Week 5 HW: Protein Design Part II
Contents
Part A: SOD1 Binder Peptide Design (from Pranam)
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
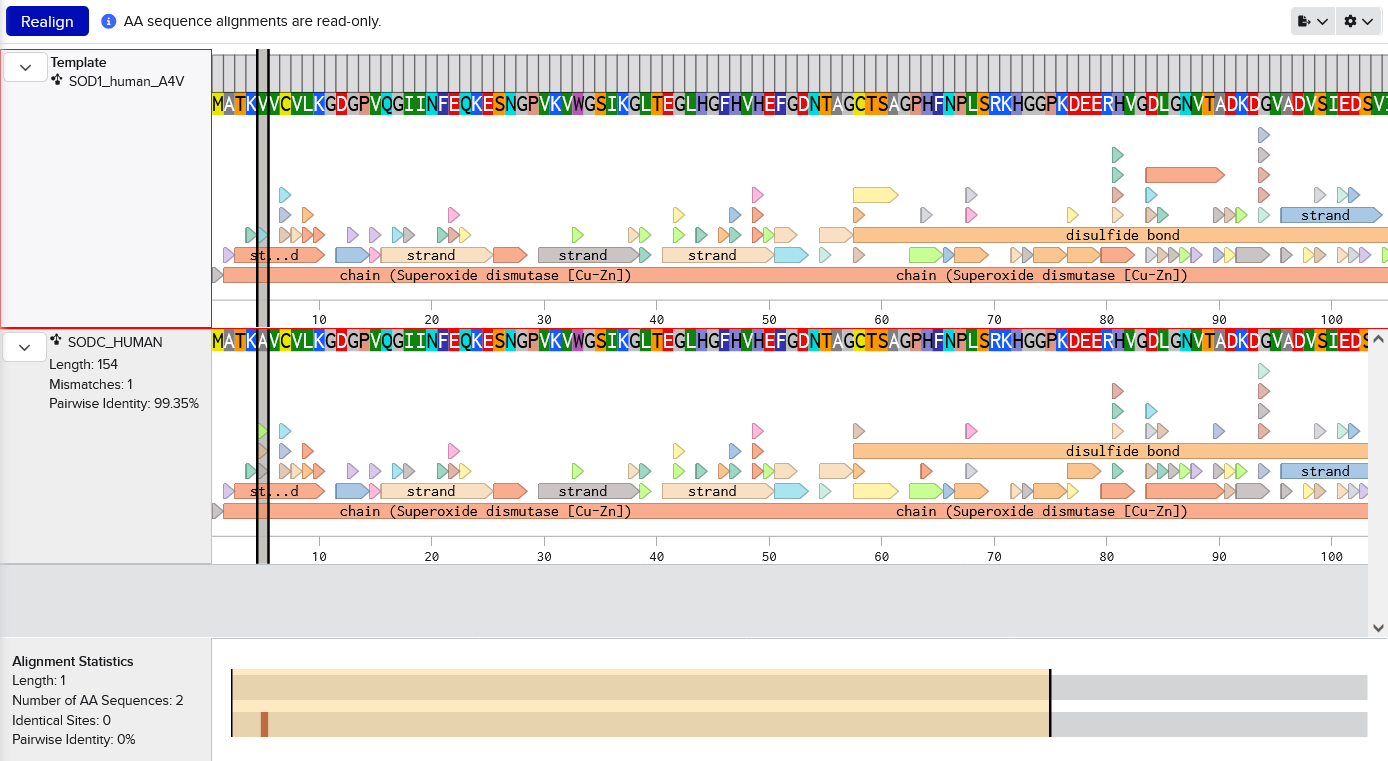
Imported from Uniprot into Benchling. Manually changed A at residue 5 to V (because this sequence includes the starting M which is not traditionally counted, I assume). Screenshot shows the mutation by aligning with the original sequence.MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Colab
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. These binders are from couple different runs because each run gives me one or more binders that contain amino acid single letter code X, which AlphaFold can’t handle because it’s non-standard.
- KRVYVVAVEHWE
- WLVPAVVLEWKK
- WRYYVAGLRWKE
- WRYYAAGARHGE
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
index Binder Pseudo Perplexity 1 KRVYVVAVEHWE 31.639343 2 WLVPAVVLEWKK 14.543342 3 WRYYVAGLRWKE 20.310199 4 WRYYAAGARHGE 9.566312 5 FLYRWLPSRRGG
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
index Binder Pseudo Perplexity ipTM score Localization 1 KRVYVVAVEHWE 31.639343 0.31 Within the beta-barrel, but not near the N-terminus. 2 WLVPAVVLEWKK 14.543342 0.34 Partially within the beta-barrel, partially within the more disordered region. Not near the N-terminus. More on the surface of the barrel, but a little buried within the disordered region. 3 WRYYVAGLRWKE 20.310199 0.29 Adjacent to the beta-barrel, but not near the N-termins. On the surface, possibly sterically interfering with the barrel because it’s an alpha-helix rather than linear. 4 WRYYAAGARHGE 9.566312 0.42 On top of beta-barrel, with one end somewhat near the N-terminus. On the surface of the barrel. 5 FLYRWLPSRRGG 0.30 In disordered region, not near the N-terminus or the beta-barrel. On the surface. AlphaFold peptide 1, highlighted residue is A4V.
