Enzyme engineering for (cyanobacterial) bioplastic production
Imagine if plastic was an environmental solution, rather than an environmental problem.
Carbon capture, utilization, and storage (CCUS) is an umbrella term for any sort of technology that pulls carbon out of the atmosphere and repurposes it into a useful product and/or moves it to long-term storage as a climate change mitigation strategy. One example is bioplastics made with photosynthesis. Plastics are polymers made up of primarily carbon, and when produced by photosynthetic organisms (such as cyanobacteria), that carbon can come directly out of the atmosphere.
![CCUS figure]
Some strains of cyanobacteria produce biopolymers called polyhydroxyalkanoates (PHAs) that can be used as drop-in replacements for conventional petroleum-derived plastics. The most common PHA is poly-3-hydroxybutyrate or PHB.
![PHA granules in cyano]
![PHB]
For my project, I wanted to use AI reasoning combined with traditional machine learning to engineer a more productive PHB synthase enzyme for cost-competitive plastic biomanufacturing.
Background
2024 was the hottest year on record, with the average global temperature over the 1.5C increase limit that environmentalists have been advocating for the last several decades (United Nations, 1.5C). Although many countries are pledging to decrease their greenhouse gas outputs, carbon will continue to be added to the atmosphere for the foreseeable future, even if at decreasing rates. Mitigation strategies, such as CCUS, will be an essential component to minimize, or even reverse course, on climate change.
In theory, cyanobacteria are a financially useful production chassis because they take advantage of ambient or waste feedstocks, such as sunlight, atmospheric carbon dioxide, non-potable water such as seawater or greywater (Schubert et al, 2024; Wlodarczyk et al, 2020). Unfortunately, the relatively low production and energy-intensive processing have prevented commercially-viable production from being realized. The visionary aim of my project is to develop a commercial-scale bioprocessing operation for a bioplastic PHB-production cyanobacteria strain, with the ultimate goal to provide a drop-in raw plastic for use in consumer goods and packaging.
Because some strains of cyanobacteria are natural PHA producers, they are a good potential production chassis for bioplastic. The ideal strain would grow well off atmospheric carbon, have high PHA production, and have other phenotypic traits that are beneficial for cheaper or easier bioproduction, such as salinity or pH tolerance for contamination prevention, nitrogen fixation for cheaper growth media, and fast settling for lower energy collection and dewatering. One possible good host strain is Cyanobacterium aponium sp. UTEX 3222 because it has planktonic growth, salinity tolerance, rapid settling, fast growth rate, and native PHA production (Schubert et al, 2024).
PHB synthase enzyme, PhaC
PHAs are produced through a biosynthetic pathway as an offshoot from sugar metabolism, believed to be for long-term energy storage. The key enzyme is the PHB synthase enzyme, PhaC (also called PHA synthase, PHB polymerase, etc.). There are four classes of PhaC enzyme, which preferentially utilize different monomers. The best studied PhaC is from Cupriavidus necator H16 (Neoh et al, 2022). PhaC has two domains: the catalytic C-domain that includes the catalytic triad His-Asp-Cys, and the N-domain. The N-domain has been demonstrated to be essential for enzymatic activity, although its exact role is still unknown - though it has been suggested to potentially play a role in substrate selectivity (Neo et al, 2022).
There has been a lot of research into increasing the substrate promiscuity of PhaC because variety in monomer composition results in different thermochemical properties of the resulting plastic (Antonio et al, 2000; Harada et al, 2021; Kane, 2021; Sivashankari et al, 2023; Timm et al, 1990; Tsuge et al, 2004; Ye et al, 2008). However, I hypothesized that increasing substrate specificity towards the 3HB monomer that results in PHB would increase the amount of PHB produced. I hoped that this also might have the secondary effects of normalizing the molecular weights of the polymer molecules produced and decreasing downstream purification costs because there would be fewer minor products to separate out.
ML/AI in protein science
My initial thought was to use machine learning to design new protein mutants; however, my early literature searches made me seriously consider the practical constraints I was operating under: namely, time (before the final project was due) and processing capability (of my personal laptop and home internet). Therefore, I expanded my original intention to include free-for-public-use large language model (LLM) agents, such as ChatGPT and Claude.ai.
Hypothesis
I hypothesized that PhaC mutants engineered for increased substrate specificity by AI and ML would have higher PHB productivity.
Project aims
Aim 1: Experimental
Generate PHB synthase mutants using LLM reasoning and traditional machine learning (ML).
Literature search
Train a free-use LLM for PHB synthase mutations.
Write prompts to minimize hallucinations by requesting suggestions be based on the data provided, describe what each suggestion is based on (including literature references to verify), and specifically identifying more speculative suggestions.
Aim 1.5
Multi-sequence alignment (MSA) of diverse bacterial PhaC of various classes.
Use Python scikit-learn library to train ML classifier for PHB synthase substrate preference based on the MSA.
Feed ML output back into LLM to generate additional mutations and hypothesized mechanisms.
Aim 2: Developmental
Test the highest-scored mutants for PHB production.
Design cell-free expression vector.
Make a library of phaC mutant sequences.
Test variants for PHB production.
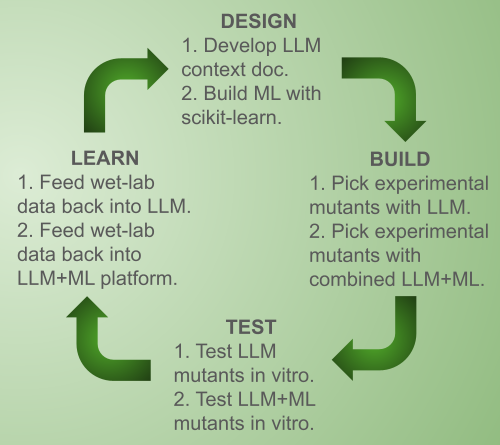
DBTL cycle(s) with LLM, ML, and in vitro testing; iterate as resources allow.
Aim 2.5
Test best-performing mutants in cyanobacterial chassis.
Literature search to identify 1-3 cyanobacterial strains to use as production chassis.
Re-design expression cassette and selected phaC mutants for cyanobacterial expression.
Test for PHB production in cyanobacterial strains.
Aim 3: Visionary
Develop a scalable bioprocessing operation that can ultimately be used for commercial plastics production.
Develop a basis for each operation step at the bench scale.
Design scale-up for a pilot plant.
Methods
Enzyme selection
I’d originally planned to use the PhaC from my intended host strain C. aponium UTEX 3222, but I was unable to find a sequence for it. I searched for the UTEX 3222 PhaC sequence in the supplemental files of the paper that said it identified PHA biosynthetic genes, but it wasn’t included (Schubert et al, 2024). It also wasn’t listed on the full annotated genome assembly (ASM3863077v1) published to the NCBI. I BLASTed the C. necator PhaC against the UTEX 3222 genome assembly, as well as a few different PhaC’s from other cyanobacterial species, but none of these produced any results.
Therefore, I decided to move forward using C. necator PhaC, hereafter called PhaC_Cn.
