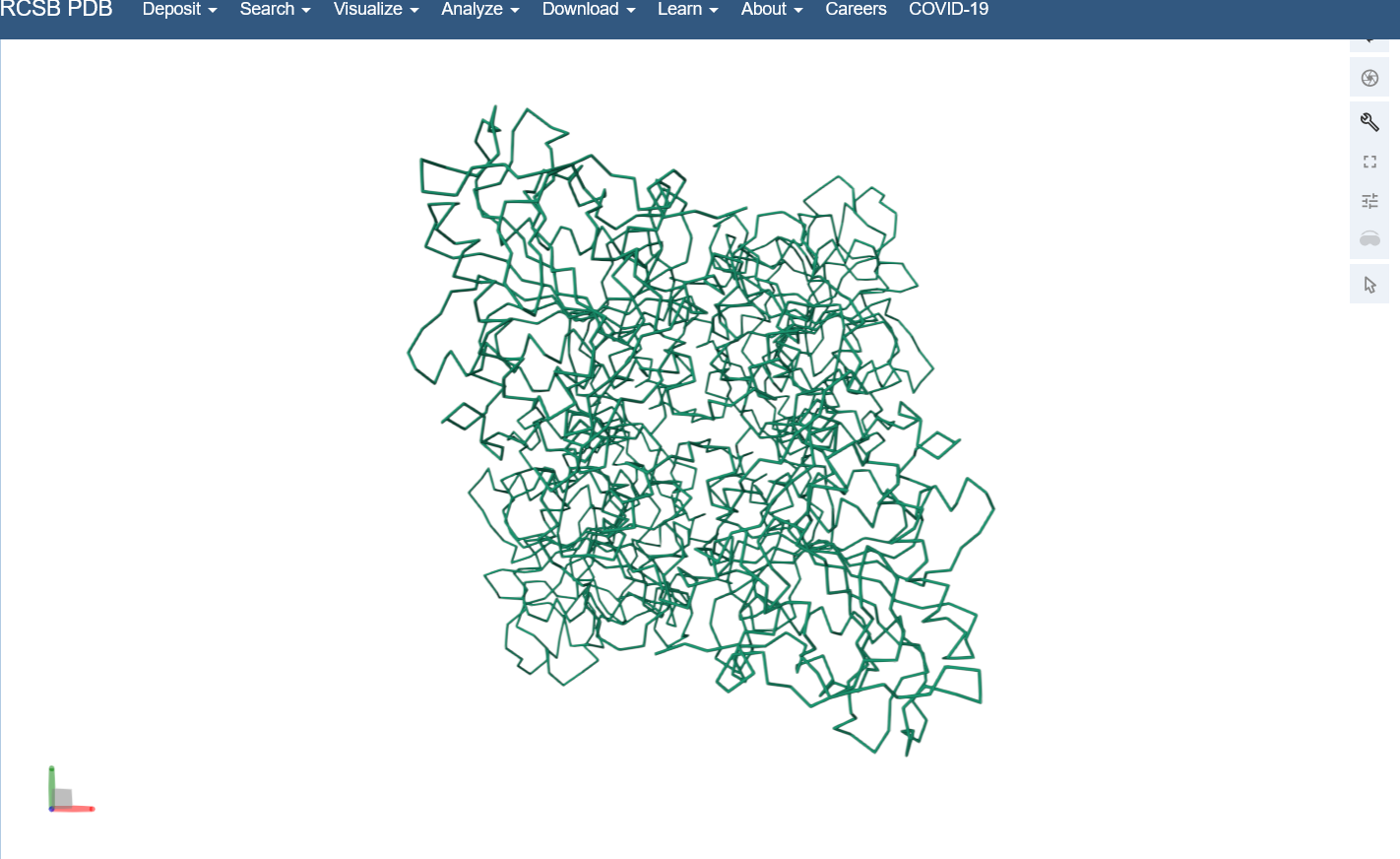

Color the protein by secondary structure. Does it have more helices or sheets?
