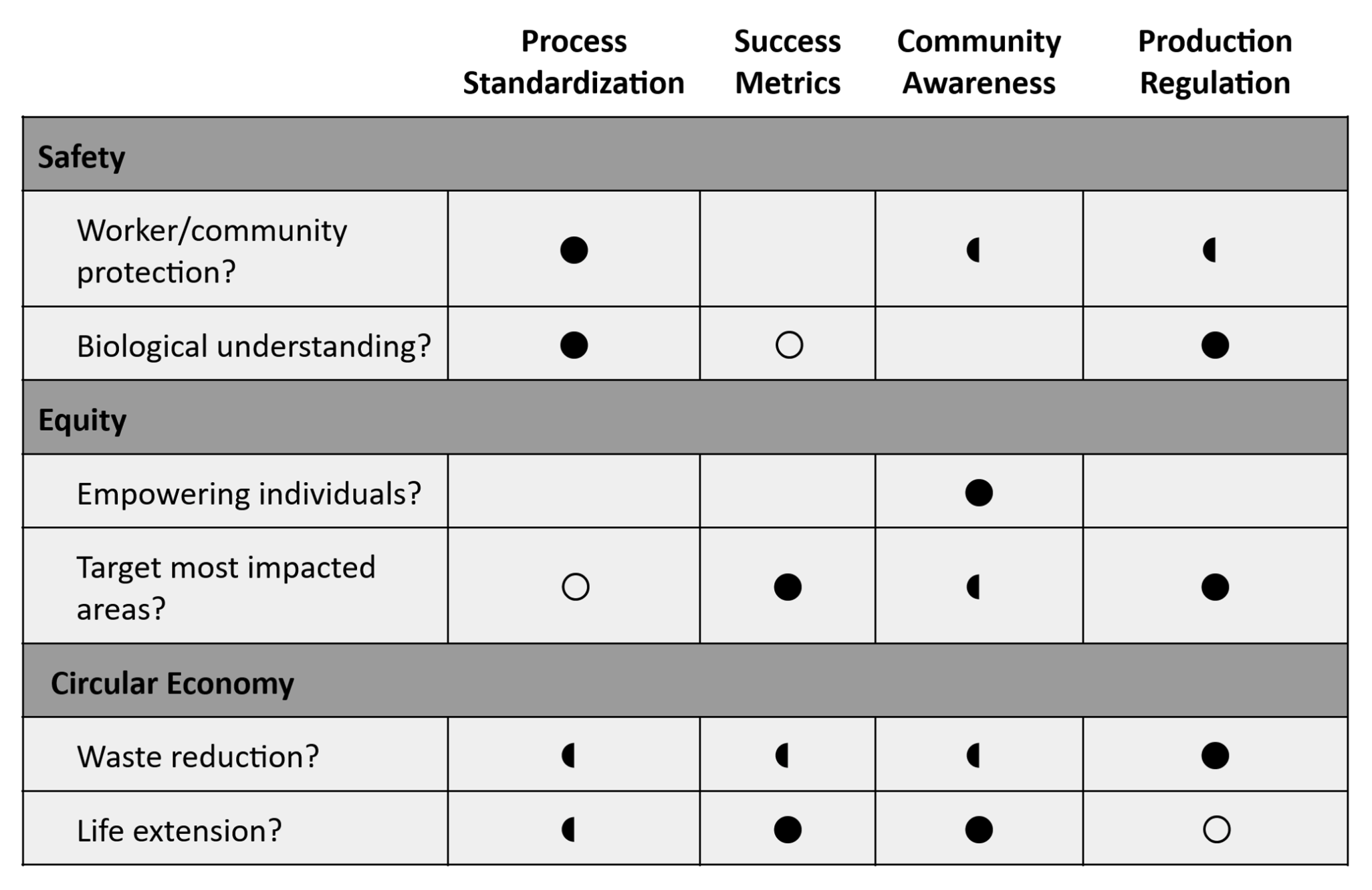
Class Assignment First, describe a biological engineering application or tool you want to develop and why. There is currently an urgent research focus on the biodegradation of plastics, due to the extremely long life cycle of synthetic polymers. Prior work has focused on a mix of exploring bacterial and microbial processes (e.g. anaerobic digestion) to break down plastics, and developing compositions that can be commercial compostable (e.g. for single use plastics). My personal interest is in fiber arts and sustainability, so I’d like to tackle this problem from a textile perspective. Fast fashion has exacerbated the volume of cheap, low quality clothes produced everyday. These clothes are often made with synthetic fibers and not for long term use (although the two are not necessarily interchangeable). I believe it’s incredibly important to find a way to biodegrade polyester, one of the most common synthetic polymers in fast fashion clothing.
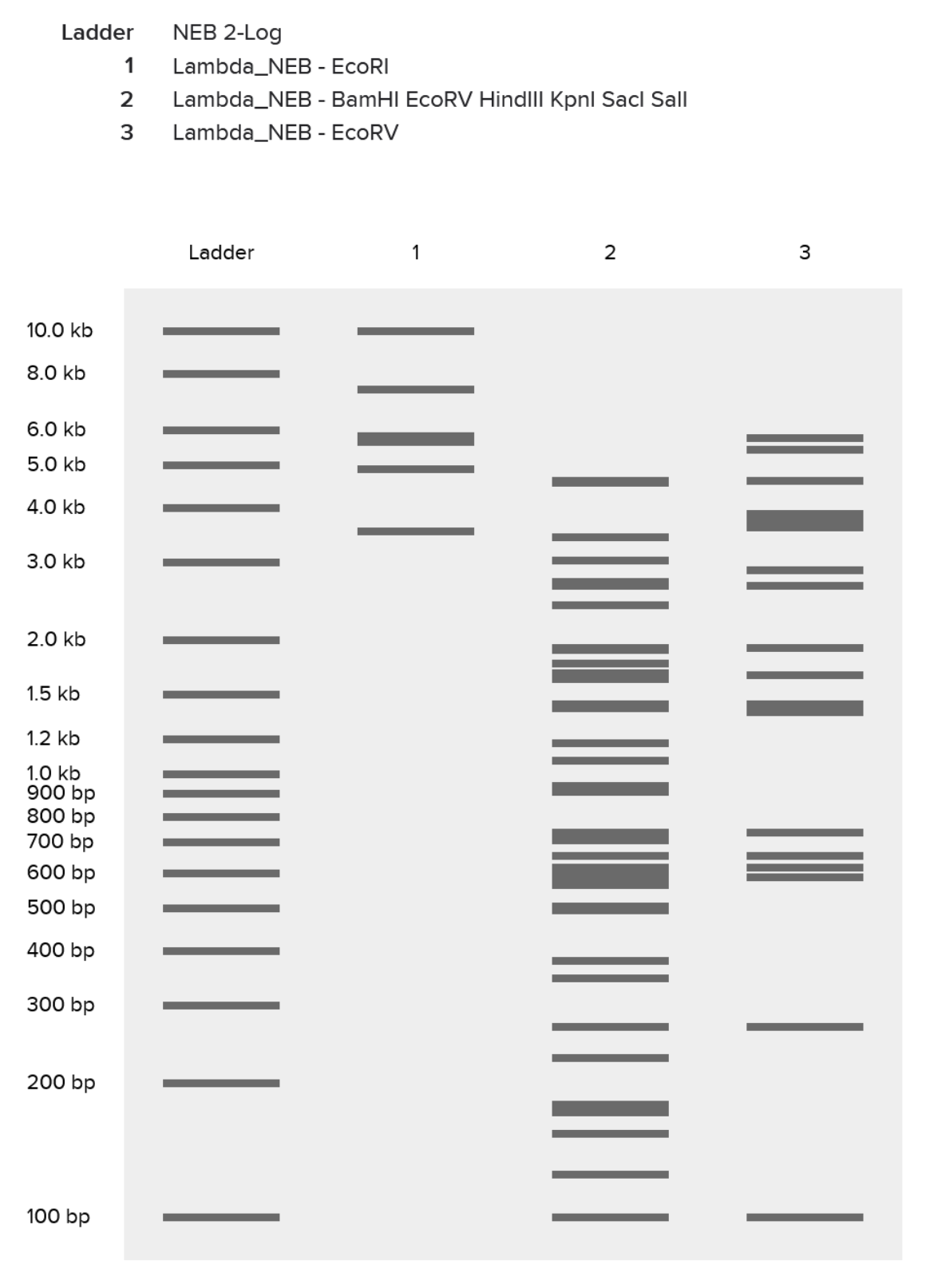
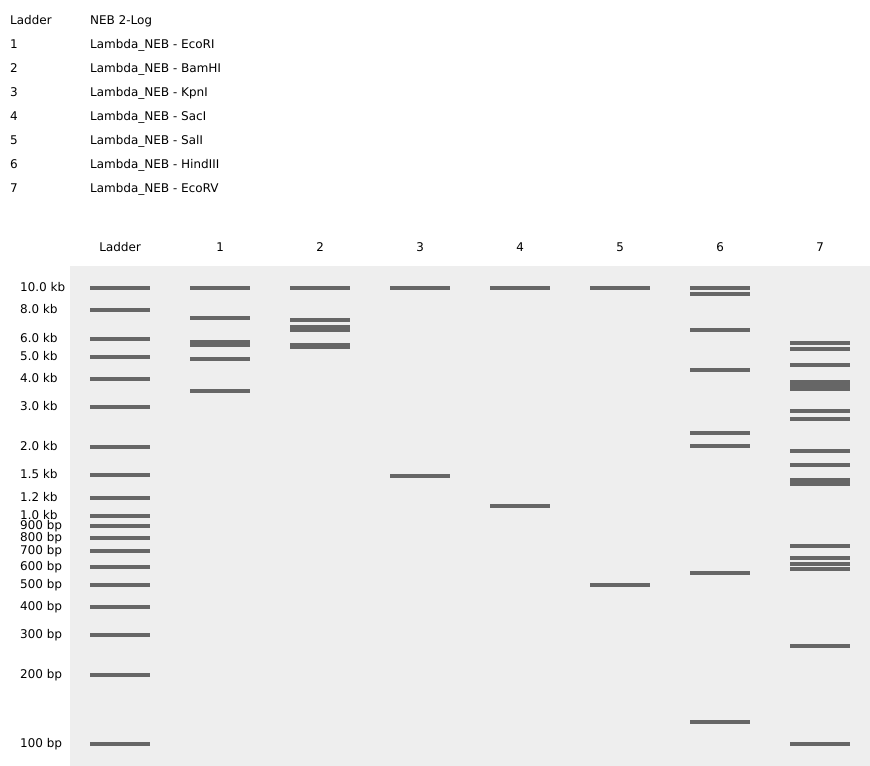
Part 1: Benchling & In-silico Gel Art Attempt Result Description 1 Had an initial mess-up where I tried to “speedrun” the process and ended up with a ladder packed with the effects of multiple restriction enzymes. 2 Finally got success with all of the listed enzymes, separately. 3 Some experimentation on Ronan’s website got me this pattern that sort of looks like a pair of pants. In hindsight I should’ve definitely explored results from a combination of enzymes (e.g. EcoRI and HindIII together), which would’ve given me a bigger range of visual results. 4 Replicated “sort-of pants” on Benchling, and my final result. Part 3: DNA Design Challenge 3.1. Choose your protein.