PhaC_Cn mutagenesis data compilation
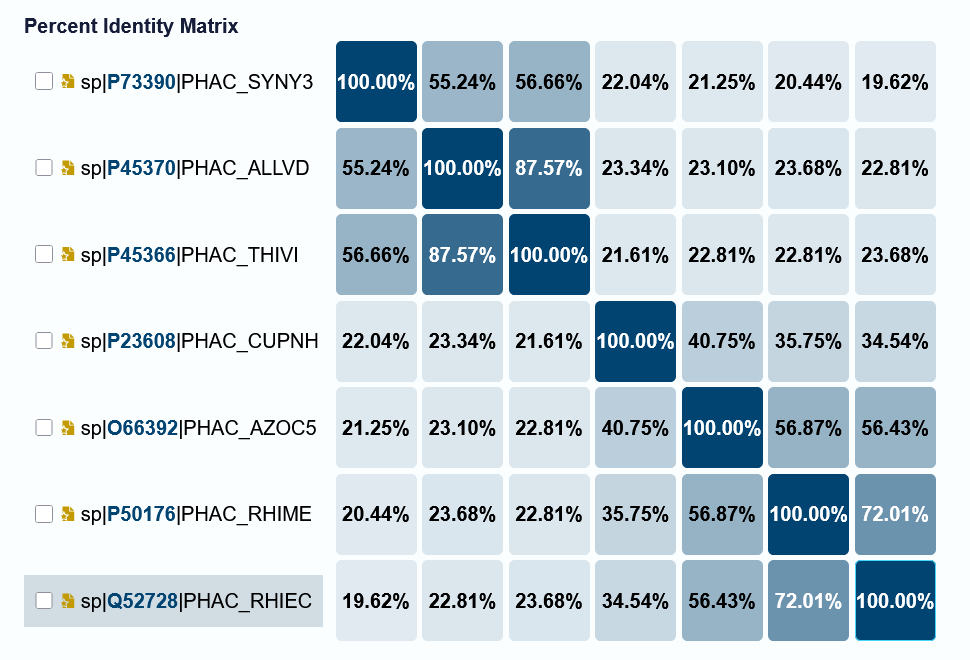
I’d originally planned to do a large MSA with diverse bacterial PhaC from all four classes. I did the following search on UniProt: (taxonomy_id:2) AND (protein_name:“Poly(3-hydroxyalkanoate) polymerase subunit PhaC”). This yielded 1,445 results. Using the internal UniProt tool, I tried to do an MSA (max 50 sequences), but this crashed my browser. I then tried to download a subset of these sequences for an MSA through Benchling or with Python code written with the help of Claude and a Python for Dummies book, but downloading hundreds of sequences froze my computer, and I gave up on the possibility of doing this on my personal laptop and home internet network. Below is the Identity Matrix of a 7 sequence MSA from UniProt that I was able to visualize, but 7 sequences is not enough to generate features or suggestions from, especially when they are so different.
So I decided to use PhaC_Cn mutagenesis data instead. I used Google Search to find peer-reviewed articles on machine learning and LLMs for protein design and PhaC_Cn, selecting the most relevant ones for further reading based ont the abstracts. From the papers I read, I additionally checked out their references for additional papers that could be helpful. Because I could only find limited mutagenesis data, I also included two other bacterial PhaC because they were listed in papers comparing the structures and sequences: PhaC from Aeromonas caviae (Harada et al, 2021) and PhaC from Chromobacterium sp. USM2 (Chuah et al, 2013). I imported these two PhaC amino acid sequences into Benchling, and aligned them with PhaC_Cn.
I started my conversation with Claude.ai by asking it about enzyme engineering with LLMs generally, to see what it knew.
i’m interested in enzyme engineering using LLMs. what can you tell me about that?
This produced a list of protein language models and tools (ESM-2, AlphaFold, ProteinMPNN, RFdiffusion, Progen2, and EVOLVEpro); practical workflows; active research topics; and limitations.
From there, I decided to ask what it thought of my hypothesis, hoping that would lead to a discussion of what the LLM needed to achieve this goal.
i’m specifically interested in mutations for substrate specificity in bacterial PhaC (polyhydroxyalkanoate synthase) enzymes. i have this idea that with the aligned protein sequences and some information on substrate preference for each one, an LLM might be able to output suggested sequences for a given substrate. Does an LLM seem like the correct tool for this?
Claude suggested that traditional ML would be a better option, but that combining LLM reasoning with ML could be a worthwhile approach. It included a warning that the biggest limitation was likely data availability. It gave four discrete options and a hybrid approach.
Option 1: Fine-tuned pLM (e.g., ESM-2) with substrate labels
Option 2: Supervised ML on alignment features
Option 3: EVE-style or EVOLVEpro unsupervised variant scoring
Option 4: Use an LLM as a reasoning layer over structured data
A hybrid pipeline:
Curate your MSA + substrate labels carefully (scl/mcl/mixed, specific monomers if known)
Identify key specificity-determining residues using mutual information or DCA (direct coupling analysis) between alignment columns and substrate labels — this gives you a reduced feature space
Use ESM-2 embeddings as features for a supervised model predicting substrate class
Score candidate mutations zero-shot using ESM-2 log-likelihood to filter for “natural-looking” sequences
Use structural context (the PhaC crystal structures / AlphaFold models) to sanity-check that suggested mutations are geometrically plausible in the binding pocket
The hybrid pipeline was had some similarities to the Protein Design II (week 5) homework, which I thought was a good sign.
I decided to move forward with options 2 and 4 since those were the approaches I’d had in mind earlier.
i’m most interested in Option 2 and Option 4. Could you please give me a very specific step-by-step instructions for both?
This prompt was a goldmine. It resulted in actually quite a lot of useful Python code for the ML approach, as well as the description of a context document that could be used for continuity between LLM reasoning sessions. I figured that between the context document and carefully worded prompts, I could also use it to minimize the chance of undetected hallucinations. Claude also suggested a pipeline for combining the two, from which I ultimately built my approach (though I didn’t exactly copy it because I thought my order worked better).
Literature (Option 4) → curated mutation DB
↓
MSA + MI analysis (Option 2) → top specificity positions
↓
LLM reasoning (Option 4) → mechanistic hypotheses about those positions
↓
ML scoring (Option 2) → rank candidate mutants quantitatively
↓
LLM (Option 4) → design wet-lab validation experiment
↓
Experimental results → feed back into both pipelines
I asked Claude for what specifically it wanted out of the context document so I could build it appropriately.
Please give me more information about the context document you would need if i wanted to go with LLM reasoning
This resulted in a list of sections, and it also offered to generate a template for me to start from. I asked it to do this.
please create a template markdown file for my context document
original context document template from Claude
While this template did contain some information on PhaC, I did not leave anything in that I did not have a reference to verify it because I wasn’t confident that it wasn’t hallucinations - and indeed, it did have some information that I suspect was hallucinations (although it may have simply been from a source I hadn’t personally read). However, going through the document, it relied heavily on an MSA that I simply wasn’t able to do, so I requested that Claude update the document to reflect the mutagenesis dataset.
i’m going to start with a dataset only containing PhaC from C. necator and known mutagenesis studies, not diverse PhaC from many different bacteria. Please update the context document template to reflect this, and let me know any particular warnings or considerations for this approach.
I filled in this context document with information that I could find in my references, and the mutagenesis data I had compiled. When I reuploaded it to Claude, it identified a few typos and questioned one of my references. I made the edits, but clarified that my reference said what I had asserted and Claude should continue to use it within my document and reasoning sessions, despite external data that Claude may have thought it had that disagreed with it. This may have been a hallucination, but I think Claude was actually just generalizing from other PhaC mutagenesis papers (not from C. necator).
I started with the warning from my context document.
Before we start, please keep in mind: My dataset consists only of C. necator PhaC1 (wild-type) and published point mutants of this single enzyme. I do not have a multi-species alignment. All positional reasoning should be grounded in (a) the experimental mutation database in Section 4, and (b) structural analysis of PDB 5T6O / AlphaFold model P23608. Do not infer specificity determinants from phylogenetic patterns — that data is not available.
Now let’s do the first reasoning session.
Claude asked for more direction, so I specificied that I wanted mechanistic reasoning.
Let’s start with mechanistic hypothesis building please.
Claude’s output for my first reasoning session started with a summary of information that I had included in my context document. While not particularly useful to me, it did confirm for me that it was drawing primarily from my context document and not as much external sources.
I refined my request with my next prompts for explicit suggestions:
That looks good - can you suggest a few single mutations within the N-terminus that would get us some data on how variation there affects substrate specificity?
Yes, and let’s include potential combinations if possible - combinations can exclude the N-terminus positions at this point since we lack data there.
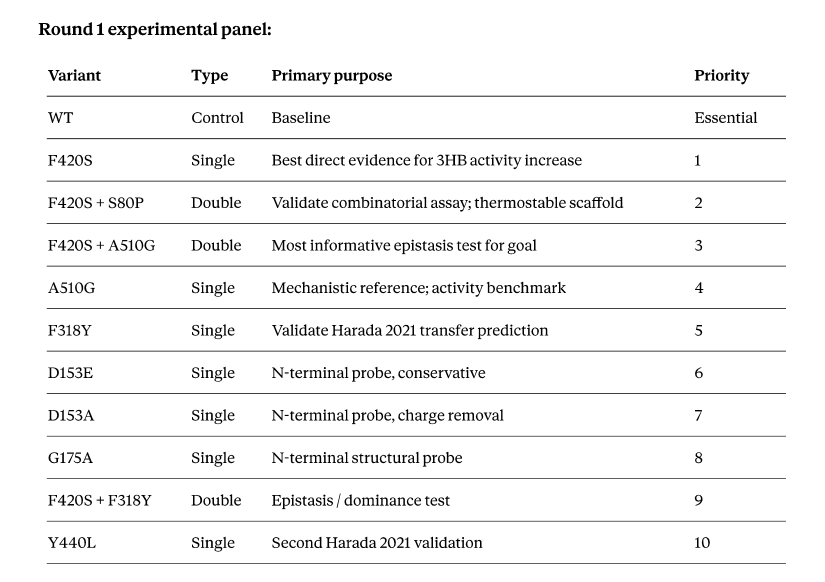
This resulted in a suggestion of a series of 11 variants for a first wet lab experimental run: the base wild-type as a control, 7 single mutations, and 3 double mutations. It was fairly conservative in its recommendations, but I suspect that’s due to the limited data.