I think it looks like it has more helices.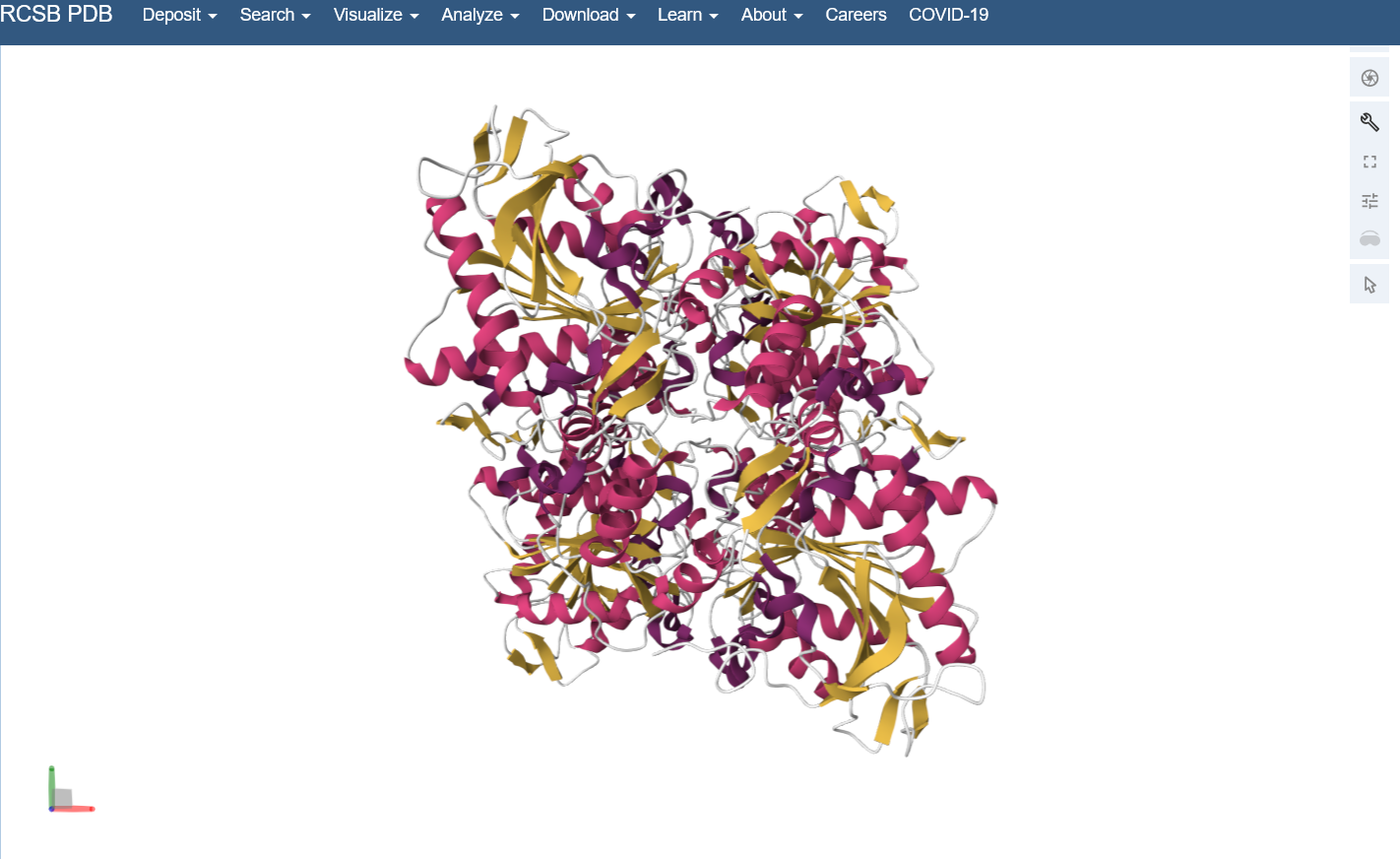
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I colored by hydrophobicity of residue in the PDB structure viewer, because it was all one color when I selected color by residue molecule type. Not sure what was up with that, but I figured hydrophobicity would let me look at the hydrophobic vs hydrophilic residues. The hydrophobic residues are more clustered towards the insides of the structure.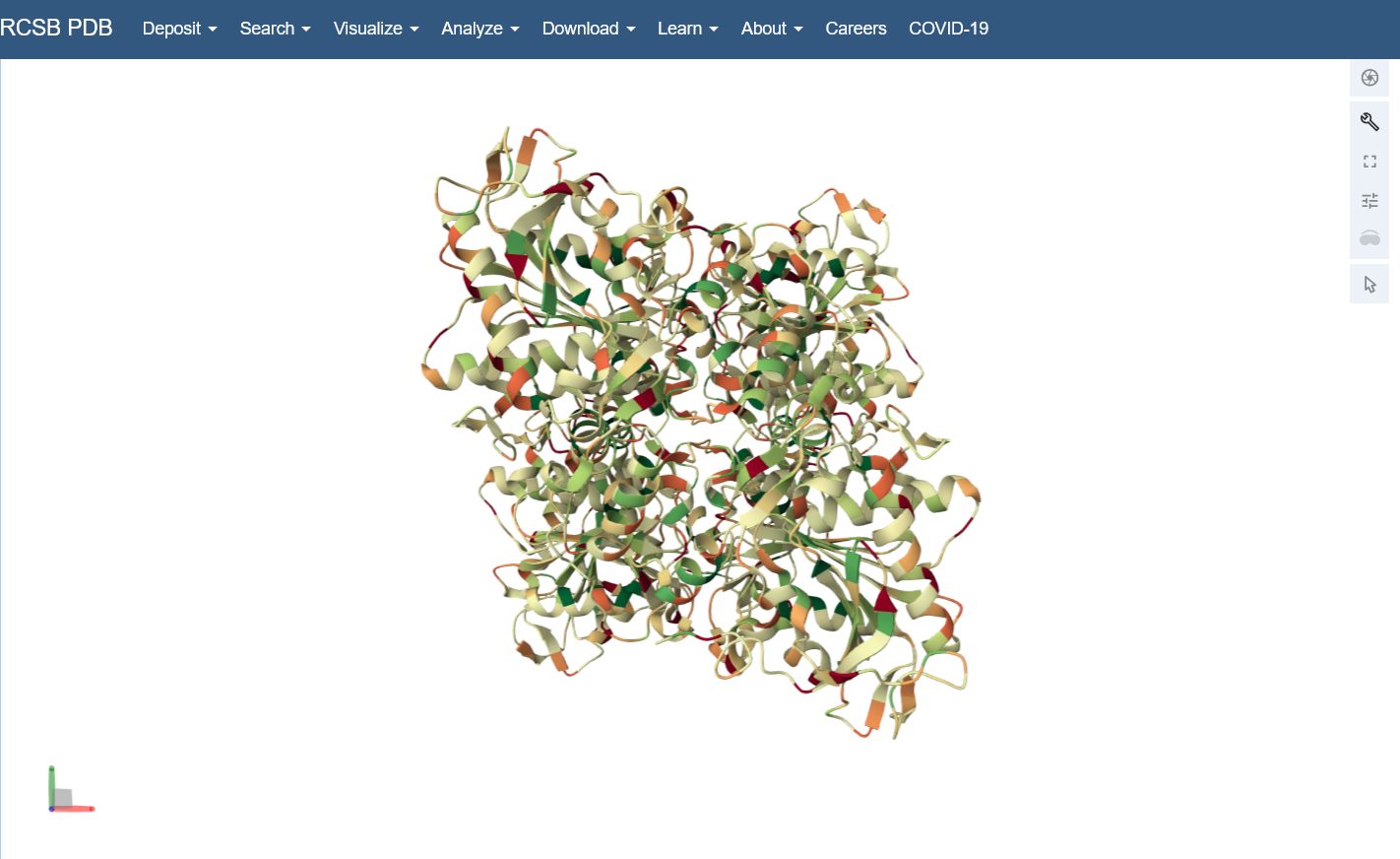