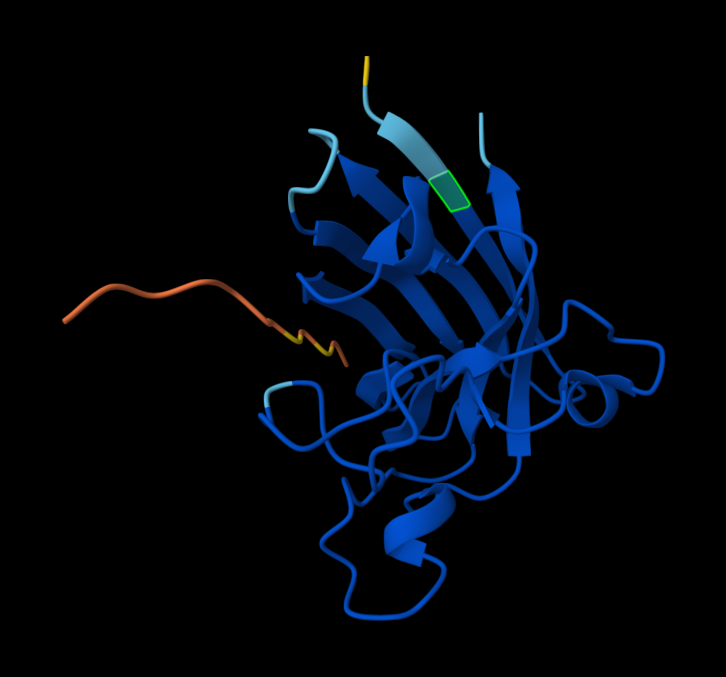
AlphaFold peptide 2, highlighted residue is A4V.
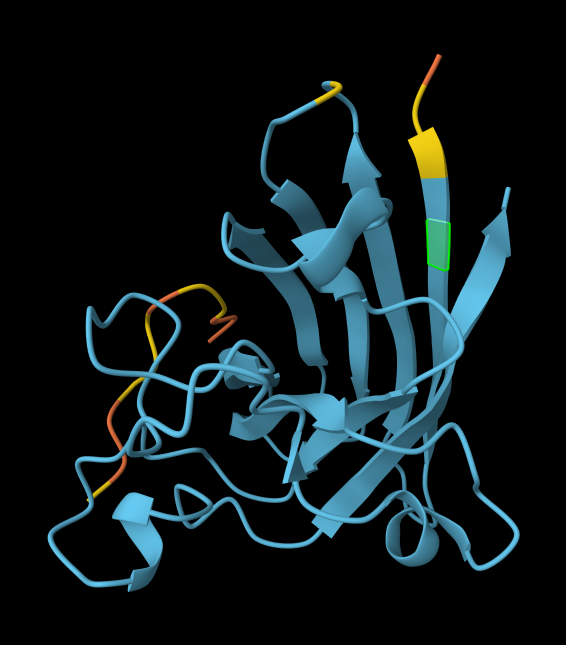
AlphaFold peptide 3, highlighted residue is A4V.
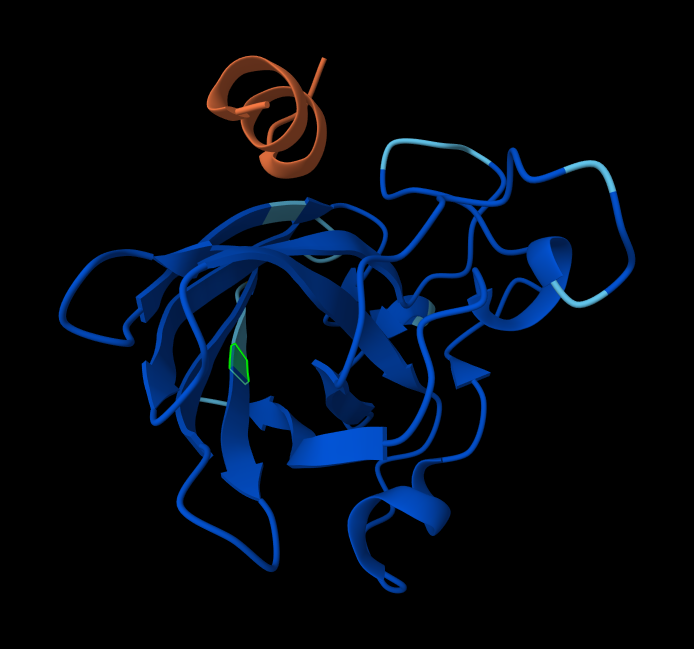
AlphaFold peptide 4, highlighted residue is A4V.