Python Script for Opentrons Artwork Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. I used https://ginkgoartworks.com/ to draw a mushroom and imported the program into Colab. Since the bacteria names don’t register as RGB colors, I had to “color-correct” well_colors to get the visualization to show up (but I assume both versions will work as long as the PCR tubes are physically in order).
Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) The Dalton is an atomic mass unit that converts from mass using Avogadro’s number. So 500 grams = 500 * 6.022 * 1023 Daltons / 100 Daltons/amino acid = 5 * 6.022 * 1023 amino acids = 3.011 * 1024 amino acids.
Part A: SOD1 Binder Peptide Design Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. P00441 can be found on the UnitProt site here. It has the following sequence:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 1: DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The master mix can be found here and contains “Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2”. The polymerase is an essential part of PCR, nucleotides are the base materials needed to form new DNA sequences, and reaction buffer lowers the energy needed to start the process.
Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Increased complexity in what it can process. Genetic circuits are capable of digital logic, but neural networks can fine tune connections and weights to represent a nuanced system with a set of many inputs and outputs. This is in particular in a cell’s ability to contain a “weighted summation” dependent on the situation it gets trained on.
Part A: General and Lecturer-Specific Questions General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free systems have an advantage in “extreme” situations, while in-vivo occurs within cells that have to be kept alive. In taking the process outside of the cell, you can handle it more roughly i.e. freeze drying the system for long-distance transport, or making a system that can be kickstarted just by adding water (in remote locations). Cell-free systems also have more control because they’re synthetic, so you can determine how big the cell is and what exactly goes in it.
Final Project For your final project: Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail. Waters Part I — Molecular Weight We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse. If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉 Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”) what you liked about the project, and what about this collaborative art experiment could be made better for next year. Unfortunately, I did not get to contribute this year, but I did discuss the project with friends in the class. I really like the concept of a collaborative (and also competitive, occasionally) project with an end result that is artistic (while also leading to the lesson next week). I think the process was very lovely, with people’s ideas growing and shifting until it reaches a fully developed design. I’m not too sure if this would lose the spirit of the assignment, but coordinating within nodes or between people might better guide us towards a final design. I feel like the end result, with the four different designs on each well, was a little lucky.
Subsections of Homework
Week 1 HW: Principles and Practices
Class Assignment
First, describe a biological engineering application or tool you want to develop and why.
There is currently an urgent research focus on the biodegradation of plastics, due to the extremely long life cycle of synthetic polymers. Prior work has focused on a mix of exploring bacterial and microbial processes (e.g. anaerobic digestion) to break down plastics, and developing compositions that can be commercial compostable (e.g. for single use plastics). My personal interest is in fiber arts and sustainability, so I’d like to tackle this problem from a textile perspective.
Fast fashion has exacerbated the volume of cheap, low quality clothes produced everyday. These clothes are often made with synthetic fibers and not for long term use (although the two are not necessarily interchangeable). I believe it’s incredibly important to find a way to biodegrade polyester, one of the most common synthetic polymers in fast fashion clothing.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future.
Ensuring safety:
What material/process is being used for plastic degradation? Any byproducts?
Can we guarantee the safety of workers throughout the process?
Can we guarantee the safety of the surrounding community?
Upholding equity:
Who/what affected areas will benefit the most from this application? More specifically, how can we prioritize places that need the most help (e.g. fast fashion landfills in Chile)?
Textile and fast fashion industries in particular have historically exploited low-cost labor. How can we counter that in our mission to combat fast fashion production?
Promoting a circular economy (avoiding greenwashing):
How can we ensure that this solution is actually helpful? How do we avoid being just another step before the landfill?
What is our end product and its use? What are the byproducts?
Next, describe at least three different potential governance “actions”.
Action 1: Standardization of Process
Purpose: A standard process, with clearly understood materials (end products, byproducts) and equipment, will better communicate the safety and efficacy of industrial facilities to the public. These exist for some products like compostable utensils but not necessarily synthetic clothing.
Design: Research scientists and regulatory safety boards must work in conjunction to develop and validate the process.
Assumptions: That the biodegradation process can be strictly controlled given the wide range of material types in clothing, and that biodegradation can scale up the same way industrial composting does.
Risks of Failure & “Success”: Restriction of the process might make the success rate lower, due to the process being less efficient.
Action 2: Polyester Tracking for Success Metrics
Purpose: Keeping tabs on where the material comes and goes, upstream and downstream, to evaluate how successful we are. Where are we getting polyester from? Where are our outputs going?
Design: We need the cooperation of all parties for data transfer.
Assumptions: That this data is currently already collected, and if not, that it’d be easy to start collecting. E.g. do most clothing landfills track the percentage breakdown of natural vs synthetic fibers?
Risks of Failure & “Success”: Our process is less effective than it appears. E.g. maybe we are primarily biodegrading high-quality athleticwear. Still good, but not the impact we want.
Action 3: Community Awareness
Purpose: Education programs about clothing and material composition can encourage more sustainable practices by the public, as well as more * engagement with our facility.
Design: Planning events, town hall discussions, etc, as well as accepting donations of clothing.
Assumptions: That community support hinges upon understanding of what we’re doing, and the negative impacts on the community are otherwise negligible.
Risks of Failure & “Success”: Education programs are ineffective if the work needed is not in the hands of the community. E.g. if we cannot accept mixed-material clothing, the community cannot necessarily separate material on our behalf. That lies with clothing production facilities.
Action 4: Regulation on Composite Material Production
Purpose: A big bottleneck in clothing recycling is the mixing of different materials (e.g. polyester fabric with cotton stitching, metal zippers, PVC coating). High-level regulations could target the production of these clothing.
Design: Regulation through fines/taxes in local textile facilities. May be harder to regulate overseas production.
Assumptions: That the volume of clothes produced locally is a significant enough % of clothes we take in. That these regulations are effective against overseas textile production.
Risks of Failure & “Success”: We have to turn away a majority of clothes and are only able to focus on a niche in synthetic fibers. Could also end up constricting the companies that choose to produce locally, leading to failure.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why.
I’d focus on Process Standardization, as safety is the absolute first priority. Following that, between Success Metrics and Community Awareness, both have potential to contribute to a circular economy, but I’d like to prioritize Success Metrics for its potential to better target impacted areas down the line. So I’d work on a more technical level to develop more effective processes and data collection (which would likely involve academic institutions/environment-focused agencies).
Ethical Concerns
I’m wary of how effective we’d be in a global setting, especially since my perceived impact with this depends on how well we can affect overseas institutions, where I believe most of fast fashion waste is made and accumulated.
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate for polymerase is 1:106, or 1 out of every 1,000,000 base pairs might be wrong. Meanwhile, the human genome spans billions of base pairs, with a diploid being about 6.3 Gigabase pairs (pr 3.2 Gigabase pairs for a haploid)1. However, the polymerase can go through a proofreading process where it uses exonuclease to remove the nucleotide through the entire monophosphate base2, essentially, allowing the sequence to “backspace” before continuing.
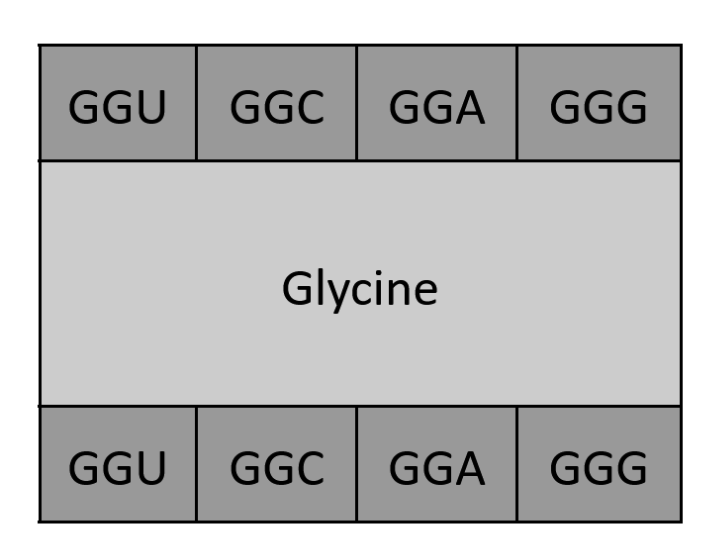
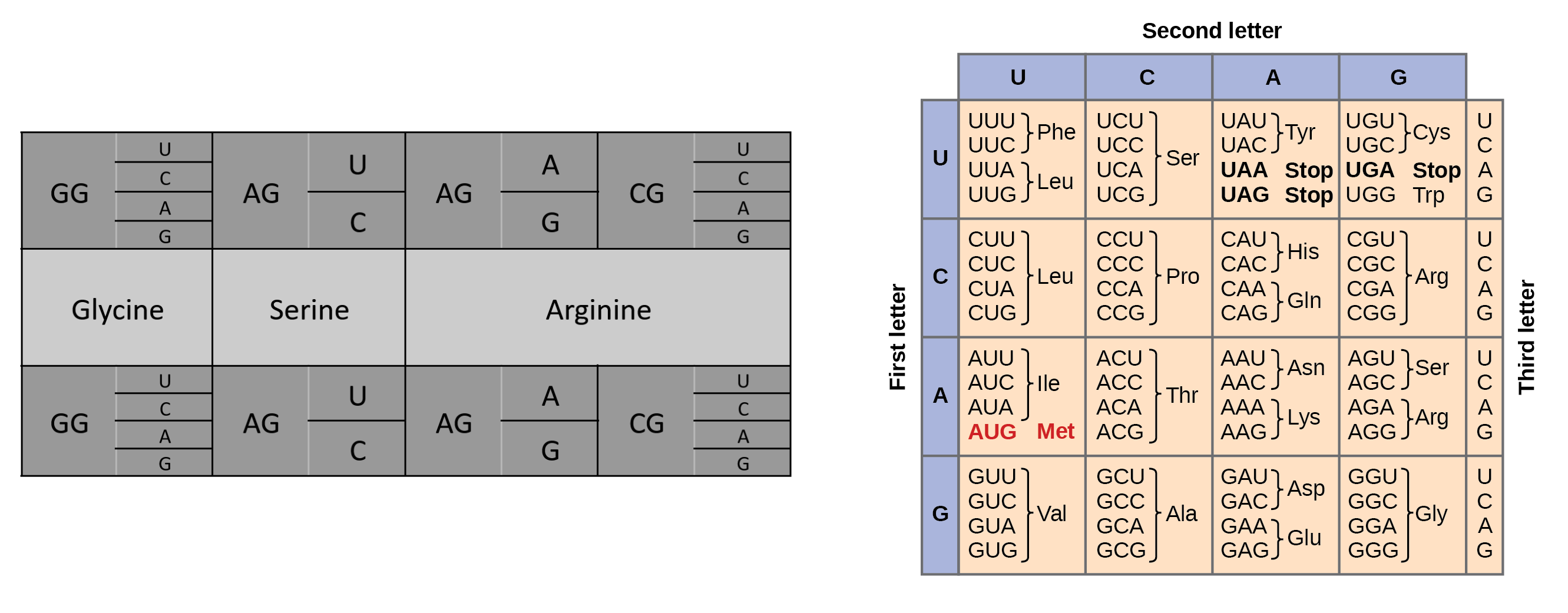
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is about 1036 bp.