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes, you can kind of see the indentation in the center of the screenshot below.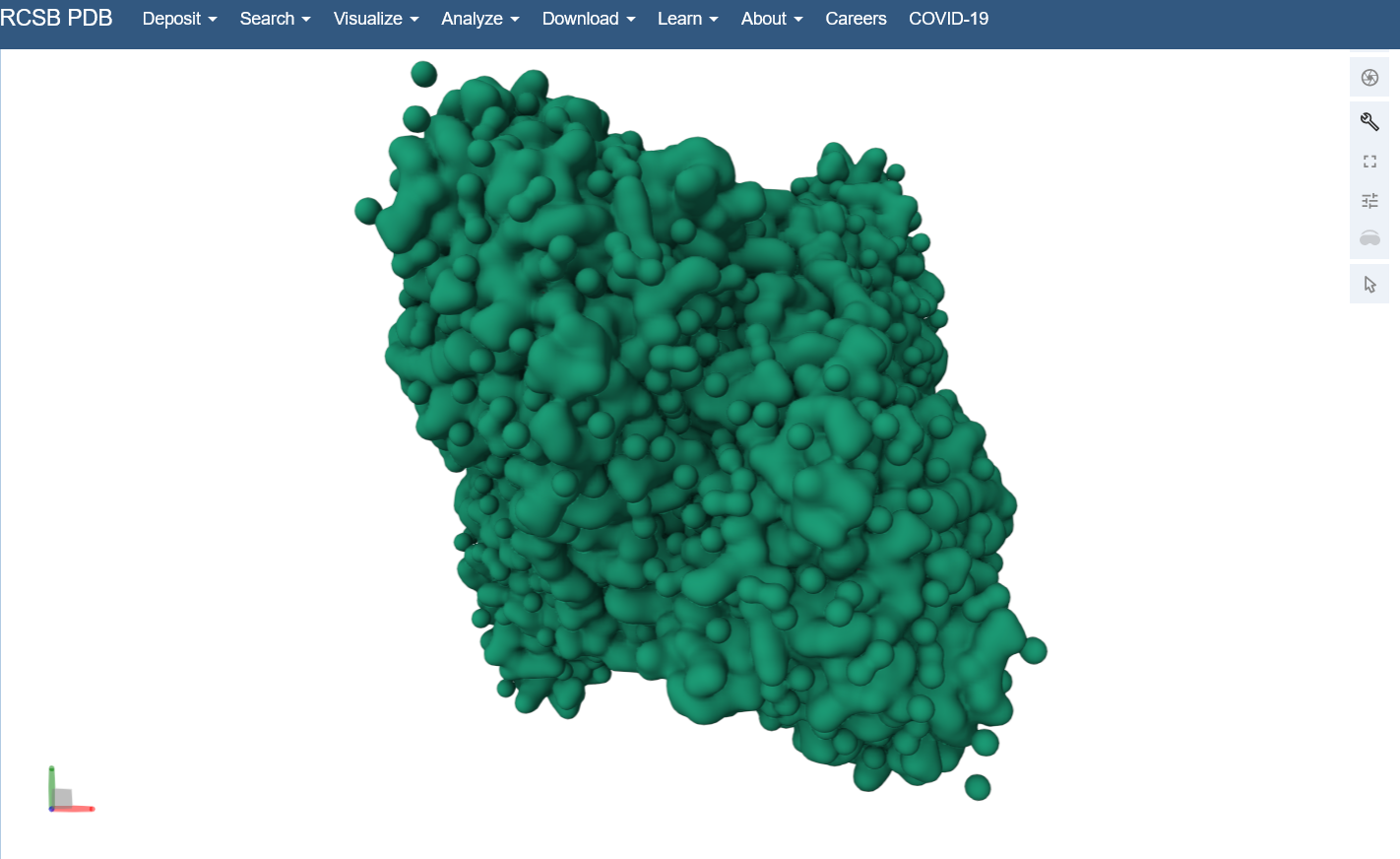
Part C: Using ML-based Protein Design Tools
I’m continuing with the C-terminal domain of PhaC, 5HZ2 in PDB. Colab notebook.
C1. Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
I copied the FASTA protein sequence from PDB into the first line of cell3 of the Colab notebook replacing the string labeled “protein_sequence”.