Finally, I asked Claude to update my context document with the log of our first reasoning session.
Yes, please log both this table and a summary of our reasoning session in the session log section.
For starters, my next steps are to continue with the ML in Aim 1.5. I hope that by using the public wifi at a library or a college campus, I’ll be able to download the sequences for the MSA. Then I can hopefully let my computer run the MSA over a long period of time. The detailed steps for aims 1.5 and onward can be found in my final report below.
Additionally, the feedback from TAs and fellow students after my presentation was to compare outputs from multiple LLMs, using my same context document and prompts. So I’d like to do that as well.
Antonio, RV; Steinbuchel, A; Rehm, BHA. Analysis of in vivo substrate specificity of the PHA synthase from Ralstonia eutropha: formation of novel copolyesters in recombinant Escherichia coli. FEMS Microbiology Letters 2000, 182(1): 111-117. https://doi.org/10.1111/j.1574-6968.2000.tb08883.x
Chek, MF; Hiroe, A; Hakoshima, T; et al. PHA synthase (PhaC): interpreting the functions of bioplastic-producing enzyme from a structural perspective. Applied Microbiology and Biotechnology 2018, 103: 1131-1141. https://doi.org/10.1007/s00253-018-9538-8
Chuah, J-A; Tomizawa, S; Yamada, M; et al. Characterization of site-specific mutations in a short-chain-length/medium-chain-length polyhydroxyalkanoate synthase: In vivo and in vitro studies of enzymatic activity and substrate specificity. Applied and Environmental Microbiology 2013, 79. https://doi.org/10.1128/AEM.00564-13
Dong, H; Yang, X; Shi, J; et al. Exploring the feasibility of cell-free synthesis as a platform for polyhydroxyalkanoate (PHA) production: Opportunities and challenges. Polymers 2023, 15(10). https://doi.org/10.3390/polym15102333
Harada, K; Kobayashi, S; Oshima, K; et al. Engineering of Aeromonas caviae polyhydroxyalkanoate synthase through site-directed mutagenesis for enhanced polymerization of the 3-hydroxyhexanoate unit. Frontiers of Bioengineering and Biotechnology 2021, 9. https://doi.org/10.3389/fbioe.2021.627082
Jossek, R; Steinbuchel, A. In vitro synthesis of poly(3-hydroxybutyric acid) by using an enzymatic coenzyme A recycling system. FEMS Microbiology Letters 1998, 168: 319-324. https://doi.org/10.1111/j.1574-6968.1998.tb13290.x
Kane, A. Toward engineering the substrate specificity of a PHA synthase (PhaC). Victoria University of Wellington, Masters thesis, 2021. https://doi.org/10.26686/wgtn.17152079
Neoh, SZ; Check, MF; Tan, HT; et al. Polyhydroxyalkanoate synthase (PhaC): The key enzyme for biopolyester synthesis. Current Research in Biotechnology 2022, 4: 87-101. https://doi.org/10.1016/j.crbiot.2022.01.002
Satoh, Y; Tajima, K; Tannai, H; et al. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis from acetate with CoA recycling and NADPH regeneration in Vitro. Journal of Bioscience and Bioengineering 2002, 95(4): 335-341. https://doi.org/10.1016/S1389-1723(03)80064-6
Schubert, MG; Tang, T-C; Goodchild-Michelman, IM; et al. Cyanobacteria newly isolated from marine volcanic seeps display rapid sinking and robust, high-density growth. Applied Environmental Microbiology 2024, 90(11). https://doi.org/10.1128/aem.00841-24
Sivashankari, RM; Mierzati, M; Miyahara, Y; et al. Exploring Class I polyhydroxyalkanoate synthases with broad substrate specificity for polymerization of structurally diverse monomer units. Frontiers in Bioengineering and Biotechnology 2023, 11. https://doi.org/10.3389/fbioe.2023.1114946
Sudesh, K; Taguchi, K; Doi, Y. Effect of increased PHA synthase activity on polyhydroxyalkanoates biosynthesis in Synechocystis sp. PCC 6803. International Journal of Macromolecules 2002, 30: 97-104. https://doi.org/10.1016/S0141-8130(02)00010-7
Taguchi, S; Nakamura, H; Hiraishi, T; et al. In vitro evolution of a polyhydroxybutyrate synthase by intragenic suppression-type mutagenesis. Journal of Biochemistry 2002, 131(6): 801-806. https://doi.org/10.1093/oxfordjournals.jbchem.a003168
Timm, A; Byrom, D; Steinbuchel, A. Formation of blends of various poly(3-hydroxyalkanoic acids) by a recombinant strain of Pseudomonas oleovorans. Applied Microbiology and Biotechnology 1990, 33: 296-301. https://doi.org/10.1007/BF00164525
Tsuge, T; Saito, Y; Narike, M; et al. Mutation effects of a conserved alanine (Ala510) in Type I polyhydroxyalkanoate synthase from Ralstonia eutropha on polyester biosynthesis. Macromolecular Bioscience 2004, 4(10): 963-970. https://doi.org/10.1002/mabi.200400075
Valentini, G; Malchiodi, D; Gliozzo, J; et al. The promises of large language models for protein design and modeling. Frontiers in Bioinformatics 2023, 3: 1304099. https://doi.org/10.3389/fbinf.2023.1304099
Wittenborn, EC; Jost, M; Wei, Y; et al. Structure of the catalytic domain of the class I polyhydroxybutyrate synthase from Cupriavidus necator. Journal of Biological Chemistry 2016, 291(48): 25264-25277. https://doi.org/10.1074/jbc.M116.756833
Wlodarczyk, A; Selao, TT; Norling, B; et al. Newly discovered Synechococcus sp. PCC 11901 is a robust cyanobacterial strain for high biomass production. Nature Communications Biology 2020, 3: 215. https://doi.org/10.1038/s42003-020-0910-8
Ye, Z; Song, G; Chen, G; et al. Location of functional region at N-terminus of polyhydroxyalkanoate (PHA) synthase by N-terminal mutation and its effects on PHA synthesis. Biochemical Engineering Journal 2008, 41(1): 67-73. https://doi.org/10.1016/j.bej.2008.03.006
Dataset scope note: This document is built around a single reference enzyme
(C. necator PhaC1) and published mutagenesis studies on that enzyme and its
close variants. It does NOT use diverse multi-species sequence alignments.
See Section 3 for implications and compensating strategies.
0. IMPORTANT
0.1 General
All sequences given or referred to here are amino acid sequences.
We are using the wild-type enzyme PhaC from Cupriavidus necator (PhaC_Cn) as the starting point.
PhaC is a polyhydroxyalkanoate synthase enzyme that polymerizes monomers into polyhydroxyalkanoate polymers.
PhaC_Cn preferred product is poly-3-hydroxybutyrate (PHB), which uses monomer 3HB.
0.2 Notation
PhaC_Cn is the wild-type sequence from Cupriavidus necator, also called Cn PhaC1.
All mutations are notated in the form of AXB, where A is the single letter code for an amino acid in PhaC_Cn, X is the amino acid position index number in PhaC_Cn, and B is the single letter code for the amino acid substituted in this mutation. Example: A510T is the C. necator wild-type amino acid sequence for PhaC with a single amino acid substitution from alanine to threonine at position 510.
C. necator is sometimes referred to as Ralstonia eutropha.
PHB is poly-3-hydroxybutyrate, sometimes also called polyhydroxybutyrate or poly[(R)-3-hydroxybutyrate].