Codons are sets of 3 base pairs, so this is about 345 amino acids.
However, many of these amino acids are redundant and can be expressed through multiple base pairs, with 6 being the highest number of variations (in Arginine). We can roughly calculate the amount of variations in a single amino acid through 64 codons/20 amino acids = 3.2 codon variations per amino acid.
This presents about 3.2 variations 345 amino acids, which gives my calculator an overflow error (about 10174).
So that’s clearly an excessive amount of variation for a single protein. However, organisms have developed something called “codon usage bias”, or preference for certain codons evolved over time. This can be due to the following reasons3:
Resource use: different tRNAs recognize different codons, so less variation means more efficient tRNA production.
Protein folding: different tRNAs for codons can translate at different rates. Codons can be deliberately chosen to have the protein fold at “fast translated” sections while waiting for the “slow translated” sections.
Gene expression: certain codons result in stronger gene expression than others. Interestingly, this can work the other way around–codon optimization is a technique that aims to increase protein expression through swapping codons4.
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Oligonucleotides are defined as DNA chains with a length under 200 nucleotides5. Oligo synthesis began with solid-phase synthesis, with additional methods (phosphodiester, phosphotriester, phosphitetriester, phosphoramidite) developed up until the 1980s. Currently solid-phase synthesis using the phosphoramidite method is the most common method; the process was leveraged to implement the first automated DNA synthesizer and has since been optimized for high DNA production volume/thermal control5.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Longer chains have reduced theoretical yield, since each additional nucleotide has an additional “elongation cycle efficiency” (think error rate) that stacks up5. This is calculated with the equation theoretical yield = elongation cycle efficiencynt. Assuming efficiency of 99%,
nt = 100 → yield = .99100 = 0.366
nt = 200 → yield = .99200 = 0.134
nt = 300 → yield = .99300 = 0.049
The phosphoramidite method in particular becomes ineffective beyond 200 base pairs. As a result, more recent alternatives (e.g. enzymatic) are being explored, as research turns to using longer sequences.
Why can’t you make a 2000bp gene via direct oligo synthesis?
If we further calculate using the above equation, the yield becomes
bp = 2000 → yield = .992000 = 0.000000002 = ~0
or effectively zero. At such a high length, the individual “error rates” compound, leaving no chance for success. Current efforts try to improve the process, i.e. increasing the elongation cycle efficiency, or use workarounds like making batches of shorter segments to link together5.
Homework Question from George Church
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
Not sure I’m fully understanding the question, but given there are 64 possible codons for amino acids yet only 20 amino acids, I’d create a code where all possible codons are inputted and outputted as “plaintext” and “ciphertext”, with the encryption “key” being the 20 amino acids they could be interpreted as. Something like the drawing below:
This could even be further streamlined for repeating letters:
(Prof. Church’s slides and paper at [6] used for reference.)
Citations
[1] Piovesan A, Pelleri MC, Antonaros F, Strippoli P, Caracausi M, Vitale L. On the length, weight and GC content of the human genome. BMC Res Notes. 2019 Feb 27;12(1):106. https://doi.org/10.1186/s13104-019-4137-z
[2] Hopfield, JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA. 1974 Oct; 71(10):4135-9. https://doi.org/10.1073/pnas.71.10.4135
[4] Athey J, Alexaki A, Osipova E, Rostovtsev A, Santana-Quintero LV, Katneni U, Simonyan V, Kimchi-Sarfaty C. A new and updated resource for codon usage tables. BMC Bioinformatics. 2017 Sep 2; 18(1):391. https://doi.org/10.1186/s12859-017-1793-7
[5] Hoose, A., Vellacott, R., Storch, M. et al. DNA synthesis technologies to close the gene writing gap. Nat Rev Chem 7, 2023 Jan; 144–161. https://doi.org/10.1038/s41570-022-00456-9
[6] Acevedo-Rocha CG, Budisa N. Xenomicrobiology: a roadmap for genetic code engineering. Microb Biotechnol. 2016 Sep; 9(5):666-76. https://doi.org/10.1111/1751-7915.12398
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
Attempt
Result
Description
1
Had an initial mess-up where I tried to “speedrun” the process and ended up with a ladder packed with the effects of multiple restriction enzymes.
2
Finally got success with all of the listed enzymes, separately.
3
Some experimentation on Ronan’s website got me this pattern that sort of looks like a pair of pants. In hindsight I should’ve definitely explored results from a combination of enzymes (e.g. EcoRI and HindIII together), which would’ve given me a bigger range of visual results.
4
Replicated “sort-of pants” on Benchling, and my final result.
Part 3: DNA Design Challenge
3.1. Choose your protein.
I recently read about snake venom and how its majority composition of proteins/enzymes make it (theoretically) edible, since it can be digested in the stomach. That was a pretty fun fact. For this assignment, I picked irditoxin, a three-finger toxin that is selectively neurotoxic towards birds and lizards (but not mammals).
Codon optimization can be useful in controlling gene expression within a sequence (both increasing and decreasing it). It can also make mRNA production more efficient and impact translation speed, which can in turn affect things like folding speed (fast-translated sequences fold while waiting for slow-translated sequences).
I tried out IDT’s Codon Optimization Tool. IDT recognized a couple different stop and start codons, so I picked the sequence between the first codon (ATG, a start) and the first stop codon (TGA at position 330). The sequence was optimized for E.coli to go for a standard, well understood, commonly used organism.
Shortened old sequence:
ATG AAA ACT CTG CTG CTG GCC GTG GCG GTG GTG GCA TTC GTG TGC CTG GGC TCA GCT GAT CAG CTG GGA CTC GGA AGG CAG CAA ATA GAT TGG GGA CAA GGC CAA GCA GTA GGT CCA CCC TAC ACA CTT TGT TTC GAA TGC AAT CGA ATG ACT TCT TCG GAT TGT TCA ACC GCT TTG AGA TGT TAC CGC GGA TCG TGC TAC ACT TTG TAC AGA CCT GAT GAG AAT TGT GAA TTG AAA TGG GCT GTC AAG GGA TGT GCT GAA ACG TGC CCT ACT GCG GGA CCT AAT GAG AGG GTG AAG TGT TGC AGA TCA CCA AGG TGC AAC GAT GAT TGA
Another mess-up: IDT denied the optimized sequence due to its complexity, which means this sequence isn’t currently manufacturable and needs to be further redesigned. Seems some of my enzyme recognition sites weren’t ideal.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
As we’re working with a neurotoxin, and not for high-volume production, I’d probably turn to a cell-free method.
Part 4: Prepare a Twist DNA Synthesis Order
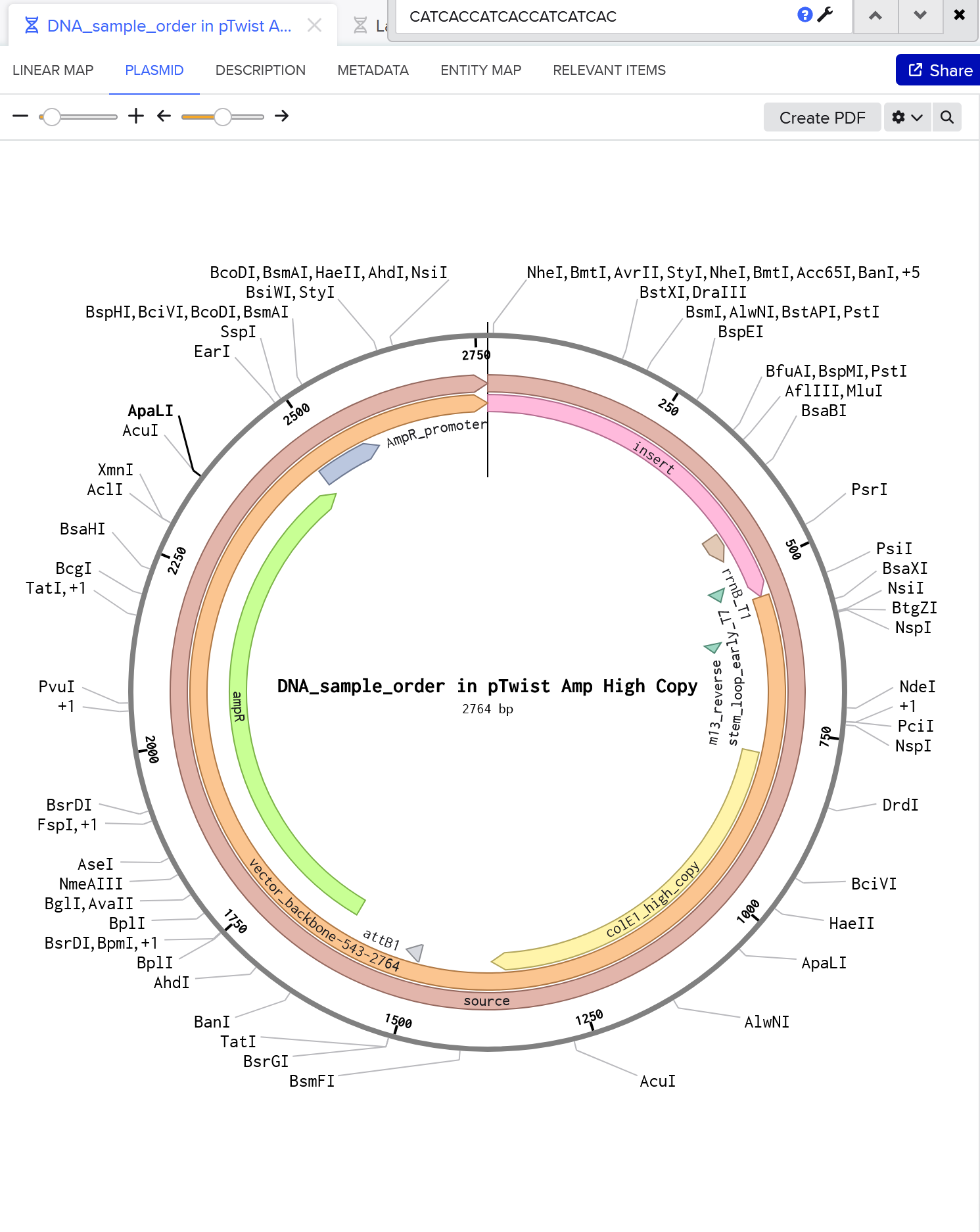
4.1-2. Create a Twist account and a Benchling account, Build Your DNA Insert Sequence
4.3-6. Select The “Genes” Option, “Clonal Genes”, Import your sequence, and Choose Your Vector
Here’s my plasmid. She’s beautiful!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I think bioindicators are an interesting group of organisms, and sequencing them could help us isolate genes that react to surroundings and get used for a more standardized, widespread environment monitoring tool. E.g. microalgae can detect wide range of water quality issues, from heavy metals to nanoparticles, yet “only a few species have been fully sequenced without any gaps”1.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
I’d likely use a NGS method to handle the sequencing of the entire genome, and of the multiple species that we come across at that. So potentially something like PacBio, which is third-generation. Both the sample used and the library need to be prepared, with the sample needing to be purified and the DNA needing to be fragmented to length and end-capped. The DNA is decoded through a polymerase that runs along the sequence. As it interacts with each nucleotide, it emits light, which is recorded live and appended onto the current sequence. The end result is a straightforward sequence of DNA nucleotides, given in a file that can be read on any notepad app.