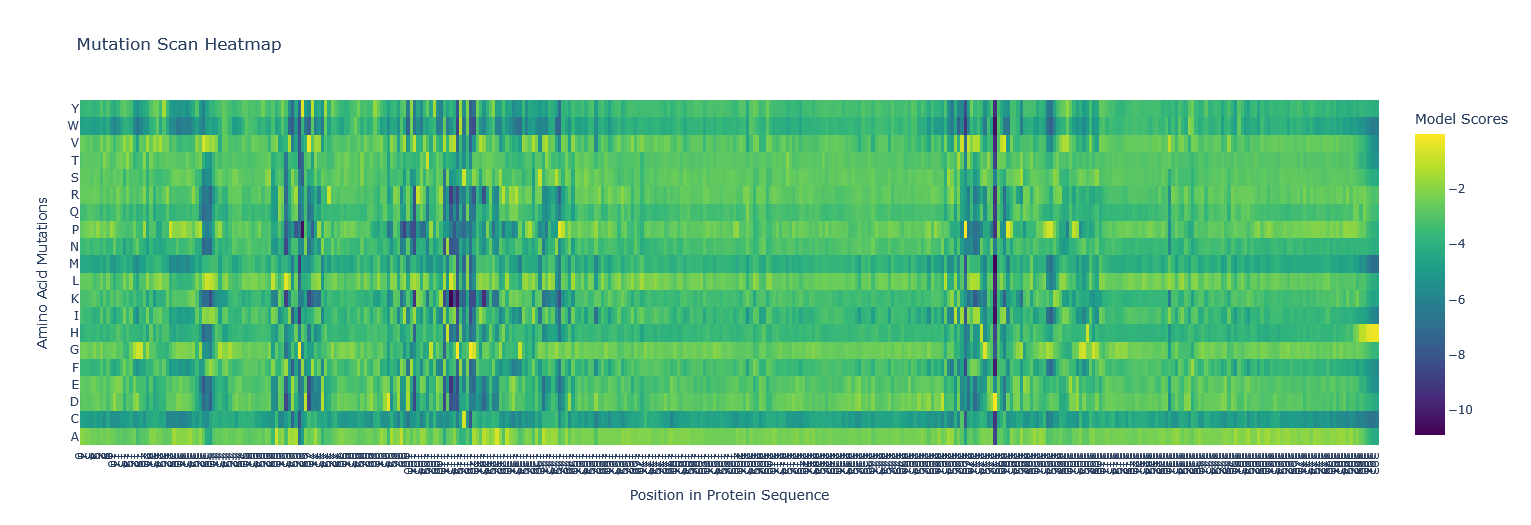
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Position 277 seems important - Aspartic acid is the only yellow/high score. Everything else is mostly dark blue, so very negative, which I think means not likely to be able to mutate. So likely, this is either important structurally or catalytically. Asp is one of the few charged amino acids, so that makes me think it might be catalytic.