Both the single letter codes and three letter codes for amino acids are used throughout this document.
PhaC_Cs is the wild-type sequence from Chromobacterium sp. USM2 (Class I, 42% pairwise identity with PhaC_Cn).
PhaC_Ps is the wild-type sequence from Pseudomonas sp. 61-3, also called Ps PhaC1 (Class II, 67% pairwise identity with PhaC_Cn).
PhaC_Ac is the wild-type sequence from Aeromonas caviae (Class I, 37% pairwise identity with PhaC_Cn).
1. Enzyme Family Background
1.1 Classification
Class
Subunit structure
Size
Native substrate preference
Example organism
I
Single subunit
~65 kDa
scl (C3–C5): 3HB, 3HV, 3HP
Cupriavidus necator H16
II
Single subunit
~60 kDa
mcl (C6–C14): 3HHx, 3HO, 3HD
Pseudomonas sp. 61-3
There are four classes, but we are not considering Classes III and IV at this point.
Class I and II share ~50% sequence identity; Class III/IV are more distantly related.
This project focuses exclusively on Class I, using Cn PhaC1 as the sole reference
1.2 Reaction chemistry
Catalyzes polymerization of (R)-3-hydroxyacyl-CoA thioesters into PHA
Ping-pong (double displacement) mechanism:
Acylation: acyl group transferred to catalytic Cys, CoA released
Transacylation: acyl group transferred to growing polymer chain
Lipase-like α/β hydrolase fold
Catalytic triad: Cys – His – Asp
C. necator PhaC1 (Cn) reference numbering: C319, D480, H508
All residue positions in this document use Cn PhaC1 numbering unless noted
1.3 Substrate scope terminology
Term
Chain length
Key monomers
Notes
scl
C3–C5
3HP, 3HB, 3HV
Native Cn PhaC1 preference
mcl
C6–C14
3HHx, 3HO, 3HD, 3HDD
Most Class II enzymes
Broad/mixed
C3–C14
scl + mcl
Often from engineered enzymes — rare
Specialty
varies
3H4MV, 3H2MB, aromatic
Non-standard monomers, almost exclusively from engineered enzymes
We are primarily interested in scl, specifically in 3HB because my hypothesis is that increasing substrate specificity for 3HB will increase PHB production.
1.4 Why substrate specificity is structurally interesting
Dimerization interface indirectly influences active site geometry (Class I/II enzymes)
N-terminal domain is not well conserved; suggested to possibly be involved in substrate selection.
2. Structural Information
2.1 Experimental structures for reference
PDB ID
Enzyme
Class
Resolution
Notes
5T6O
C. necator PhaC1
I
1.8 Å
Primary reference, this structure contains the catalytic domain only
5XAV
Chromobacterium sp. USM2 PhaC
I
1.48 Å
additional Class I reference
2.2 AlphaFold model for Cn PhaC1
UniProt accession: P23608
Overall pLDDT: 85.94
Confidence notes: high confidence in core domain, low in N-terminal region residues 1–61
Use AF model for: loop conformations, surface regions not in crystal structure
Prefer crystal structure (5T6O) for: active site geometry, tunnel dimensions
2.3 Key structural regions (Cn PhaC1 numbering)
Region
Residues
Function
Notes
N-terminal domain
1–200
Regulatory, dimerization, possibly involved in substrate selection
Less conserved, lower structure confidence; changes in positions 153 and 175 could affect substrate selection
Core catalytic domain
200–400
Contains Cys319
High confidence
C-terminal domain
401–589
Contains Asp480, His508
High confidence
Substrate-binding tunnel
Arg398, His481
Selectivity determinant
channel is ∼18 Å in length, leading into C319.
Dimer interface
70-88
Stability
Avoid mutations here
Product-egress route
Ser201, Asp421
Avoid mutations here
product channel lined by a series of hydrophobic residues leading from the active site to the surface of the protein at a ∼95° angle to the proposed substrate entrance channel, extending ∼12.5 Å long away from the β-sheet core of the catalytic domain and widens into a small solvent pocket near the surface of the protein by the two noted residues
2.4 Substrate-binding tunnel residues
(Fill this table carefully — it is the core of your structural reasoning)
Position
WT residue
Role in tunnel
Notes
398
Arg
Entrance region
strictly conserved in Class I enzymes
481
His
Entrance region
highly conserved in Class I enzymes; mutagenesis study showed that H481Q lost 80% activity of wild-type
How to fill this table: Open 5T6O in PyMOL or ChimeraX. Select C319.
Run: select tunnel_res, byres (all within 10 of resi 319). List those
residues here with distances. This is worth spending 1–2 hours on — it
will substantially improve LLM reasoning quality.
NOTE : fill section 2.5 when you have the time to go through PyMol. 2.3 and 2.4 were filled from literature.
2.5 Tunnel geometry notes
Estimated tunnel constriction in WT Cn PhaC1: ~[X] Å (from structural analysis)
Residues that form the constriction point: [list]
Estimated minimum cavity volume for 3HHx-CoA accommodation: [X ų if known]
[Add MD simulation or docking results here as they become available]
3. Dataset Scope, Limitations, and Compensating Strategies
This section is critical. Read before every LLM session.
3.1 What your dataset contains
Reference enzyme: C. necator H16 PhaC1 (wild-type)
Variants: Published point mutants from the mutagenesis literature; published point mutants predicted to impact substrate specificity from PhaC_Cs and PhaC_Ac mutagenesis data - these mutants have been formatted into PhaC_Cn sequence through alignment identification.
Labels: Substrate incorporation data from those studies
What it does NOT contain: Homologous PhaC sequences from other species, Class II sequences, or unlabeled natural variants
3.2 Implications and honest limitations
Issue
Explanation
Impact
No evolutionary signal
Without a multi-species alignment, you cannot use co-evolutionary analysis (MI, DCA) to identify specificity-determining positions
Cannot compute MI scores; Section 3.4 of the original template is not applicable
Narrow sequence space
All data points are close variants of one sequence (1 mutation from WT)
Model cannot extrapolate to distant sequence space; suggestions far from WT are unreliable
Sparse coverage
Published mutagenesis studies cover only a small fraction of all possible positions
Many positions have no experimental data; reasoning about them is purely structural/hypothetical
Publication bias
Literature overwhelmingly reports positive results (mutations that did something interesting)
Negative results (mutations with no effect) are underrepresented; hard to learn what doesn’t matter
Lab-to-lab variability
Different studies use different assay conditions, hosts, carbon sources
Quantitative comparisons across studies are unreliable
Limited combinatorial data
Few studies systematically explore epistatic interactions
Combining individually beneficial mutations may not be additive
3.3 What this dataset IS good for
The LLM can reason very effectively about:
Mechanistic hypotheses — why does mutation X change specificity, based on
structure and chemistry?
Interpreting your experimental results — what does an unexpected outcome tell
you about the mechanism?
Experimental design — which mutations to test next given what is known?
Identifying gaps — which positions have never been mutated but are
structurally important?
Literature synthesis — connecting observations across papers into a
coherent mechanistic model
3.4 Compensating strategies
To partially offset the lack of multi-species alignment data:
Lean heavily on structural reasoning (Section 2) — fill in the tunnel
residue table as completely as possible; this replaces alignment signal
as your primary source of positional hypotheses
Include Class II reference data explicitly — even if not in your training
set, you can add a “comparative note” section describing which Cn PhaC1
positions correspond to Class II residues (from manual alignment of just
Cn PhaC1 vs. Ps PhaC1). This gives the LLM evolutionary context without
requiring a full MSA.
Weight negative results equally to positive — if you can find papers
reporting mutations that failed to shift specificity, record them in
Section 4.3. They are highly informative and rare in the literature.
Be explicit about data gaps in prompts — tell the LLM “position X has
never been mutated in the literature” so it flags its reasoning as
structural/hypothetical rather than evidence-based.
Use the LLM to propose positions to structurally analyze — ask it which
tunnel residues it would prioritize examining in the crystal structure,
then verify those manually before including them in subsequent prompts.