5.2 DNA Write
What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I’m continuously interested in biomaterials as an alternative to things like plastic. There’s been a lot of work on getting plastic that is biodegradable, and I’m wondering if there’s a way to go about it from the opposite direction, like fortifying kombucha leather to last longer.
What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Solid-phase synthesis using the phosphoramidite method seems to be the go-to method, so I’d stick with that. The steps are coupling the base with phosphoramidite, capping unreacted sites, oxidating the phosphate, deblocking, and then repeating as needed. The limitation to this process is that it decreases in efficiency past a certain number of bp (~200 as discussed last week) so could potentially be difficult as the needed sequence becomes longer.
5.3 DNA Edit
What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
This feels like a touchy subject in line with our ethical considerations from last week. The responsibility of human genome sequencing seems enormous, so I’ll consider other organisms. I’m thinking of how filter feeders play an important role in the water ecosystem, essentially “purifying” the water. Could something like that be intentionally edited into plants (or other microorganisms) to boost its “purifying” effect on the air?
What technology or technologies would you use to perform these DNA edits and why?
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I’m unsure of this part as I’m not too familiar with the work needed to introduce this behavior into land organisms. I believe CRISPR-Cas9 is also a go-to for gene editing, so would probably be a popular approach regardless. The process begins with a guide RNA finding the target sequence in the DNA, at which Cas-9 “cleaves” the segment. Then either new DNA can be added (replacing the segment), or the DNA strands repair (deleting the segment). For preparation, the appropriate guide RNA needs to be sourced. A limitation of CRISPR-Cas9 is in its effectiveness; it has the potential to insert DNA incorrectly, which can lead to mutations when applied to human genome sequencing.
Citations
[1] Evangelia Stavridou, Lefkothea Karapetsi, Georgia Maria Nteve, Georgia Tsintzou, Marianna Chatzikonstantinou, Meropi Tsaousi, Angel Martinez, Pablo Flores, Marián Merino, Luka Dobrovic, José Luis Mullor, Stefan Martens, Leonardo Cerasino, Nico Salmaso, Maslin Osathanunkul, Nikolaos E. Labrou, Panagiotis Madesis, Landscape of microalgae omics and metabolic engineering research for strain improvement: An overview,
Aquaculture, Volume 587, 2024, https://doi.org/10.1016/j.aquaculture.2024.740803.
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
I used https://ginkgoartworks.com/ to draw a mushroom and imported the program into Colab. Since the bacteria names don’t register as RGB colors, I had to “color-correct” well_colors to get the visualization to show up (but I assume both versions will work as long as the PCR tubes are physically in order).
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
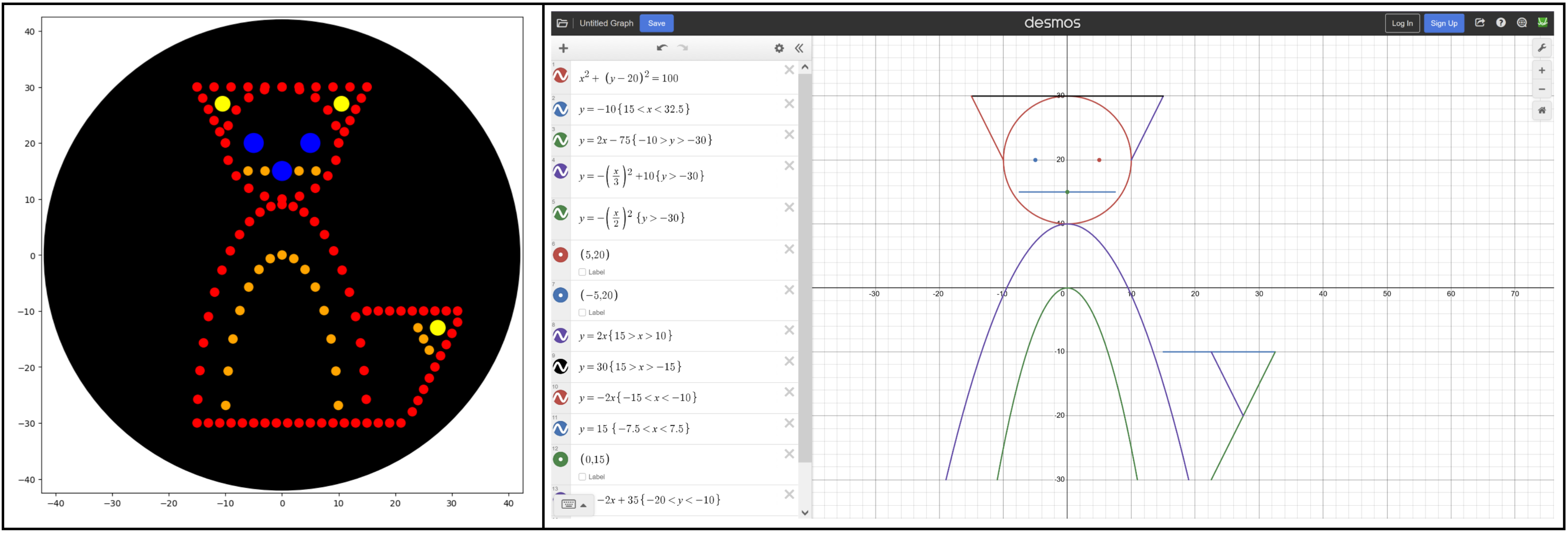
I tried to play around with math functions to create a new design, like the Mathematical Heart sample. I drew up a cute fox in Desmos graphing calculator using the following functions, making sure to scale them to the 40 mm limit. Transferring that to Colab was a bit more difficult, and I had to play around with the functions, ranges, and dispense volume to find something that looked good.
Some notes from the process:
Polar functions are ideal for spacing out the points. I used polynomial equations (y=x2) for the body but ended up switching to ellipses (y2 + x2 = r2).
I had trouble picking up/dropping tips, especially with the color changes in each part of the fox. So my program appends all points to lists sorted by color and then dispenses each list in a draw() function (like the Mathematical Heart).
I had a lot of trouble tracking volume to aspirate, especially as some for loops go into the negatives, so my refill is based on the actual volume of the pipette (pipette_20ul.current_volume) and is handled all at once in the draw() function.
Colab link for both projects here, including the color-corrected version for the mushroom.
Extra: Nebula art
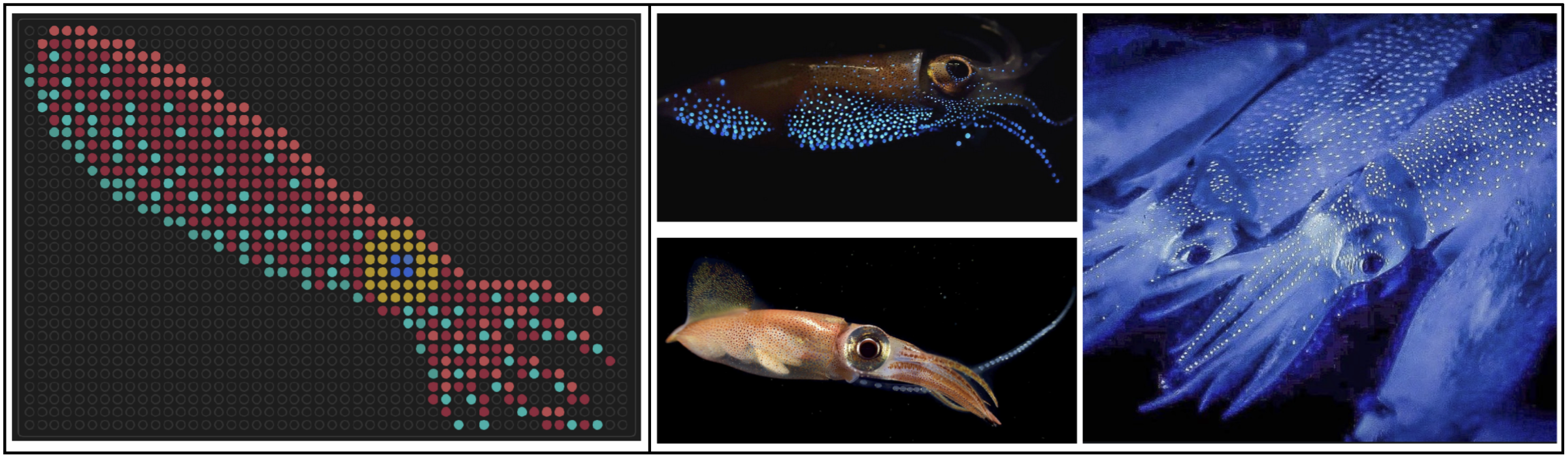
I designed some artwork for the 1536-well plates on the Nebula, which were made during the Saturday 2-6 pm Cloud Lab session. The first one was a firefly squid, inspired by bioluminescent photos I’ve seen of them underwater. Link to gallery image here.
I also made a second one resembling the Chinese jianzhi for Lunar New Year. I experimented with two color sets, to see how bacteria with similar coloring would contrast against each other. Link to gallery images here and here.
Here’s all the fluorescent artwork from that session!