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
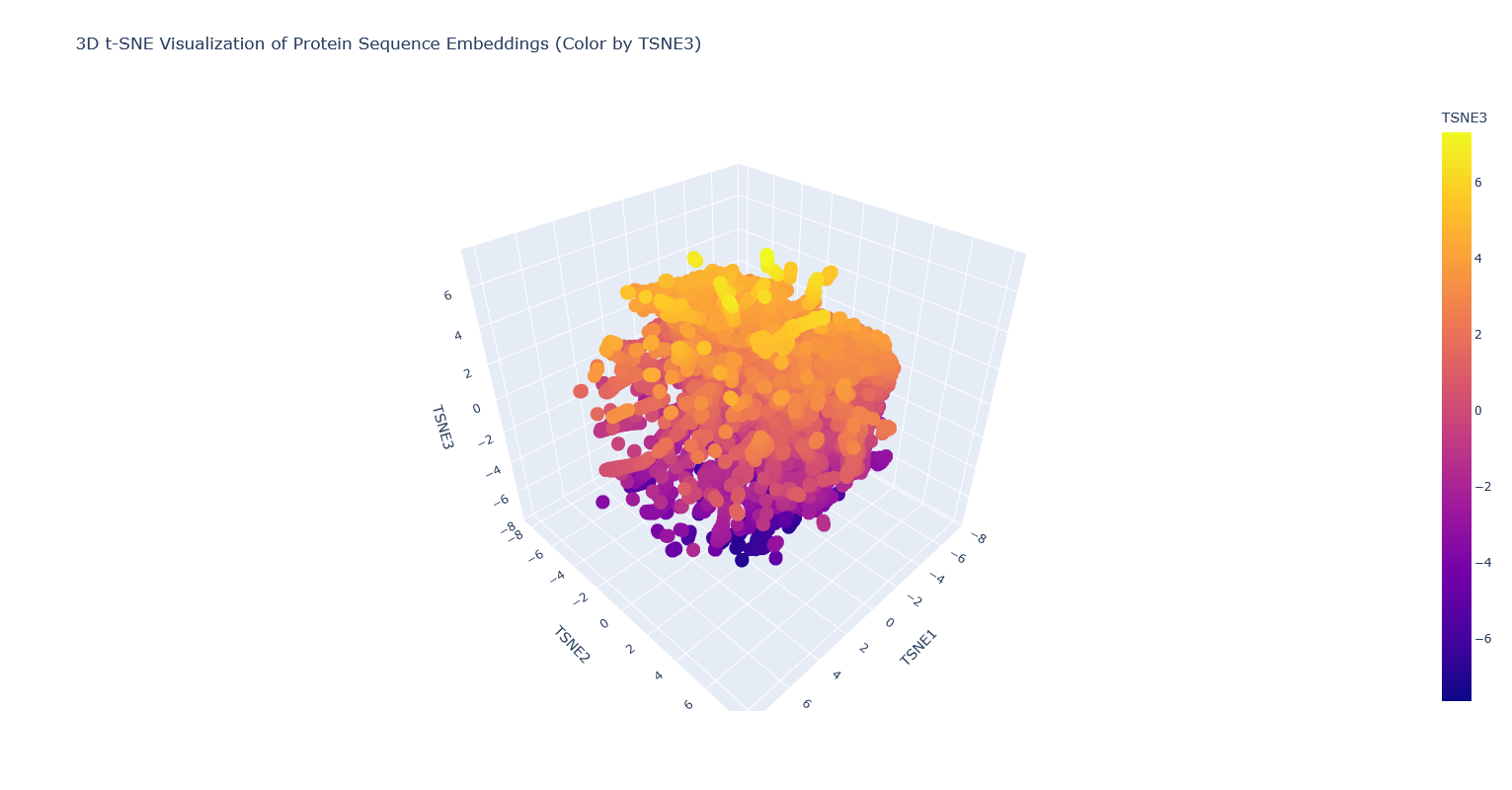
- Analyze the different formed neighborhoods: do they approximate similar proteins?