3.5 Class II reference comparison
(Manual alignment of just Cn PhaC1 vs. one Class II enzymes —
fills in some evolutionary context without a full MSA)
Cn PhaC1 residue
Cn AA
Ps PhaC1 equivalent residue
Ps AA
Significance
153
D
130
E
N-terminal position predicted to affect selectivity
175
G
151
G
N-terminal position predicted to affect selectivity
201
S
179
G
PHB egress channel
319
C
296
C
catalytic triad residue C
398
R
370
R
Substrate tunnel entrance
421
D
393
D
PHB egress channel
480
D
451
D
catalytic triad residue D
481
H
452
H
Substrate tunnel entrance
508
H
479
H
catalytic triad residue H
How to fill this: Use a pairwise alignment tool (e.g. EMBOSS Needle at
https://www.ebi.ac.uk/Tools/psa/emboss_needle/) with Cn PhaC1 (UniProt P23608)
and Ps PhaC1 (UniProt Q9Z3Y1). This takes ~10 minutes and is worth doing.
I used Benchling
4. Experimental Mutation Database
(This is the heart of your dataset — populate as completely as possible)
4.1 Key literature to mine for Cn PhaC1 mutations
Tsuge et al. (2003) Macromolecules — F420 region, systematic Class I
Amara et al. (2002) — systematic Class I mutagenesis panel
Rehm et al. — early mechanistic mutagenesis
Nomura et al. — broad-specificity engineering attempts
Insomphun et al. — 3HHx incorporation focus
Hiroe et al. — combinatorial mutagenesis
[Add others as you find them — search PubMed: “PhaC mutagenesis” OR
“polyhydroxyalkanoate synthase substrate specificity”]
Mining tip: For each paper, extract: (1) every mutation tested,
including ones with no effect — these are just as valuable, (2) exact
assay conditions, (3) quantitative data where reported. Even a table
footnote saying “A300G showed no change in specificity” belongs here.
4.2 Gain-of-function mutations (change in substrate specificity or activity)
Mutation
Effect
Activity vs WT
Reference
Notes
F420S
Increased 3HB specificity
increase
Taguchi et al 2002
2.4-fold increase in specific activity towards 3HB; this differs from studies on other PhaC enzymes but is correct here
F318Y
Increased mcl incorporation
no data
Harada et al 2021
predicted from PhaC_Ac mutagenesis
Y440L
Increased mcl incorporation
no data
Harada et al 2021
predicted from PhaC_Ac mutagenesis
R101L
Allowed aromatic monomer incorporation
no data
Kane 2021
predicted from PhaC_Cs mutagenesis, possible false positive
A510D
Increased molecular weight of polymer produced
no data
Tsuge et al 2004
A510E
Increased molecular weight of polymer produced
no data
Tsuge et al 2004
A510M
Increased mcl incorporation
no data
Tsuge et al 2004
A510Q
Increased mcl incorporation
no data
Tsuge et al 2004
A510C
Increased mcl incorporation
no data
Tsuge et al 2004
A510G
Increased mcl incorporation
increased activity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510W
no change
increased activity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510S
Increased mcl incorporation
no change
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510T
Increased mcl incorporation
no change
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
4.3 Neutral mutations (no significant effect on specificity)
(Underrepresented in literature but critically important — record every
instance you can find)
Mutation
Region
Why tested
Outcome
Reference
Note
A510H
A510
A510 mutations known to have effect
No change in specificity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510I
A510
A510 mutations known to have effect
No change in specificity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510P
A510
A510 mutations known to have effect
No change in specificity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510V
A510
A510 mutations known to have effect
No change in specificity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510Y
A510
A510 mutations known to have effect
No change in specificity
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
Deletion of residues 2-65
N-terminal
Little existing data on N-terminal
No change in specificity
Ye et al 2008
slight increase in activity
4.4 Deleterious mutations (loss of activity or expression)
Mutation
Effect
Reference
Note
A510F
Inactive
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510K
Inactive
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510L
Inactive
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510N
Inactive
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
A510R
Inactive
Chuah et al 2013
predicted from PhaC_Cs mutagenesis
H481Q
Reduced to 20% of wild-type activity
Wittenborn et al 2016
attributed to substrate binding loss
4.5 Combinatorial / double mutants
Mutations
Effect vs. singles
Reference
F420S + S80P
Reduced to 79% of wild-type activity, but with better thermostability
Taguchi et al 2002
4.6 Thermostability mutations
(Relevant when stacking specificity mutations)
Mutation
ΔTm
Effect on activity
Effect on specificity
Reference
S80P
increase in thermostability
Reduced to 27% of wild-type activity
none observed
Taguchi et al 2002
4.7 Positions that have NOT been mutated in literature
(Fill as you read — these are candidate positions for novel exploration)
Position
WT AA
Structural role
Why interesting
153
D
N-terminal
predicted to affect substrate specificity
175
G
N-terminal
predicted to affect substrate specificity
398
R
Substrate tunnel entrance
substrate binding
4.8 Data quality notes
Substrate specificity data given qualitative only due to differences in experimental conditions
In vitro CoA-release assays (DTNB) give intrinsic kinetic data but don’t
fully reflect in vivo selectivity under substrate competition
Some older studies used racemic substrates — stereospecificity may confound
apparent chain-length specificity
[Add specific notes about inconsistencies you notice across papers]
5.5 Catalytic and key residue positions (for quick reference)
Residue
AA
Role
C319
Cys
Catalytic — nucleophile; DO NOT MUTATE
D480
Asp
Catalytic triad; DO NOT MUTATE
H508
His
Catalytic triad; DO NOT MUTATE
R398
Arg
Tunnel entrance; strictly conserved - mutate with caution
H481
His
Tunnel entrance; highly conserved - mutate with caution
D153
Asp
N-terminal position predicted to affect selectivity; mutational target
G175
Gly
N-terminal position predicted to affect selectivity; mutational target
S201
Ser
PHB egress channel
D421
Asp
PHB egress channel
6. Engineering Target
6.1 Primary goal
[State precisely, e.g.:]
Increase substrate specificity towards 3HB specifically or scl generally with combinatorial mutations in the N-terminal and elsewhere, with the hypothesis that this will increase overall PHB production.
6.2 Secondary goals
Avoid total loss of activity
6.3 Acceptable tradeoffs
Up to 30% reduction in activity acceptable
6.4 Hard constraints — DO NOT VIOLATE
Do NOT mutate catalytic triad: C319, D480, H508
Avoid dimer interface residues: 70-88
6.5 What has already been tested
(Update after every experiment round — prevents redundant suggestions)
Mutation(s)
3HHx result
Other notable effects
Date
Notes
WT control
~0 mol%
Baseline
[date]
7. Production and Assay Context
7.1 Expression system
Host: Cell-free expression, E. coli BL21(DE3) lysate
Assay type: visual inspection for insoluble PHB granules
Substrate(s): 3HB-CoA
Buffer conditions: HEPES-KOH pH 7.5
7.4 PHA analysis
Extraction method: TBD
Monomer analysis: GC-MS for identification]
Quantification standard: LC-MS
Throughput: TBD
8. Reasoning Guidelines for LLM
8.1 Dataset context — tell the LLM explicitly at session start
Always include this statement at the top of each session prompt:
“My dataset consists only of C. necator PhaC1 (wild-type) and published point mutants of this single enzyme. I do not have a multi-species alignment. All positional reasoning should be grounded in (a) the experimental mutation database in Section 4, and (b) structural analysis of PDB 5T6O / AlphaFold model P23608. Do not infer specificity determinants from phylogenetic patterns — that data is not available.”
8.2 Prioritization criteria (in order, adjusted for this dataset)
Direct experimental evidence — mutations in Section 4 with measured outcomes
Structural/mechanistic reasoning — based on 5T6O crystal structure and
tunnel geometry (Section 2)
Analogy to Class II — using the pairwise comparison in Section 3.5,
noting explicitly when this is being used
Chemical intuition — physicochemical rationale for a substitution,
flagged as [SPECULATIVE] if no structural or experimental support
8.3 Required output format for mutation suggestions
For every suggested mutation, provide:
(a) Mutation in standard notation (e.g. A149F, Cn PhaC1 numbering)
(b) Primary evidence basis: Experimental / Structural / Class II analogy /
Chemical intuition [SPECULATIVE]
(c) Mechanistic rationale — specific, not generic
(d) Consistency with existing data in Section 4 — does it contradict anything?