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
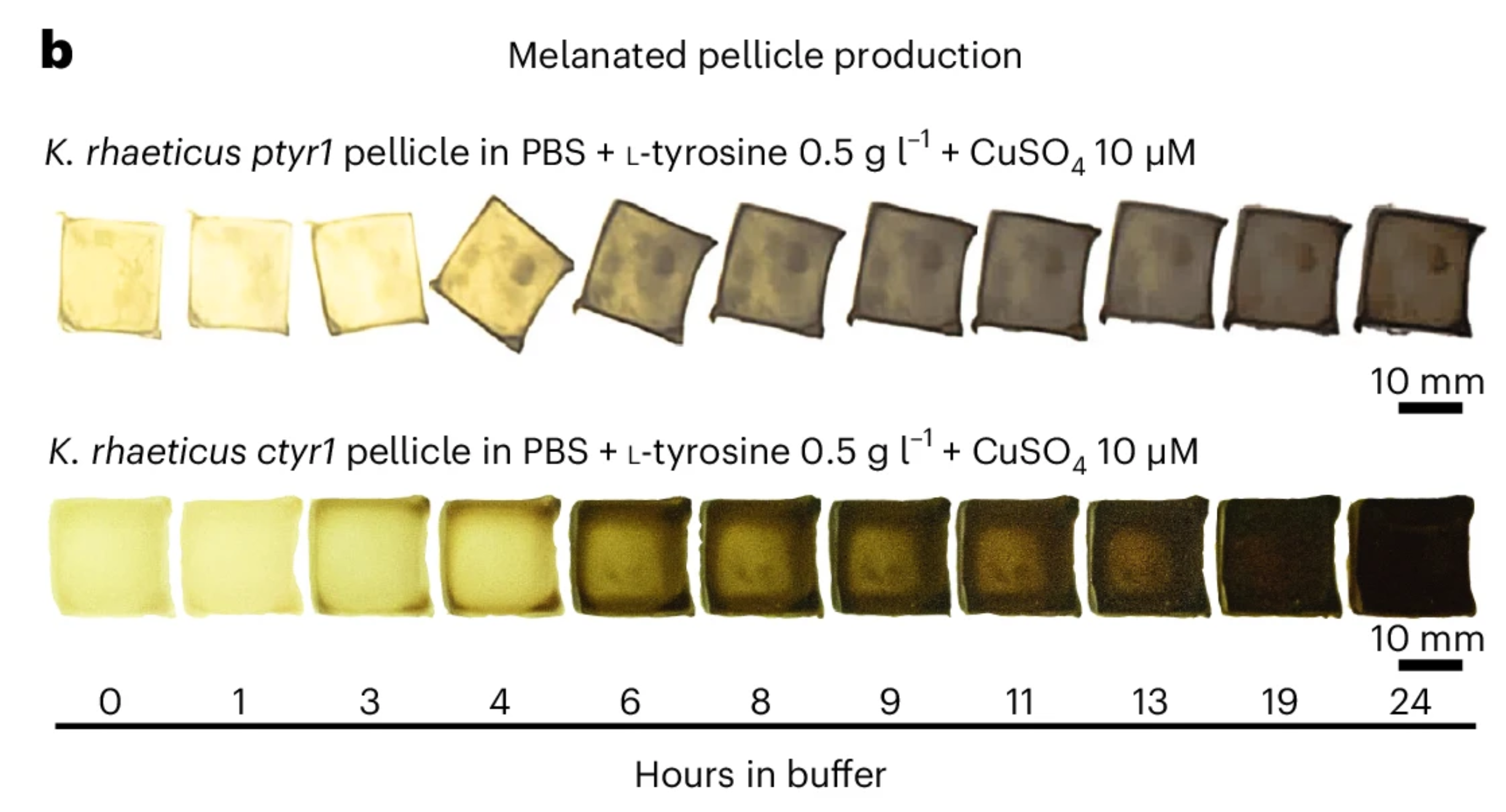
This paper explores “dyeing” bacterial cellulose, a bioplastic alternative to leather, which I found pretty interesting. Instead of applying an independent biodegradable dye, the researchers engineered Komagataeibacter rhaeticus to develop eumelanin (dark melanin), which gives a range of shades seen in the photo above. Opentrons is used in the production of the eumelanin development buffer, in which the K. rhaeticus pellicle is incubated to “dye” itself. The robot mixes the buffer, cells, and supernatant within a reaction plate while maintaining a constant low temperature to prevent initial eumelanin growth.
Walker, K.T., Li, I.S., Keane, J. et al. Self-pigmenting textiles grown from cellulose-producing bacteria with engineered tyrosinase expression. Nat Biotechnol 43, 345–354 (2025). https://doi.org/10.1038/s41587-024-02194-3
Write a description about what you intend to do with automation tools for your final project.
More research is needed on my part for this, but I’d like to explore scaling up or going in depth with the range of results for my project (e.g. if self-pigmenting, then trials to develop swatches of colors). This would require a lot of samples, and liquid handlers like the Opentrons would be necessary for producing all the samples indentically.
Coloring Bioplastic/Biotextiles (with an art-focused approach)
Self-pigmenting Bacterial Cellulose: Building on the above paper, further development with dyed bacterial cellulose using pheomelanin instead of eumelanin for a different color range. Likely this is already being explored, so as an ambitious goal Komagataeibacter rhaeticus could be edited to express both pheomelanin and eumelanin, allowing you a 2D range of colors.
Structural Color on Textiles as Biopigment: Naturally occuring structural color is tied to the genome, so we could intentionally induce colors as a natural, biodegradable dyes for textiles. This paper highlights a bacteria that naturally form to output structural color, and this paper explores gene knockout to change the color expressed by one of the bacterias Flavobacterium IR1. A potential project could explore different colors on these bacteria, or find a way to introduce the bacteria to textiles without affecting its formation.
Environmental Sensors (Algae biosensors?)
I’d like to explore fluorescence through engineering algae to detect toxic pollutants. I’d be interested in getting a biosensor to detect a singular type of pollutant (e.g. presence of heavy methods), which would require multiple strains of microalgae versus just one general strain that reacts uniformly to all pollutants. So part of this work would probably also include characterizing different responses to find the one that points to that pollutant.
Polyester Biodegradation
Integration with Byproduct Biodegradation: Ideonella sakaiensis is a bacteria that can break down PET plastic through a two step process involving PETase and MHETase enzymes. The end products include carbon dioxide, water, and methane, which in itself a pollutant. Methane itself is a subject of research, with methanotrophs being a type of bacteria that metabolize methane. I’m wondering if the bacteria used for breaking down plastic can be somehow integrated with the added function of breaking down methane through gene engineering.
Polyester-Eating Enzymes: This is a less familiar topic for me, but current work on enzyme degradation focuses on improving the performance of natural enzymes, e.g. its thermostability, pH control, etc. Since there’s such a wide range of work being done, I’m sure there’s some further testing that could be done on an underfocused bacteria/performance metric/modification method.
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
The Dalton is an atomic mass unit that converts from mass using Avogadro’s number. So 500 grams = 500 * 6.022 * 1023 Daltons / 100 Daltons/amino acid = 5 * 6.022 * 1023 amino acids = 3.011 * 1024 amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Our bodies don’t interact with the meat that way. It’s a matter of absorbing the energy and nutrients from the cells, not carrying the cells over to reuse them. The protein also goes through our stomach and digestive track, which is a process more intended to break down what we eat and extract from it.
Why are there only 20 natural amino acids?
The natural amino acids are determined by codons, which are determined by three nucleotides (of which can be adenine, uracil, guanine, cytosine). This gives 4 x 4 x 4 = 64 total codons, but redundancy among codons produces only 20 unique amino acids. More is definitely possible, but 20 seems to encapsulate all the amino acids we need.
Can you make other non-natural amino acids? Design some new amino acids.
Natural amino acids come from the range of nucleotides across 3 spaces, 4 * 4 * 4 = 64. We can design new amino acids in a variety of ways, one notable way being reversing the chirality (from L-amino acids to D-amino acids), which could sort of be applied to any amino acid in existence for a “new” design.
Where did amino acids come from before enzymes that make them, and before life started?
The 1953 experiment making “Primordial Soup” proved that amino acids (and other organic compounds) could come from natural reactions under ideal conditions with inorganic compounds (methane, ammonia, and hydrogen). It seems like there’s a couple of different ways organic compounds could be created abiotically.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If the helices are usually right handed, then this one will be left handed (opposite to what is normal).
Can you discover additional helices in proteins?
Additional helices can always form in new proteins, though I don’t think you can “introduce” new helices to a protein whose folding pattern is known. But since new proteins are made all the time, yes, there will frequently be helices in their shapes, which can be figured out through protein modeling/prediction.
Why are most molecular helices right-handed?
Not exactly sure why it became this way, but it definitely stays this way for ease of transcription and translation. One type of chirality tends to be dominant so that all templates and future proteins/molecules can be identical and interact with each other. Otherwise, left- and right- handed molecules have the exact same properties, but being able to piece together requires the same handedness.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Initially, you have multiple β-strands that can arrange laterally. These strands can sort of reach left and right and “link arms” with neighboring strands through strong hydrogen bonds. The repeated structure in each strand allows for repeated patterning.
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
β-sheets stem from a presence of strands in the protein with hydrogen backbones, so I’m guessing amyloid diseases often have that chemical makeup. Then it’ll tend to form the sheets just because it’s a very stable configuration.
Design a β-sheet motif that forms a well-ordered structure.
Motifs appear to be ways to arrange the strands laterally against each other. This is easy to do in concept but might not reflect motifs that can occur naturally. I’m imagining a potential concept where you take a sheet and fold two ends to make a cylinder–that’s the current beta barrel design. If you imagine a long, rectangular sheet, though, there’s a way to fold it to make sort of a ribbon (like alpha helices’ shape). I wonder if it’s possible to get to that shape with very long but few beta strands.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I picked crystallin, which is a protein in the eye responsible for the movement of your iris as you focus. It’s notably transparent, being part of the eye lens, and water-soluble, which was a callback to our lecture. I picked the protein because I was interested in how cataracts were formed.
The specific protein I went with for the following questions is P02511, or Alpha-crystallin B (in humans).
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Using the Colab notebook, the protein is 175 amino acids long with the most common amino acid being P (and appearing 17 times).
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
According to UniProt, it’s part of the small heat shock protein (HSP20) family, along with all other Alpha-crystallin B proteins. However, according to the Transporter Classification Database, it’s part of the α-Crystallin Chaperone (CryA) family (where other Alpha-crystallin B proteins don’t appear).
Does your protein belong to any protein family?
Homology refers to protein sequences that likely have a common ancestor (identified through having similarities in sequence/structure?). Using the BLAST software gives 250 results for similar proteins, with results primarily appearing to be Alpha-crystallin B in different animals.
Identify the structure page of your protein in RCSB
This step was particularly difficult for me, as I didn’t always understand how to get to the answer based on what I had on the screen.
When was the structure solved? Is it a good quality structure?
The structure seems to be initially solved in 2009 but has increased members up until 2025. Some particularly high resolution structures were identified in 2012 and 2014 through X-ray diffraction, with a resolution of 1.0 - 1.5 Å.
Are there any other molecules in the solved structure apart from protein?
I’m not entirely sure how to identify this…at least visually, I didn’t identify any other components that that seemed to stick out as an entirely different molecule.
Does your protein belong to any structure classification family?
Using the Structural Classification website, it belongs to the “Alpha crystallin-like” family, further within the “Hsp20 chaperone-like” family.