I think they probably mostly do, but it’s kind of hard to tell, because there are so many proteins that it’s hard to visually see which are clustered vs overlapping clusters, and also many of the proteins are just labeled “automated matches” which isn’t really helpful for identification. - Place your protein in the resulting map and explain its position and similarity to its neighbors.
It’s nearest to a lipase, a few esterases/thioesterases, and some acetyl-transferases. These are all also from bacteria. I think this makes sense, because these are all kind of involved in biosynthesis of (sometimes long) carbon-containing molecules.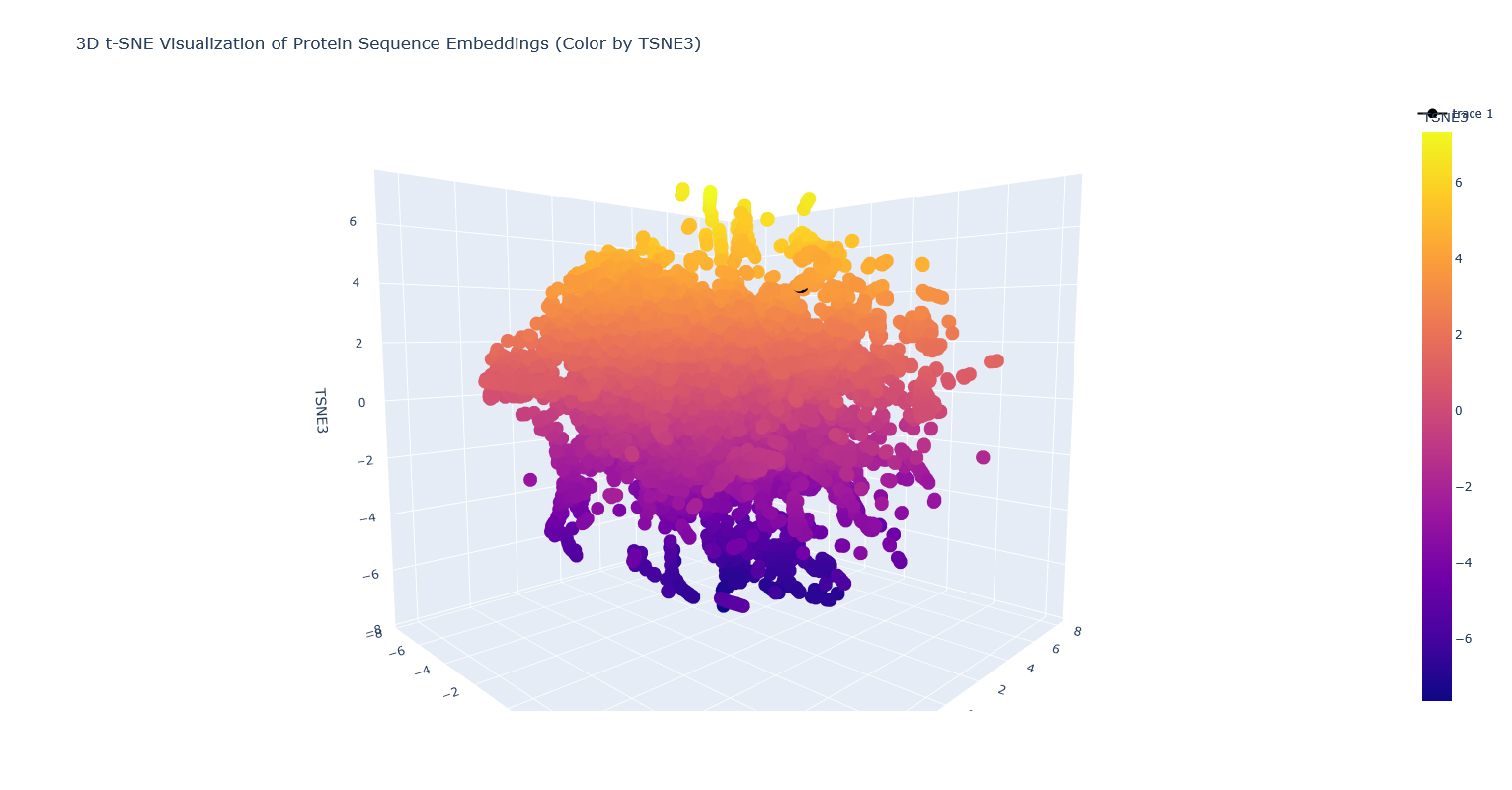 Note: PhaC is the partially covered black dot surrounded by orange-yellow dots.