(e) Confidence: High (direct experimental support) / Medium
(structural + analogy) / Low (chemical intuition only)
(f) Predicted risk: stability, expression, activity loss
8.4 Reasoning I do NOT want
Statements like “this position is conserved in mcl enzymes” — you do not
have alignment data to support this; use only the pairwise comparison in 3.5
Quantitative predictions of mol% outcomes
Suggestions violating hard constraints in Section 6.4
Suggestions already in Section 6.5 “already tested” table
Filling data gaps with plausible-sounding inventions — flag uncertainty explicitly
8.5 Especially useful prompts for this dataset type
Given the single-enzyme focus, these prompt types will be most productive:
Gap analysis: “Which tunnel-lining residues (Section 2.4) have never
been mutated in the literature (Section 4.7)? For each, give a structural
rationale for whether they are likely to affect specificity.”
Mechanistic interpretation: “Mutation X gave unexpected result Y.
Given the structural context of position X (distance to C319, neighboring
residues, tunnel role), propose 2–3 mechanistic explanations.”
Epistasis prediction: “Given that A149F and S325A are individually
beneficial, reason about whether their combination is likely to be additive,
synergistic, or antagonistic, based on their structural relationship.”
Experimental prioritization: “I can test 12 variants. Given the
mutation database and structural data, design a 12-variant panel that
maximizes information gained about specificity determinants.”
8.6 My background
PhD in bioengineering. Comfortable with molecular biology,
enzyme kinetics, and microbial fermentation. Less experienced with
structural biology and bioinformatics — please explain structural reasoning clearly but
do not oversimplify the biochemistry.
9. Session Log
(Prepend full context document + append this log to every session)
Mechanistic hypothesis building for PhaC_Cn scl selectivity
Suggest single N-terminal mutations to probe that region
Suggest single and combinatorial mutations outside the N-terminus
Consolidate all suggestions into a Round 1 experimental panel
Key outputs / hypotheses:
Hypothesis 1 — Two-point selectivity model:
scl selectivity in PhaC_Cn is established at two structural points:
Tunnel constriction (A510): Ala510 acts as a steric gate physically excluding acyl chains longer than ~C5 from reaching C319. This is the primary passive size filter. WT Ala at this position is already near-optimal for scl — bulkier substitutions (F, K, L, N, R) are inactive, and smaller substitutions (G, S, T) widen the tunnel and permit mcl incorporation.
Active site geometry (F420): Residues near the base of the active site, including F420, optimize catalytic geometry for the C4 acyl-enzyme intermediate. F420S gives a 2.4-fold increase in 3HB-specific activity, consistent with relief of steric strain in the binding pose for 3HB-CoA.
Hypothesis 2 — R398 and H481 are positioning/catalytic residues, not selectivity gates:
Both are strictly or highly conserved across Class I enzymes regardless of substrate preference. R398 likely contacts the CoA moiety to position the acyl chain for nucleophilic attack; H481 likely contributes to transition state stabilization. Neither is a primary chain-length selectivity determinant. Both are high-risk mutation targets.
Hypothesis 3 — Residual mcl leakiness has an uncharacterized structural origin:
WT PhaC_Cn incorporates very low levels of 3HO and 3HDD. Since A510 appears near-optimal for scl exclusion and F420S already improves 3HB activity, the residual mcl leakiness likely originates from dynamic flexibility elsewhere in the tunnel — possibly in the uncharacterized region between R398/H481 (entrance) and A510 (constriction). This region is not yet described in Section 2.4 and should be a priority for PyMOL analysis.
Hypothesis 4 — N-terminal domain role is genuinely unclear:
Deletion of residues 2–65 (Ye et al. 2008) shows no specificity change, suggesting the extreme N-terminus is dispensable. D153 and G175 are proposed as candidate positions based on Class II pairwise comparison only. G175 is conserved as Gly in both Class I and II, suggesting structural rather than selectivity role. D153 differs conservatively between classes (Asp vs. Glu) and is the stronger candidate. All N-terminal reasoning is [SPECULATIVE].
12 total variants including WT. Fits standard 12-well format with no spares — consider dropping Y440L (priority 10) to keep a spare well if expression failures are anticipated.
Flagged uncertainties / SPECULATIVE tags:
All N-terminal suggestions (D153E, D153A, G175A) are [SPECULATIVE] — grounded only in AlphaFold model (low pLDDT in this region) and Class II pairwise comparison. No structural data from 5T6O available for this region.
F318Y and Y440L are predictions transferred from PhaC_Ac (37% identity) — transfer reliability is unknown and should be treated as unvalidated until tested directly in PhaC_Cn.
F420S mechanistic basis (Hypothesis A: steric relief vs. Hypothesis B: electrostatic contribution) is not resolved by available data — both remain plausible.
The identity of tunnel residues between R398/H481 and A510 is not characterized in Section 2.4 — this gap limits mechanistic reasoning about the constriction region.
Epistasis between all combinations is unknown; F420S + A510G is the combination with the most interpretable expected outcome.
Action items:
Complete PyMOL tunnel analysis (Section 2.5) — residues within 10 Å of C319, focusing on the R398-to-A510 region
Verify structural position of F420 relative to C319 and catalytic triad in 5T6O
Identification of PhaC analog in Cyanobacterium aponium UTEX 3222 and overproducing or engineering for increased efficiency
BLAST/align with known PHA-synthases
Compare efficiency / mutations that improved turnover in other PhaC - test analogous mutations (aligned location, similar or different AAs). improved substrate specificity?
Site-specific saturation mutagenesis? Would be good use for automation
Quorum sensing based killswitch (i.e. cell dies if it escapes bioreactor)
Has to have some kind of inducible element or won’t grow after initial transformation
What’s good at quorum sensing already?
Something else??? Something in E coli that can be done on Opentron
Because it’s more convenient for a final project to be executed in Victoria remotely
Cyanobacterial expression plasmid across multiple cyano species
needs to include E coli machinery for manipulation and production (and conjugation, for relevant species)
Ideas:
PhaC protein engineering
Short term aim: Design small library of PhaC variants with expected improvement
Medium term aim: Generate library and test in chassis strain
Long term aim: Develop PHB bio-manufacturing cyanobacterial strain for carbon-neutral/carbon-negative plastic (depending on biodegradation).
Quorum sensing based circuit for biocontainment
Short term aim: Design killswitch with genetic circuit to trigger based on quorum sensing.
Medium term aim: Build genetic circuit with expression based on quorum sensing with a measureable output; test circuit in E. coli.
Long term aim: Optimize circuit sensitivity and test with killswitch expression; integrate into bio-manufacturing chassis strains for population-linked biocontainment.
Broad cyanobacterial expression plasmid
Short term aim: Design plasmid backbone based off native cyanobacterial plasmids and established E. coli machinery.
Medium term aim: Test expression in multiple cyanobacterial strains (including some previously considered genetically intractable with classic broad-host-range vectors).
Long term aim: Establish protocol for domestication of newly prospected, wild-type cyanobacterial strains using the cyanobacterial plasmid.
Mar 31, 2026
Leaning towards quorum sensing killswitch because it’s more aligned with my prior experience and knowledge, so i think it will take less research on my behalf. since i’m already falling behind on homeworks, i’m worried about how much time it would take to optimize a protein since i have no prior machine learning experience.
Quorum sensing notes:
auto-inducer: triggers expression
keep in mind phenolic compounds and other naturally occurring quorum quenching
also keep in mind the potential for auto-inducer production from other related bacteria; like if biomanufacturing strain escapes bioreactor but lands in soil with existing microbiome - we still want the escaped cells to die
maybe we could make a synthetic quorum sensing system for orthogonality: would require biosynthetic pathway for auto-inducer, auto-inducer recognition (inducible promoter, transcription factor, riboswitch, etc.), auto-inducer export pathway to preferentially or rapidly diffuse out of the cell
for killswitch activation at low population (cells escaped from bioreactor), maybe consider secondary/back-up activation from an environmental signal
potentially test circuit with fluorescence or colorimetric output first before killswitch/toxin-antitoxin genes
References
Miguel, CMTS; Santos, CA; Lima, EMF; et al. Quorum Sensing in Bacteria: From Mechanisms to Applications in Foods. 2026. Current Opinion in Food Science: 101394. DOI: 10.1016/j.cofs.2026.101394.
maybe autoinducer can also trigger expression of toxin repressor
killswitch trigger for population low (like Paula’s idea for targeted drug delivery):
low constitutive expression of antitoxin
autoinducer-triggered expression of toxin
maybe autoinducer can also trigger expression of antitoxin repressor
Apr 2, 2026
In Victoria node recitation last night, Derek suggested a possibility for cell-free testing of the system to be able to use the Gingko cloud lab instead. Originally i had figured that because i want it to be a killswitch system, that it needs to be in a living cell. Also because traditional quorum sensing systems are dependent on autoinducer concentration within vs outside the cell membrane. But Derek suggested considering instead how to design a quorum sensing system that would be cell-free like on a paper biosensor: that triggers when at a minimum concentration, rather than triggering at the first cell it sees.