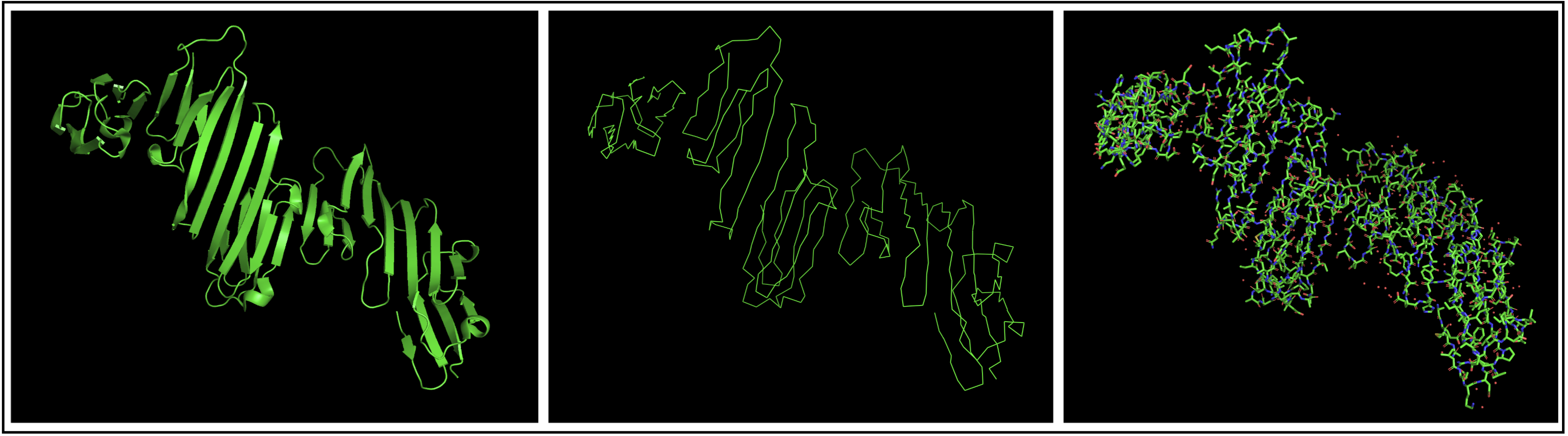
Open the structure of your protein in any 3D molecule visualization software:
I chose to use PyMol to open my structure, getting the structure below.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
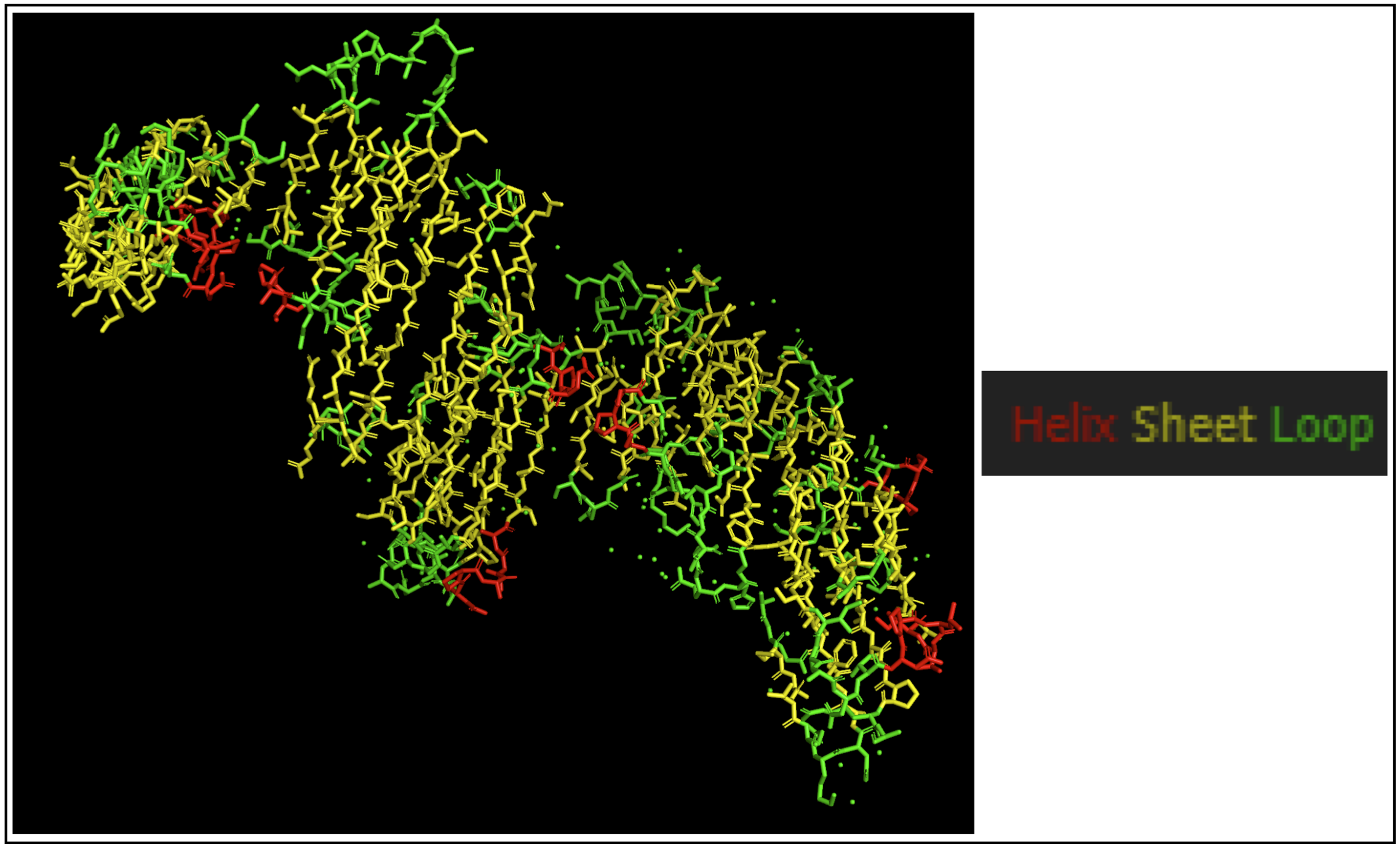
Color the protein by secondary structure. Does it have more helices or sheets?
The protein seems to mostly be composed of sheets with some helices.
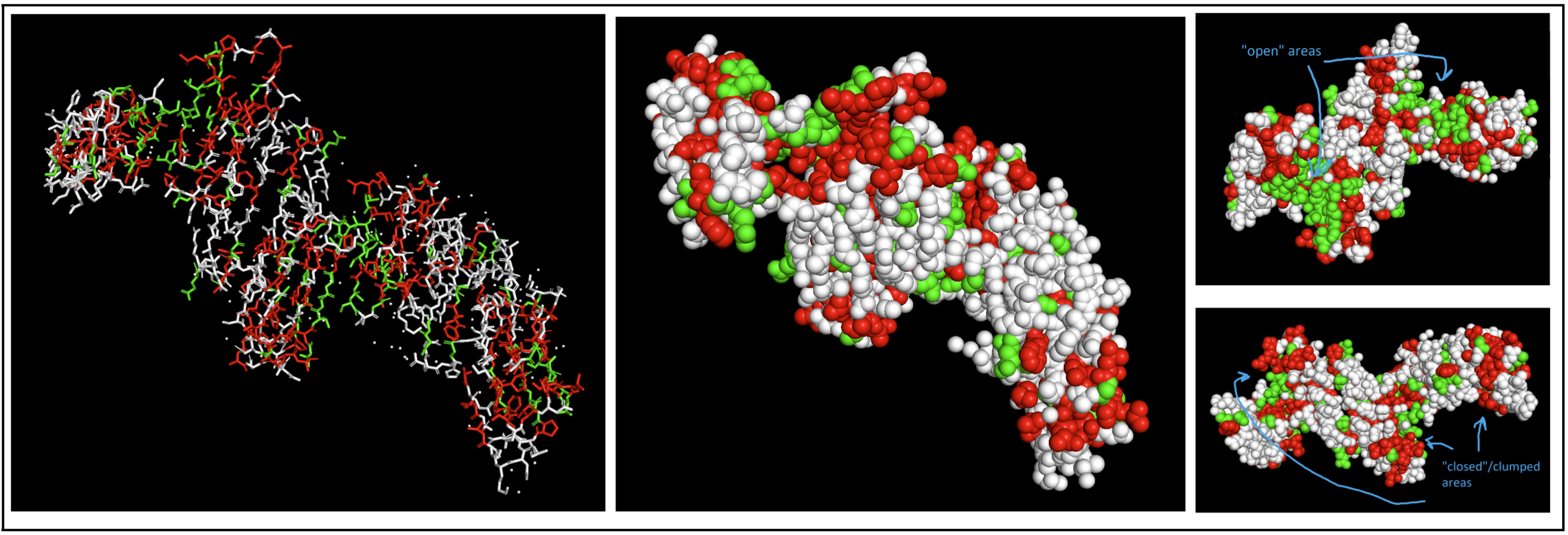
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I colored hydrophobic residues in red and hydrophillic residues in green.
I had to switch to a spheres visualization to better see how molecules were interacting. It was a little hard for me to see a significant pattern, but I do feel like the hydrophilic residues have more “open” facing areas, whereas the hydrophobic residues were more clumped (both together and with neighboring residues).