Note: PhaC is the partially covered black dot surrounded by orange-yellow dots.
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
Code for visualization: New cell after cell53 of the Colab. i wrote the following code based off existing Python knowledge, and mostly looking at the prior couple cells.
Then ran former cell 54 (currently cell 55 since i added a new one) as usual. Separated out the visualization generation code into a separate cell. Ran the initial dataframe creation. Made a new cell to confirm what my sequence descriptor was:
Then visualized with the following code in a single cell. The chunk that was added is after the fig_3d.update_layout and before fig_3d.show(). This chunk was adapted from the bit that was posted by Noureldin Rihan on the Discourse forum.
C2. Protein Folding
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
This looks like a smaller and less intricate structure than the solved structure. I’m not sure what’s up with that.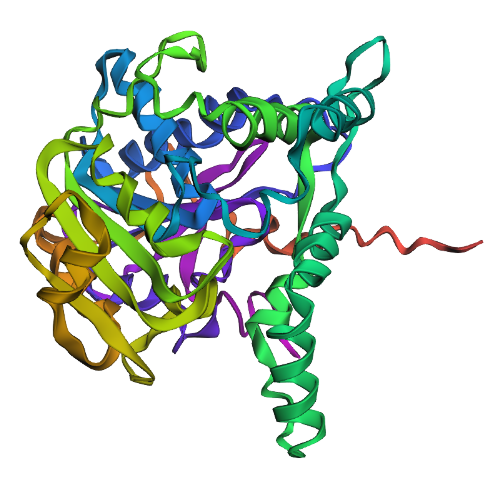
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I replaced all the Es with Ds and removed the His-tag at the end of the sequence. This yielded the following structure: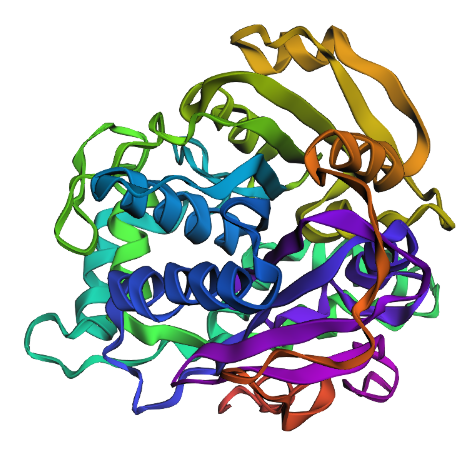 I think it looks similar. So at least with the small mutations it’s resilient. larger mutations probably not.
I think it looks similar. So at least with the small mutations it’s resilient. larger mutations probably not.
C3. Protein Generation
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The output from the third cell after the Inverse Folding with MPN heading:
Based on the heatmap, it has far less flexibility in sequence than the original.