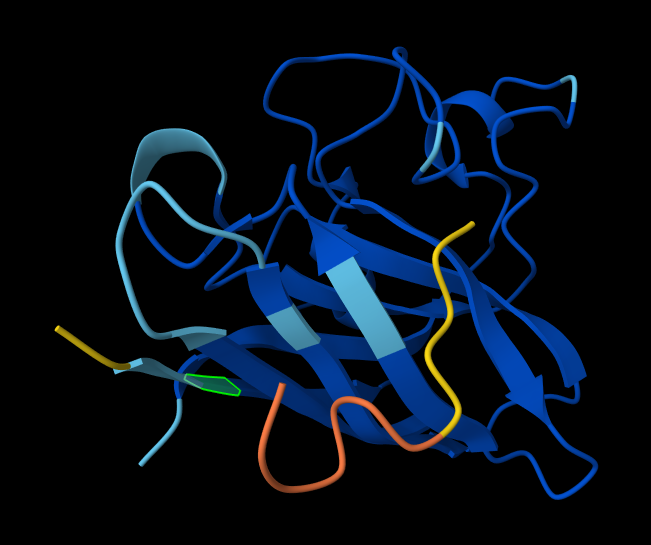
AlphaFold known peptide, highlighted residue is A4V.
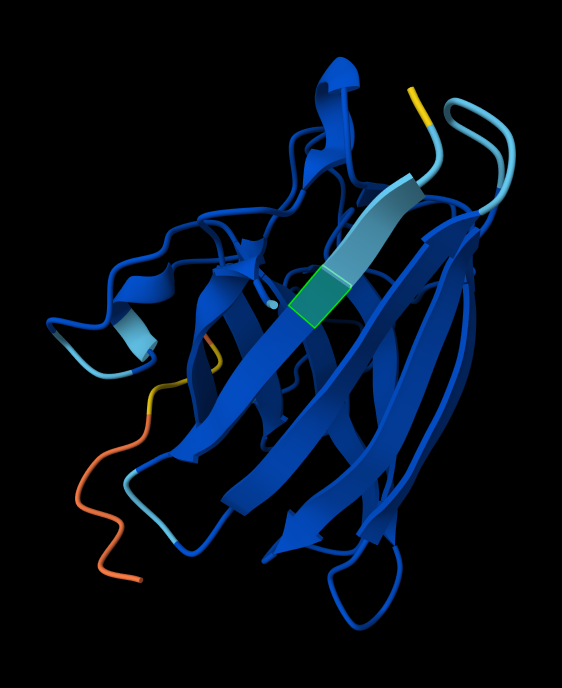
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Three of my 4 peptides have ipTM values above the known binder. Even my one peptide that has a lower value is almost the same (0.29 vs 0.3). Three of my peptides have very similar values, but one standout is much higher (0.42 vs 0.3). This would suggest that at least that peptide, if not all of them, is worth pursuing further.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes: Predicted binding affinity; Solubility; Hemolysis probability; Net charge (pH 7); Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
index Binder Pseudo Perplexity ipTM score Binding affinity 1 KRVYVVAVEHWE 31.639343 0.31 6.739 2 WLVPAVVLEWKK 14.543342 0.34 6.450 3 WRYYVAGLRWKE 20.310199 0.29 6.637 4 WRYYAAGARHGE 9.566312 0.42 6.401 5 FLYRWLPSRRGG 0.30 6.361
Actually for my peptides, the higher ipTM scores tend to have lower binding affinities predicted by PeptiVerse. The highest ipTM score was 0.42 from peptide 4 - it had the lowest pseudo perpexity score and one of the lower binding affinities. The second lowest ipTM score was 0.31 from peptide 1 - it had the highest psueo perplexity score and the highest binding affinity. The known peptide had a similar binding affinity as the rest of my peptides: 6.361. It’s actually lower than two of them and pretty close but slightly lower than the other two.