Phrasing it that way made me think of the analog computing of the neuromorphic circuits: where inputs are additive (positive or negative). So to reach a minimum concentration rather than the very first thing present, there has to be a counter-actor to the sensed thing present; something that degrades or binds to the autoinducer or signalling metabolite that is expressed constitutively at a low level. So when the signalling molecule concentration is low (low population), it will be all used up by the counter-actor before it can trigger expression of the QS-controlled genes. When the signalling molecule concentration is high (high population), it will outnumber the counter-actor, so it can still trigger the QS-controlled genes.
For example, a riboswitch that recognizes a small molecule metabolite. The metabolite is produced and exported by cells, and when present, activates the gene of interest (in my switch a killswitch or fluorescent protein). We’d also want to express the riboswitch as an aptamer that is unconnected to the gene of interest to bind the metabolite at low concentrations, until a high concentration of the metabolite is reached and the metabolite outnumbers the loose aptamer and can trigger the riboswitch to activate gene expression.
Apr 3, 2026
Brainstorming and design
Drafts
Title:
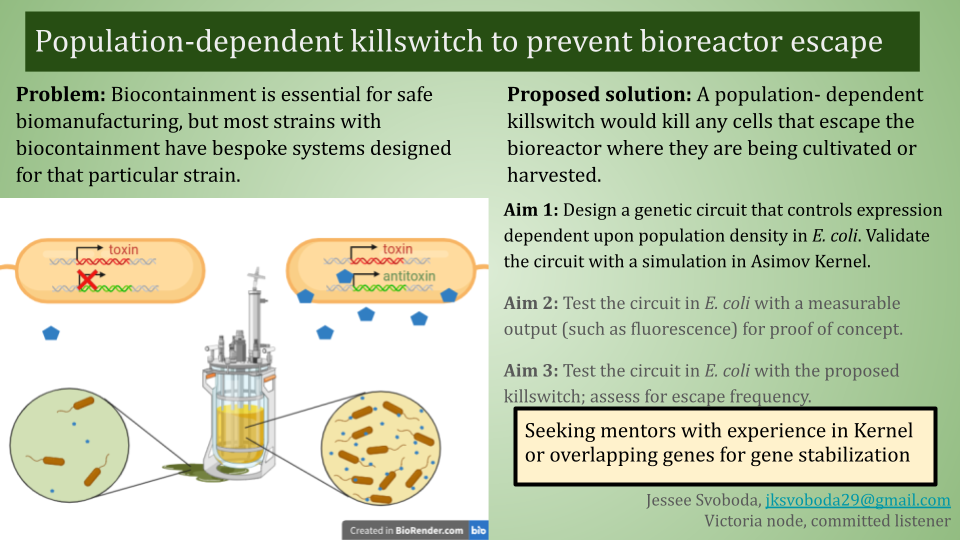
Population-dependent killswitch to prevent bioreactor escape
Short description:
Biocontainment is essential for safe biomanufacturing, but most strains with biocontainment have bespoke systems designed for that particular strain. A population-dependent killswitch would kill any cells that escape the bioreactor where they are being cultivated or harvested. My initial idea is a toxin-antitoxin system expressed under control of a quorum-sensing circuit. Future considerations: safeguards against biocontainment escape through mutation, multiple levels of regulation.
Aims:
Design a genetic circuit that controls expression dependent upon cell population density in E. coli. The circuit will be designed with the intent for a final use with a killswitch, but fluorescent or colorimetric outputs might be used for initial design and validation. Validate the circuit with a simulation in Asimov Kernel.
Test circuit in E. coli with a measurable output (such as fluorescence).
Test circuit with killswitch; integrate into a biomanufacturing chassis strain for population-linked biocontainment.
Companies:
Asimov - I plan on using Kernel to design and simulate my genetic circuit. Basecamp Research - Maybe their AI can help me design overlapping genes to prevent killswitch escape via toxin gene mutation. Cultivarium - If successful, quorum-based biocontainment could be a useful genetic tool to port to new potential chassis microbes.
Project idea slide
Project description: Biocontainment is essential for safe biomanufacturing, but most strains with biocontainment have bespoke systems designed for that particular strain. A population-dependent killswitch would kill any cells that escape the bioreactor where they are being cultivated/harvested. Initial idea is a toxin-antitoxin system expressed under control of a quorum-sensing circuit. Need to consider safeguards against biocontainment escape through mutation, multiple levels of regulation.
Asimov: I plan on using Kernel to design and simulate my genetic circuit.
Basecamp Research: Maybe their AI can help me design overlapping genes to prevent killswitch escape via toxin gene mutation.
Cultivarium: if successful, quorum-based biocontainment could be a useful genetic tool to port to new potential chassis microorganisms.
References:
Leonard SP; Halvorsen TM; Lim B; et al. Synthetic overlapping genes stabilize genetic systems. 2026. mBio, 17(3):e0272525. DOI: 10.1128/mbio.02725-25.
Blazejewski, T; Ho, H-I; Wang, HH. Synthetic sequence entanglement augments stability and containment of genetic information in cells. 2019. Science, 365(6453): 595-598. DOI: 10.1126/science.aav5477
Last night in the Victoria node recitation, Derek was really talking about how cool the Kernel-Twist-Nebula-Waters pipeline is, and he mentioned that he was a little disappointed that not many projects seemed like they were fully utilizing it. Especially since he still isn’t back in Victoria, it seems like the only way i’ll be able to get any actual lab data unfortunately, so that’s different from my original plan with the quorum sensing. i ended up messaging Derek on Discourse to ask if it was too late to change my mind, and he said that while technically yes, since no one had signed up on my slide as a mentor yet, i could change it. So i went ahead and came up with a new slide and replaced my original response.
Description:
Current bioplastic production is too expensive to compete commercially with petroleum plastics. Bioplastics, such as PHA, produced by cyanobacteria are one possible solution because they require minimal feedstocks (atmospheric carbon dioxide, sunlight, non-potable water), but processing can be expensive (harvesting, dewatering, purification). Cyanobacterium UTEX 3222 is a new potential chassis for PHB bioplastic with lower processing intensity due to its phenotypic properties (planktonic grown and settling, potential native PHB production). This strain can be further improved by engineering PHA synthase for increased substrate specificity, to preferentially produce PHB (to decrease purification steps). Existing data on PHA synthase substrate preference and specificity can be used to train an ML tool, which could recommend mutations that could be tested in a cell-free expression system.
Companies:
Basecamp Research: Machine learning expertise and datasets
Biofabricate: experience with biomaterials marketing and maybe scale-up
Boltz.bio: ML for protein design (but it’s for drug discovery)
Cultivarium: specifically for aim3, UTEX 3222 is currently considered genetically intractable
Ginkgo: Cell-free protein expression system
Twist: DNA synthesis for my mutant library
Waters: MS to identify PHA (general) and PHB (specific) production
References
These are for using mass spectrometry for PHA analysis, for the Waters step of the pipeline.
Khang, TU; Kim, M-J; Yoo, JI; et al. Rapid analysis of polyhydroxyalkanoate contents and its monomer compositions by pyrolysis-gas chromatography combined with mass spectrometry (Py-GC/MS). 2021. International Journal of Biological Macromolecules, 174: 449-456. DOI: 10.1016/j.ijbiomac.2021.01.108
Johnston, B; Radecka, I; Chiellini, E; et al. Mass spectrometry reveals molecular structure of polyhydroxyalkanoates attained by bioconversion of oxidized polypropylene waste fragments. 2019. Polymers, 11(10):1580. DOI: 10.3390/polym11101580
Conners, EM; Bose, A. State-of-the-art methods for quantifying microbial polyhydroxyalkanoates. 2025. ASM Applied and Environmental Microbiology, 91(9):e00274-25. DOI: 10.1128/aem.00274-25
These are for machine learning for enzyme engineering.