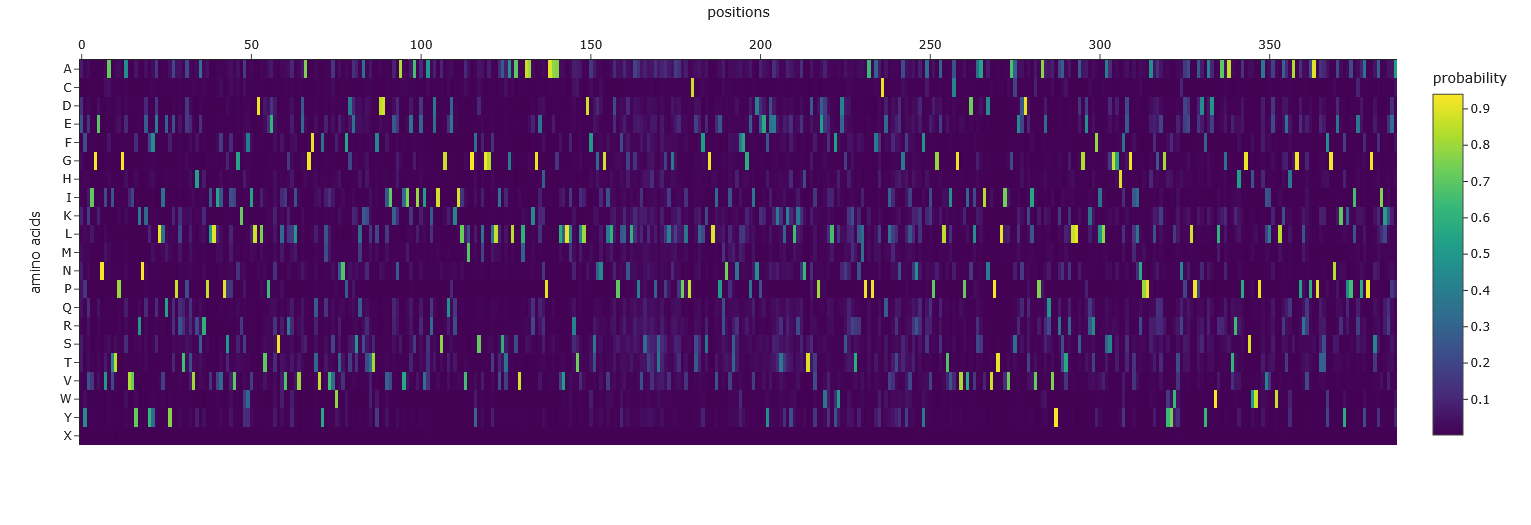
Then after the heatmap cell, there was the last cell that gave a different output that also looked like a predicted sequence, so I’m unclear which one we should look at:
- Input this sequence into ESMFold and compare the predicted structure to your original.
Replacing the original 5HZ2 protein sequence with the new sequence from the last cell into ESMFold (cell 54) gives us this predicted structure below. Which I guess looks kind of similar to the original predicted structure, but still to me does not look like the PDB structure. The new sequence doesn’t have a His-tag at the end, but it does kind of look like it has a linear tail like a His-tag, which is neat.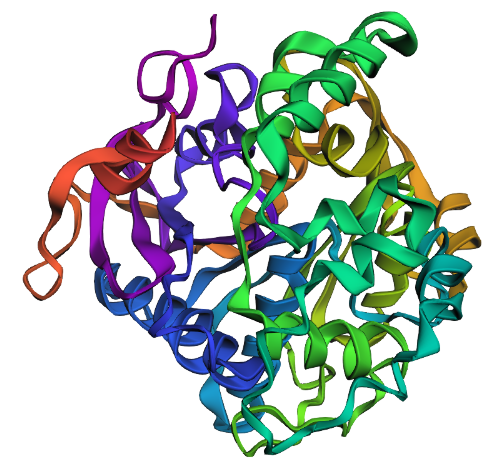
Part D: Group Brainstorm on Bacteriophage Engineering
What do we know:
- E. coli DnaJ binds to denatured proteins to prevent/disassemble aggregates (native function in heat-shock).
- DnaJ binds to the hydrophilic tail of MS2-L protein.
- point mutation of highly conserved proline in DnaJ results in no lysis (so maybe no more binding of MS2-L tail?)
- removal of MS2-L tail recovers lysis function (meaning DnaJ is only necessary when tail exists)
- suggests hydrophilic tail aggregates in some way that prevents lysis except in presence of DnaJ to stop aggregation
- so stability should be improved if we can figure out how the tail is interacting with the tail of other MS2-L molecules, and then mutating that away so there is no aggregation and dependence on DnaJ
graph TB; A[sequence and structure of MS2-L] -->|if geometry and chemical interactions are known| B[view interactions between MS2-L copies] A -->|if geometry and interactions are not known| C[model interactions with AlphaFold or something that can do protein interactions] B -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions] C -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions] D -->|use knowledge of hydrophobicity/charge/etc. OR use ESM2 mutational scan and select ones that it finds unlikely| E[Select dissimilar AAs to substitute in interacting residues] E -->|AlphaFold or similar| F[model protein folding in new AA sequence with selected mutations] F -->|something that can model protein interactions| G[model interactions between mutant MS2-L copies] G -->|select mutations that have similar hydrophilicity as original tail but less interaction with each other and maybe also with DnaJ| H[test mutations in lab]
Potential problems:
- don’t know what can model protein-protein interactions
- we might have covered this in class but i don’t remember. i can rewatch the lectures
- what if modeling doesn’t show interactions between the tails? we know there probably has to be one…
- might have to simplify by only modeling the tail section, but that is probably known already (will have to model folding and interactions with full protein sequence in later steps probably)
- could start with DnaJ, what in MS2-L binds with the essential proline in DnaJ, and assume that it’s spatially close to that. then test various mutations of nearby residues