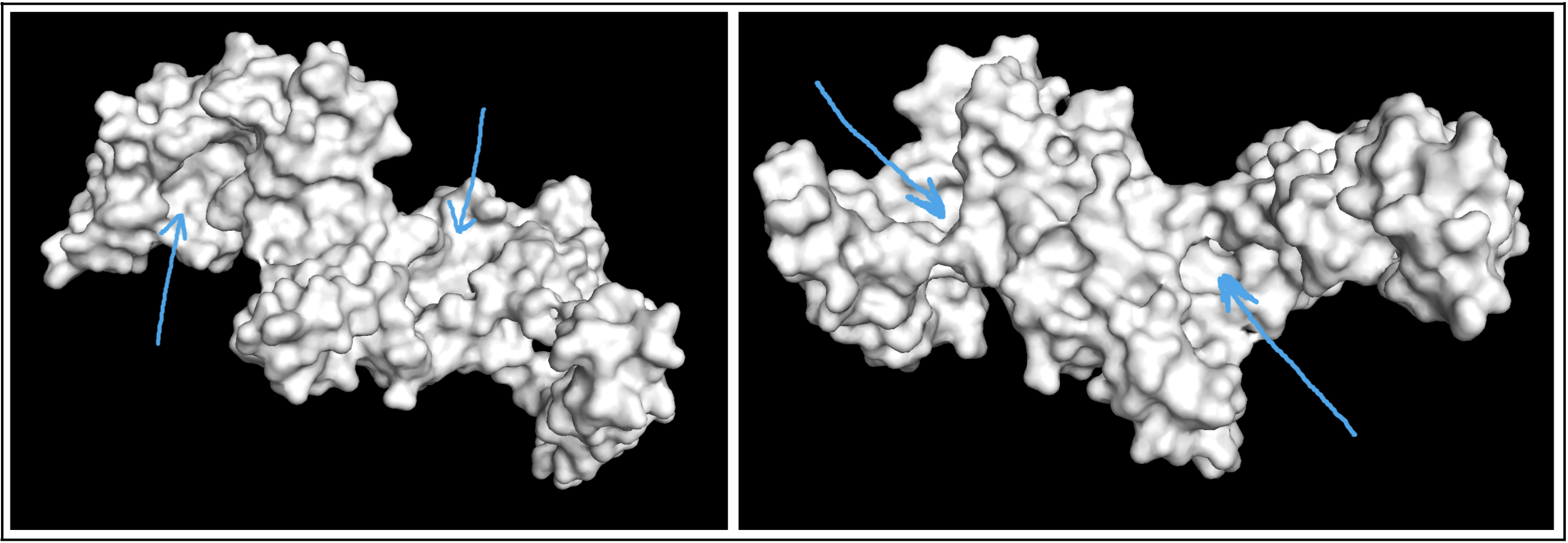
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Visualizing the protein as a surface was really helpful! I could easily find a couple areas that could be binding pockets. It’s a little difficult to show it accurately in a photo, but I indicated potential areas below:
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
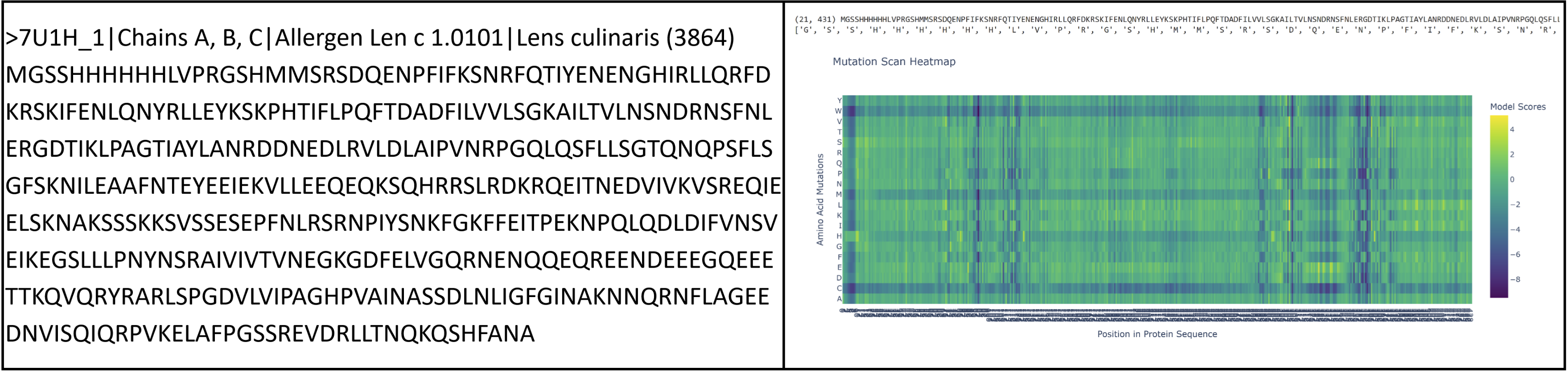
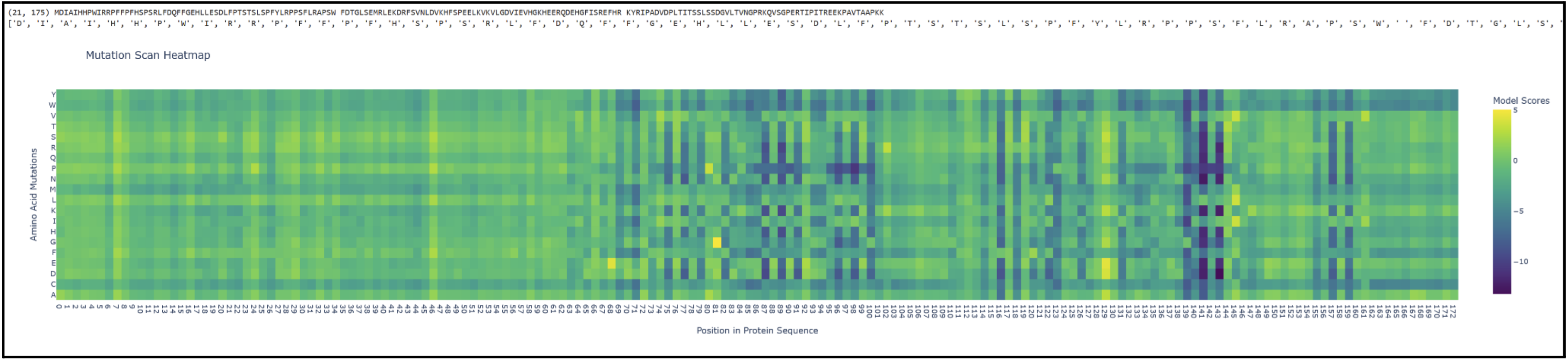
I had to pivot here as I originally chose vicilin from lentil beans (Lens culinaris), which was a type of globulin storage protein. It has a rather long sequence (shown below) and as a result made reading some results pretty challenging. I ended up switching back to crystallin from Part B.
C1. Protein Language Modeling
Deep Mutational Scans
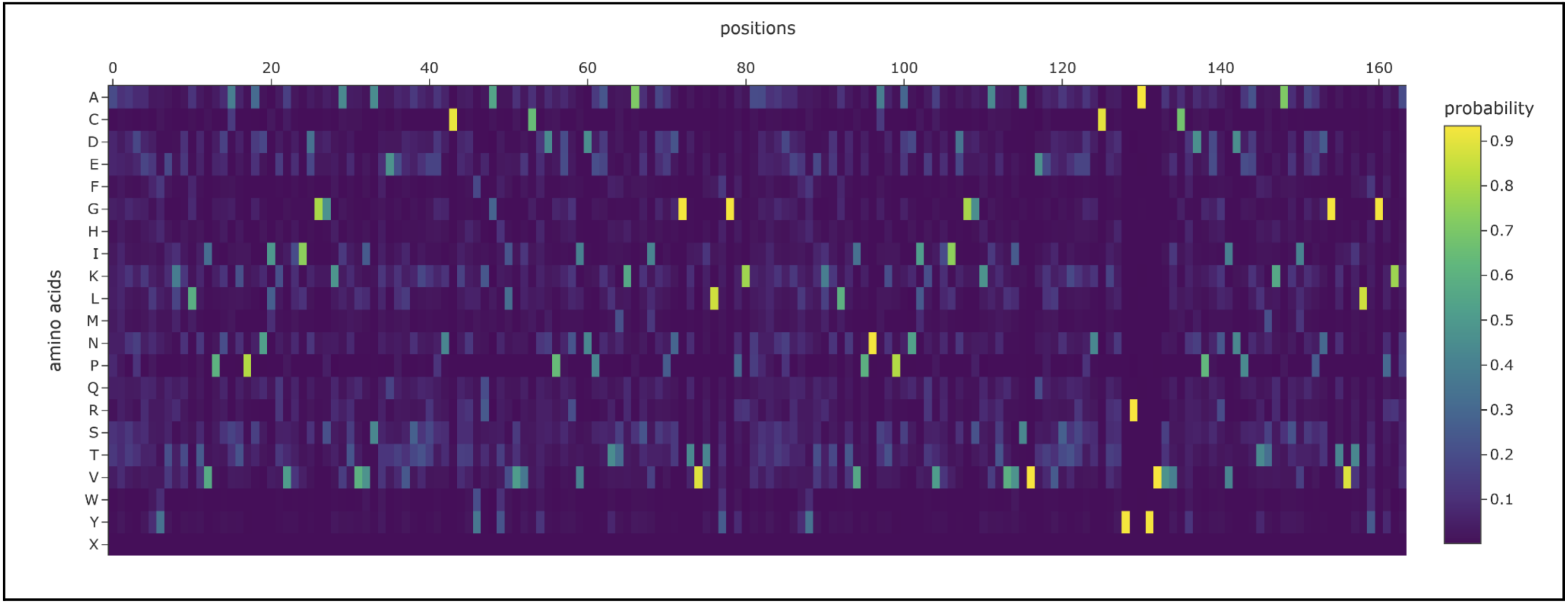
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Proteins in the latter ⅔ of the sequence, particularly 87 (L), 89 (V), 96 (I), 98 (V), 139 (G), 141 (L), and 143 (V), seem subject to severely detrimental mutations (looking at the dark blue streaks). This indicates to me that Leucine and Valine are rather important residues that should avoid mutation.
Meanwhile, the first ⅓ of the sequence seems pretty tolerant to any changes. Notably, protein 129 seems like it can be mutated with mostly beneficial outcomes.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
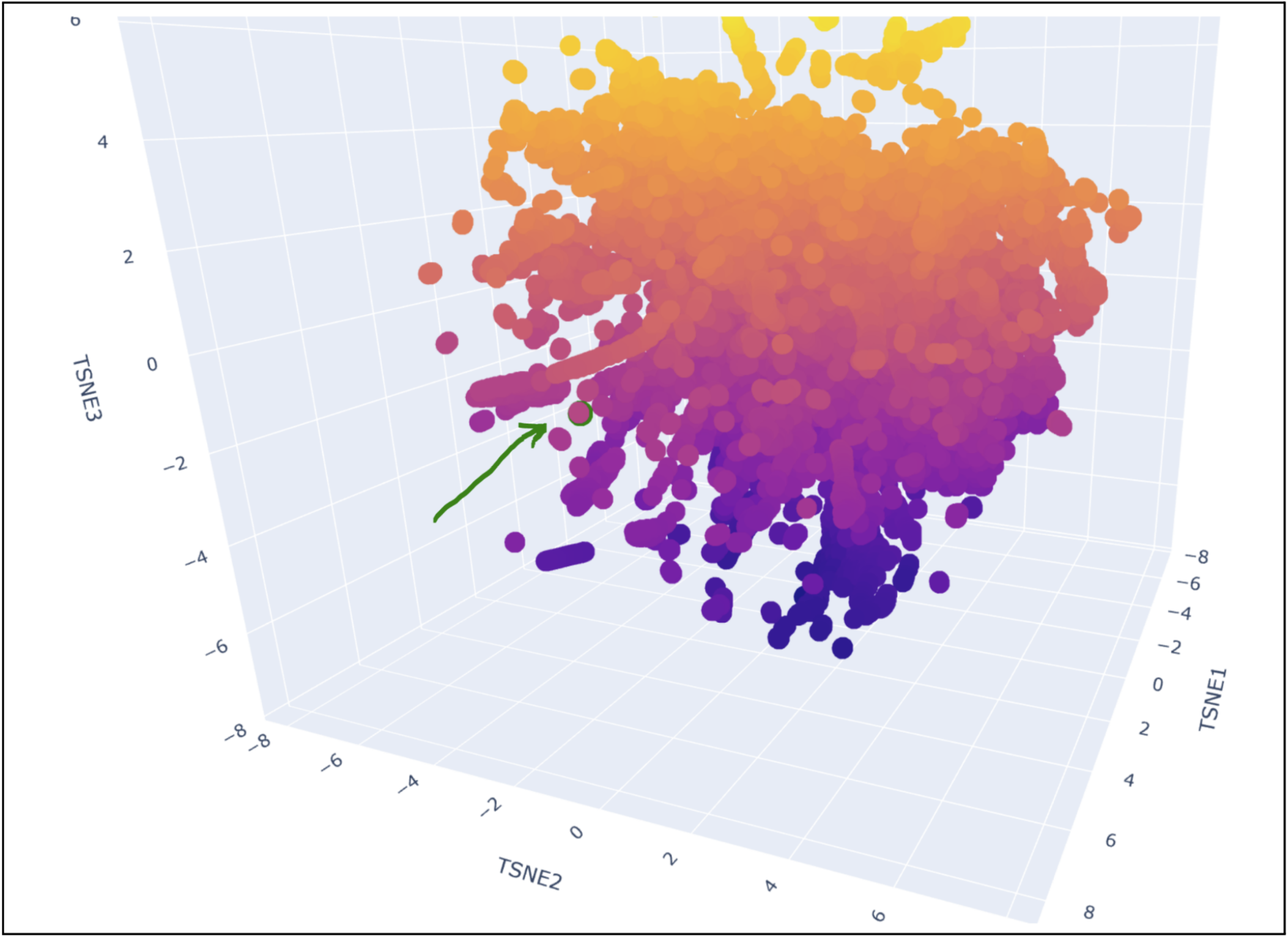
Place your protein in the resulting map and explain its position and similarity to its neighbors.
