Group Final Project
Group: Jessee Svoboda, Paula Carrodeguas, Iman Karibzhanova, Peter Hanna
From homework 4: Group Brainstorm on Bacteriophage Engineering
What do we know:
- E. coli DnaJ binds to denatured proteins to prevent/disassemble aggregates (native function in heat-shock).
- DnaJ binds to the hydrophilic tail of MS2-L protein.
- point mutation of highly conserved proline in DnaJ results in no lysis (so maybe no more binding of MS2-L tail?)
- removal of MS2-L tail recovers lysis function (meaning DnaJ is only necessary when tail exists)
- suggests hydrophilic tail aggregates in some way that prevents lysis except in presence of DnaJ to stop aggregation
- so stability should be improved if we can figure out how the tail is interacting with the tail of other MS2-L molecules, and then mutating that away so there is no aggregation and dependence on DnaJ
graph TB; A[sequence and structure of MS2-L] -->|if geometry and chemical interactions are known| B[view interactions between MS2-L copies] A -->|if geometry and interactions are not known| C[model interactions with AlphaFold or something that can do protein interactions] B -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions] C -->|visual analysis and mutation modeling| D[Identify important residues in MS2-L tail interactions] D -->|use knowledge of hydrophobicity/charge/etc. OR use ESM2 mutational scan and select ones that it finds unlikely| E[Select dissimilar AAs to substitute in interacting residues] E -->|AlphaFold or similar| F[model protein folding in new AA sequence with selected mutations] F -->|something that can model protein interactions| G[model interactions between mutant MS2-L copies] G -->|select mutations that have similar hydrophilicity as original tail but less interaction with each other and maybe also with DnaJ| H[test mutations in lab]
Potential problems:
- don’t know what can model protein-protein interactions
- we might have covered this in class but i don’t remember. i can rewatch the lectures
- what if modeling doesn’t show interactions between the tails? we know there probably has to be one…
- might have to simplify by only modeling the tail section, but that is probably known already (will have to model folding and interactions with full protein sequence in later steps probably)
- could start with DnaJ, what in MS2-L binds with the essential proline in DnaJ, and assume that it’s spatially close to that. then test various mutations of nearby residues
From homework 5: Final Project: L-Protein Mutants
We didn’t get to this part of the project unfortunately. But we did have some planning discussion.
My assumption was that DnaJ stabilizes the L-protein by preventing aggregation that would otherwise occur with the long tail.
Peter suggested:
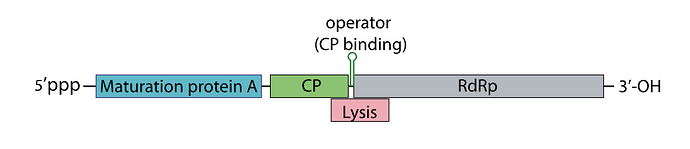
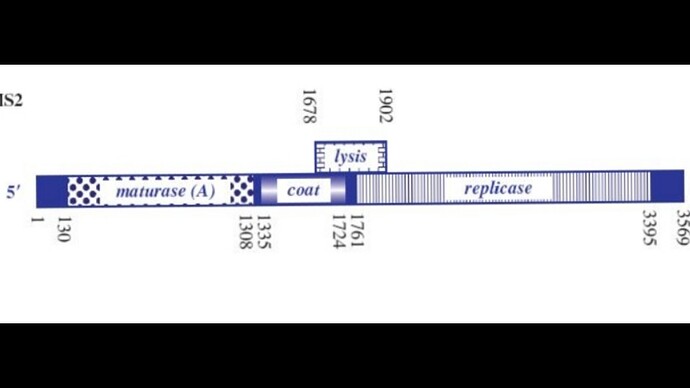
Sooo, the phage genome is very tightly regulated, I decided to take a look on how this regulation work, and it’s mainly based on RNA secondary structures How the lysis protein is regulated: The start codon and the shine-Dalgarno sequence are buried in an RNA hairpin, rendering virtually inaccessible to the ribosome, only when a ribosome slips during Coat protein’s translation termination does it get get translated, this has a very rare 5% chance of occuring How the replicase protein is regulated: There’s a 19 nt hair called the operator or TR (translation repression) located upstream of the replicase protein, as the CP is translated, dimers form, that binds the TR hairpin, repressing replicase translation and signaling the beginning of the capsid assembly One of the things I noticed, the TR hairpin overlaps with the lysis protein too, so in theory, it does repress it too I’ve attached a linear map of the MS2 genome to follow along, here is its source too: Emesvirus ~ ViralZone