Satoh, Y; Tajima, K; Tannai, H; et al. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis from acetate with CoA recycling and NADPH regeneration in Vitro. 2003. Journal of Bioscience and Bioengineering, 95(4): 335-341. DOI: 10.1016/S1389-1723(03)80064-6
Apr 9, 2026
Talking to Derek about the timeline in our Victoria node recitation last night, he suggested that everything for lab work will probably need to be ordered in the next two weeks to get data in time for final project presentations. He also said that if we want to run anything on the Ginkgo Nebula cloud lab, we need to talk with Ronan and see if he has the capacity for it. Given this timeline, i am almost definitely not going to be able to figure out any ML-guided protein engineering before the final ordering. What i’m thinking instead is to design initial constructs of PhaC from C. necator, PhaC from UTEX 3222, and a rational design for a UTEX 3222 PhaC mutant, all designed for cell-free expression. The reaction will probably include the monomer, since it’s simpler than using the full 5-enzyme cell free system from Satoh et al (reference 8 above) that used acetate as the feedstock, but I will need to double check the energy and CoA regeneration.
Apr 10, 2026
Enzyme sequence choices
PhaC enzyme from Cupriavidus necator was chosen as my wild-type. I used the amino acid sequence from Uniprot and codon-optimized it in Benchling for Escherichia coli. This was from homework 2 i think, and i just used that one.
For the mutant: I found a review paper (Ref1) that identified Ala510 in PhaC_Cnecator as having a role in substrate specificity: with A510M, A510Q, and A510C all increasing promiscuity (M, C both sulfur-containing residues; Q, C both polar residues; M, Q both larger residues); a related PhaC from Chromobacterium sp. USM2 found that changing the analogous A to M/W/V (all non-polar residues, larger than A) increased promiscuity; and the same PhaC from Chromobacterium sp. USM2 found that changing the analogous A to S (similar size, but polar) increased substrate specificity (towards short-chain-length PHAs, like PHB). It’s surprising that A->S had the oppposite effect as A->C, for these two different PhaC variants from different bacteria. But since I didn’t have time to read a lot more, I figured A510S was a good construct to test against the PhaC_Cnecator wild type to start with.
I tried to identify the PhaC sequence from UTEX 3222 to test as well, but I was unable to, as of yet. While the paper in which UTEX 3222 was prospected said the authors identified the genes encoding the PHA biosynthesis enzymes (Ref2), the genes weren’t annotated on the full genome sequence assembly, and I got no results BLASTing either C. necator PhaC or cyanobacterial PhaC from Synechocystis sp. PCC 6803 or the more closely relatated Microcystis aeruginosa sp. PCC 7608SL. I also tried BLASTing PhaE (another PHA biosynthetic enzyme) from a few different cyanobacterial strains as well, with still no results. All BLAST searches were tBLASTn to search for the nucleotide gene sequence within the genome assembly from the amino acid sequences from the various known PhaC
protein sequences. The genes were also not listed amongst the biosynthetic genes listed in the Supplementary Information from the UTEX 3222 paper. I was out of ideas at this point, and on a time constraint, so to get constructs added to the order list today, I decided to move forward just with the PhaC_Cnwt and PhaC_CnA510S for now. If/when I get my ML design program working, maybe I can email George Church (or whoever on the author list did the genome annotation) to try to run it through, but I can definitely start with the C. necator one.
Construct design
Derek sent me a message asking me to order constructs today. So I went into Kernel to design PhaC_Cnecator with T7 promoter, RBS and terminator for cell-free expression because the E. coli based cell-free expression kits I found online from both NEB and Thermo Fisher both used T7 polymerase. In Kernel, I used a T7 promoter, T7 RBS, and T7 terminator from the iGEM repository. I chose promoter Bba_Z0251 from the many options because it had a lot of documentation on its iGEM registry page, and matched the full consensus sequence (from the T7 promoters iGEM page). I chose RBS Bba_Z0261 from the many options because it was analyzed by the same iGEM team as the promoter I used. I used T7 terminator Bba_K731721 from the many options because it most closely matched a quick google search for the T7 terminator sequence. While Kernel did have a genetic part for PhaC_Cnecator in the Uniprot repository, it actually didn’t have a nucleotide sequence associated with it. So i copied the promoter, RBS, and terminator sequences from Kernel into Benchling, where I used the previously codon-optimized PhaC_Cnecator sequence from homework 2. I used Benchling’s translation tool to identify the Ala at position 510, and changed a single nucleotide to change A510 to A510S (Ala: GCC; Ser: TCC) for the mutant.
Then I exported the FASTA files for both constructs and uploaded them into the Twist portal for clonal genes. I couldn’t remember if it mattered using linear gene fragments or clonal genes within a plasmid, but I went with clonal plasmid because previous experience with linear fragment orders from Twist were pretty low concentration. I remembered from lecture that Ronan preferred us to use chloramphenicol for an antibiotic marker if needed, so I decided to use the pTwist-Chlor-HighCopy cloning vector. Twist’s interface found both genes to be complex, so I used its internal codon optimization to fix this issue: I identified the organism as E. coli, did not omit any restriction enzyme recognition sites, and selected the promoter, RBS, and terminator regions as sequences that should not be changed. To my surprise, these sequences were not identical except the one point mutation; they were optimized differently, but I suppose it doesn’t really matter. Then I exported the full constructs (including plasmid) GenBank files from Twist and re-uploaded into Benchling to generate the link for adding the spreadsheet.
After meeting with Derek to explain, he suggested using linear gene fragments instead of clonal, so I re-did the Twist ordering bit to generate prices and optimized fragment GenBank files. I elected to leave the adaptors on because I assume those will give long enough arms, but I’m not really sure. Derek said he’d check with Ronan.
Experimental design
I checked that Millipore Sigma does in fact carry my substrate, I think. The substrate being the PHB monomer: 3-hydroxy-butyryl-CoA. However, it’s very expensive, so I’ll see about also ordering the DNA and cheaper substrates for the 5-enzyme biosynthetic pathway with CoA recycling that was in one of the papers I found (Ref3). After comparing the even just of all the substrates I’d still need for the full pathway, it’s cheaper just to order the original substrate (since I’d still need at least a little bit of CoA, which is still expensive on its own) then to also get a bunch of additional DNA. Derek mentioned that I need to figure out what kind of purification is needed for Waters to analyze my PHB product at the end of the reaction.
Reference
Chek, MF; Hiroe, A; Hakoshima, T; et al. PHA synthase (PhaC): interpreting the functions of bioplastic-producing enzyme from a structural perspective. 2018. Applied Microbiology and Biotechnology, 103: 1131-1141. DOI: 10.1007/s00253-018-9538-8
Schubert, MG; Tang, T-C; Goodchild-Michelman, IM; et al. Cyanobacteria newly isolated from marine volcanic seeps display rapid sinking and robust, high-density growth. 2024. ASM Applied and Environmental Microbiology, 90: e00841-24. DOI: 10.1128/aem.00841-24
Satoh, Y; Tajima, K; Tannai, H; et al. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis fro macetate with CoA recycling and NADPH regeneration in Vitro. 2003. Journal of Bioscience and Bioengineering, 95(4): 335-341. DOI: 10.1016/S1389-1723(03)80064-6
Apr 13, 2026
After Derek talked to Ronan, he suggested going back to clonal genes. I used my original PhaC_Cn construct, but then I decided to just have the point mutation for the mutant and have the rest of the sequence be identical. So I copied the PhaC_Cn-pTwist construct into a new DNA sequence in Benchling, and made the point mutation (GCA->TCA), and then verified quickly in Twist that this sequence is still simple and the same price.
Updated idea slide for cell-free synthesis Aim 1 instead of LLM/ML
Apr ?, 2026
Wanted to get started on the AI/ML aspect, so i started looking into that. My original plan had been to use this as an opportunity to expand my Python capabilities and play around with scikit-learn, but unfortunately, i realized i just didn’t have the time for that before the end of the course. After hearing Victoria node TAs Derek and Piyush talk about the AI tutor they developed for the course, I thought about maybe working with an LLM like Claude instead since that would require less initial coding on my end, and could still work because proteins are not dissimilar to language.
Apr 27, 2026
Did some reading, and it turns out that people have used LLMs for protein design before.