Peptide 1:
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 0.549 | Probability |
| Hemolysis | Non-hemolytic | 0.099 | Probability |
| Binding affinity | Weak binding | 6.739 | pKd/pKi |
| Net charge (pH 7) | -0.14 |
Peptide 2:
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 0.904 | Probability |
| Hemolysis | Non-hemolytic | 0.091 | Probability |
| Binding affinity | Weak binding | 6.450 | pKd/pKi |
| Net charge (pH 7) | 0.76 |
Peptide 3:
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 0.598 | Probability |
| Hemolysis | Non-hemolytic | 0.052 | Probability |
| Binding affinity | Weak binding | 6.637 | pKd/pKi |
| Net charge (pH 7) | 1.77 |
Peptide 4:
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 0.982 | Probability |
| Hemolysis | Non-hemolytic | 0.023 | Probability |
| Binding affinity | Weak binding | 6.401 | pKd/pKi |
| Net charge (pH 7) | 1.85 |
Known peptide:
| Property | Prediction | Value | Unit |
|---|---|---|---|
| Solubility | Soluble | 0.608 | Probability |
| Hemolysis | Non-hemolytic | 0.047 | Probability |
| Binding affinity | Weak binding | 6.361 | pKd/pKi |
| Net charge (pH 7) | 2.76 |
Choose one peptide you would advance and justify your decision briefly.
I’d probably choose either peptide 1 or peptide 4.
- Peptide 1: has the highest pseudo complexity score. It has a similar ipTM as the known peptide, and a higher binding affinity. It also has good solubility, hemolysis, and charge predictions. AlphaFold predicted it to be within the beta-barrel.
- Peptide 4: has the lowest pseudo perplexity score. It has a higher ipTM than the known peptide, and a similar binding affinity. It also has good solubility, hemolysis, and charge predictions. AlphaFold predicted it to be near the N-terminus.
I’d move forward with peptide 4 because of it has similar properties as the known peptide, but has possible binding location near the A4V mutation.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card. Colab
Make a copy and switch to a GPU runtime: T4 GPU runtime
In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
I chose the first 10 residues, roughly centered around the A4V mutation. - Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
index Binder Binding affinity Hemolysis Solubility 1 CTRDYPVCRACR 7.1381 0.0499 1.0000 2 ACRGRRFAFFRV 6.8598 0.0189 1.0000 3 GSRRWWVYWHWR 7.5707 0.0225 1.0000 4 VWAAIWRREYGK 6.4160 0.0222 1.0000
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
These peptides are different from the PepMLM peptides. I’d go through the same process I did with the PepMLM peptides to evaluate these peptides: modeling with AlphaFold and and evaluate with PeptiVerse.
Part C: Final Project: L-Protein Mutants
We didn’t get to this part of the project unfortunately. But we did have some planning discussion.
My assumption was that DnaJ stabilizes the L-protein by preventing aggregation that would otherwise occur with the long tail.
Peter suggested:
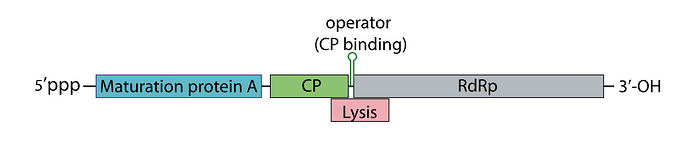
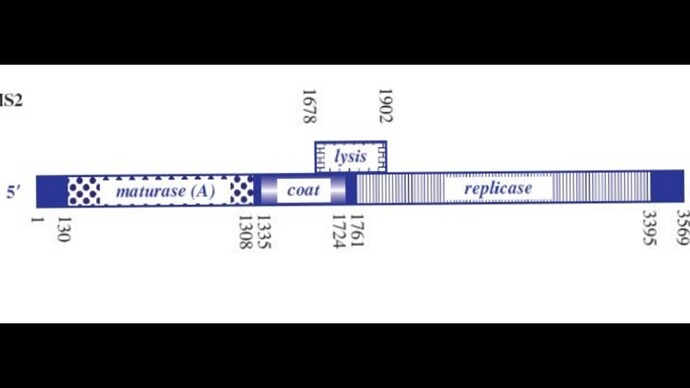
Sooo, the phage genome is very tightly regulated, I decided to take a look on how this regulation work, and it’s mainly based on RNA secondary structures How the lysis protein is regulated: The start codon and the shine-Dalgarno sequence are buried in an RNA hairpin, rendering virtually inaccessible to the ribosome, only when a ribosome slips during Coat protein’s translation termination does it get get translated, this has a very rare 5% chance of occuring How the replicase protein is regulated: There’s a 19 nt hair called the operator or TR (translation repression) located upstream of the replicase protein, as the CP is translated, dimers form, that binds the TR hairpin, repressing replicase translation and signaling the beginning of the capsid assembly One of the things I noticed, the TR hairpin overlaps with the lysis protein too, so in theory, it does repress it too I’ve attached a linear map of the MS2 genome to follow along, here is its source too: Emesvirus ~ ViralZone