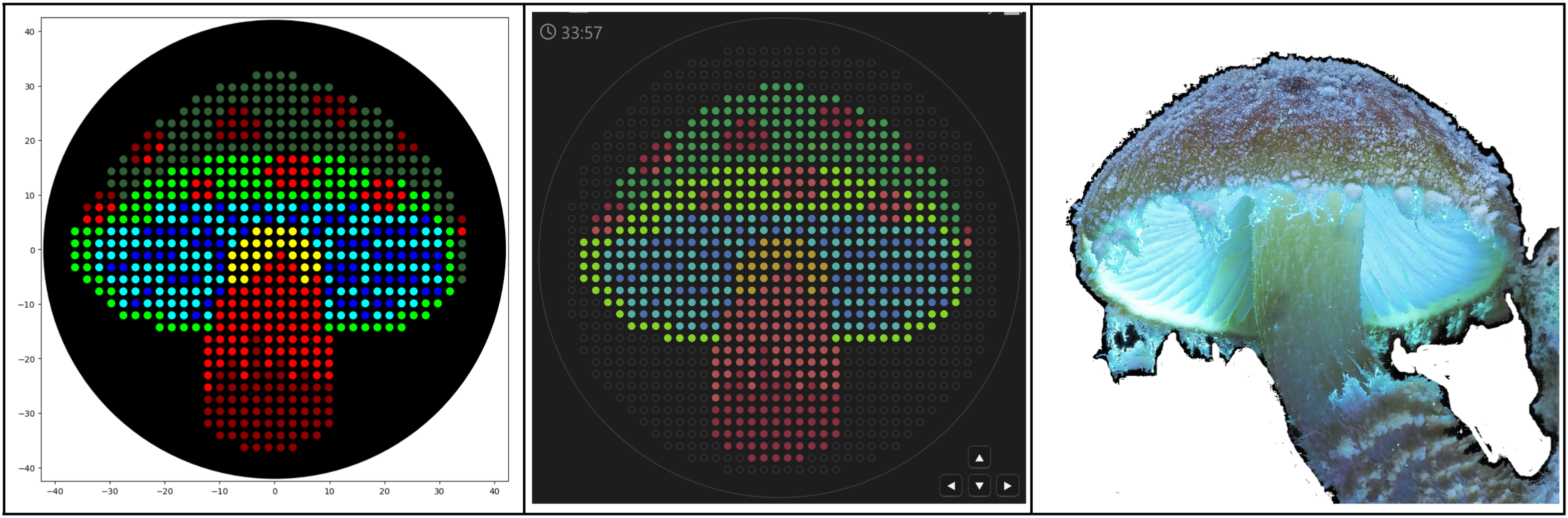
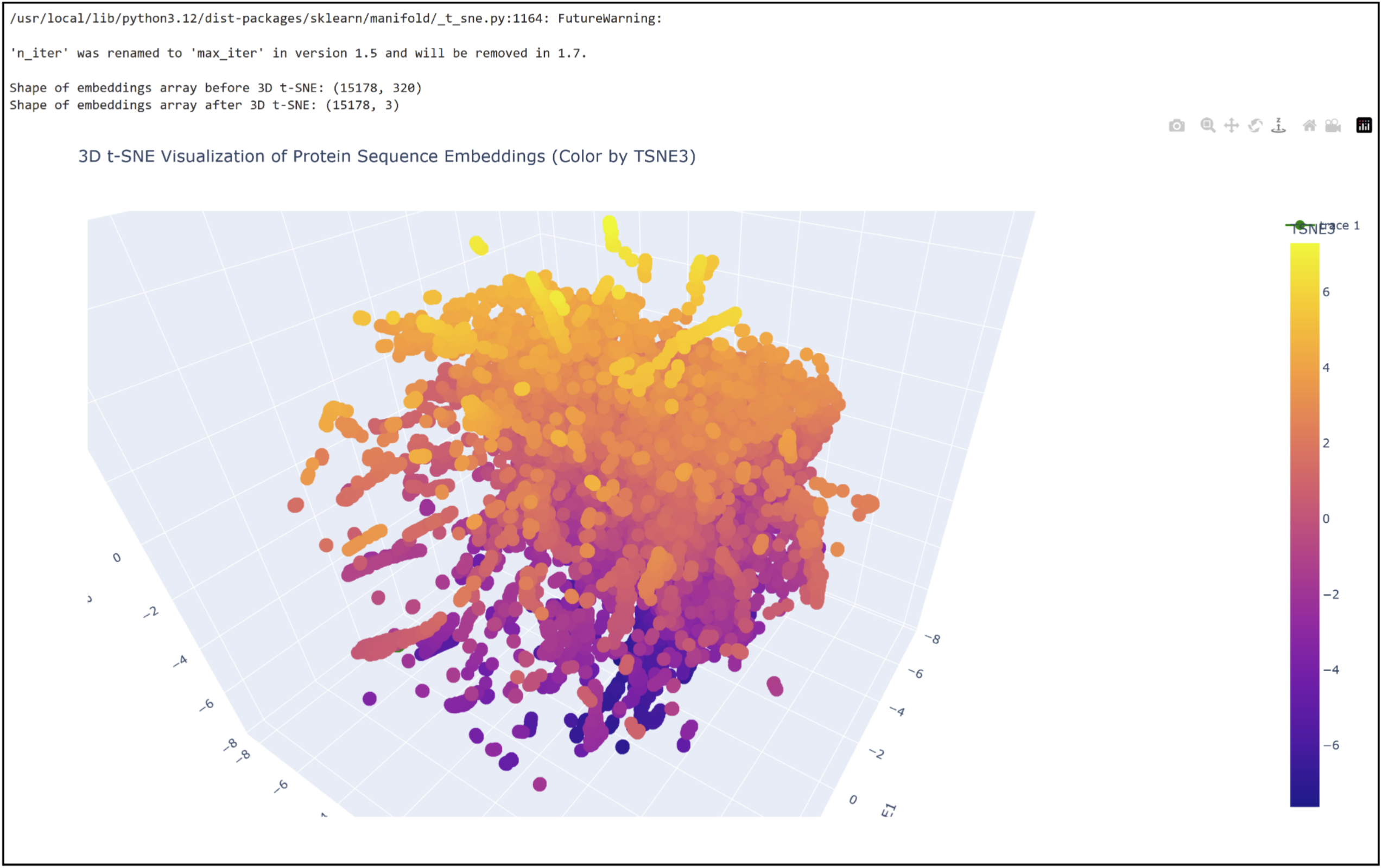
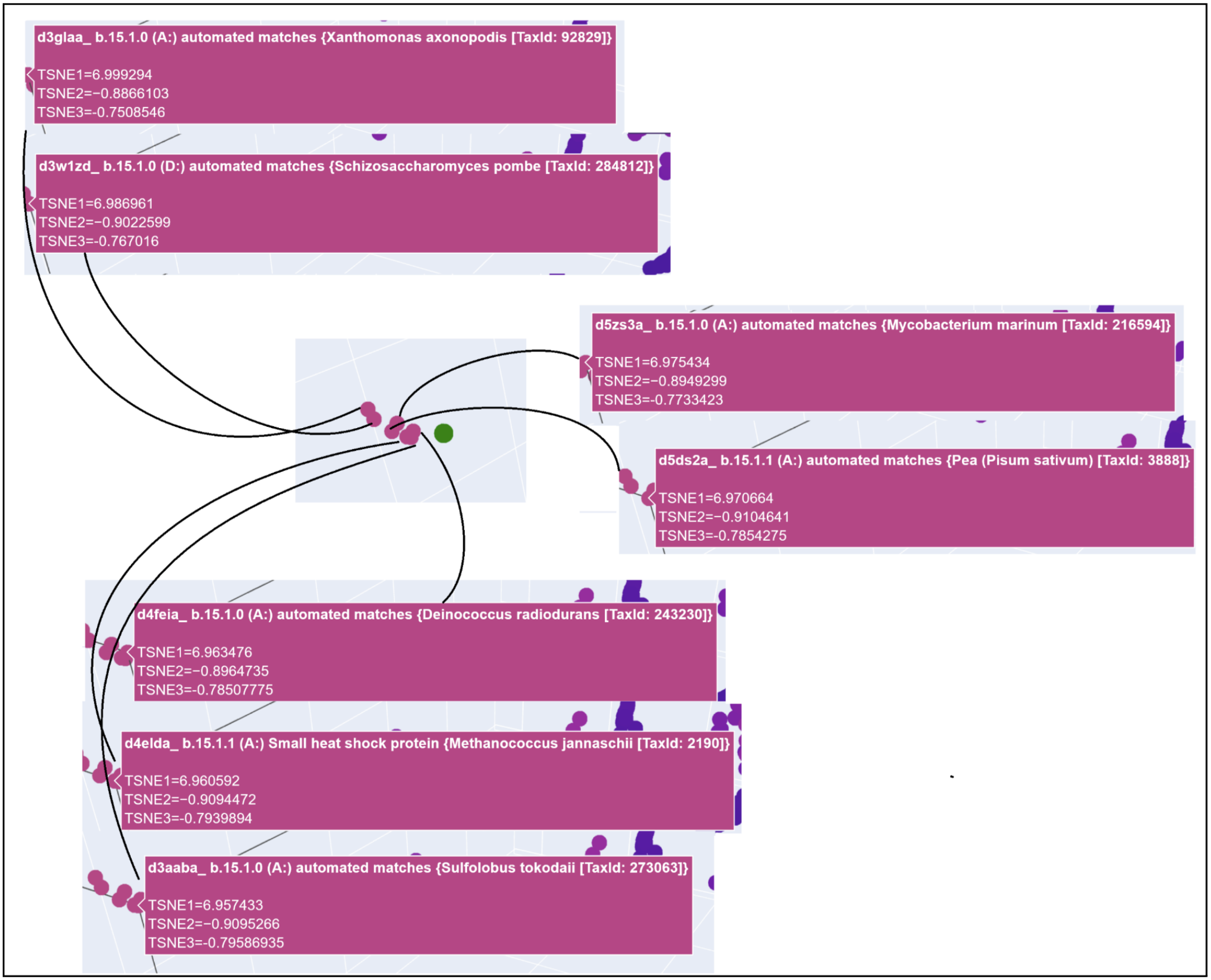
3D t-SNE Visualization (done with Plotly) in the Colab.
Shoutout to Nourelden Rihan for his helpful guide on the forum! I was able to plot my protein pretty easily thanks to him.
My protein was pretty close to a cluster of other proteins, seen in the photo below.
C2. Protein Folding
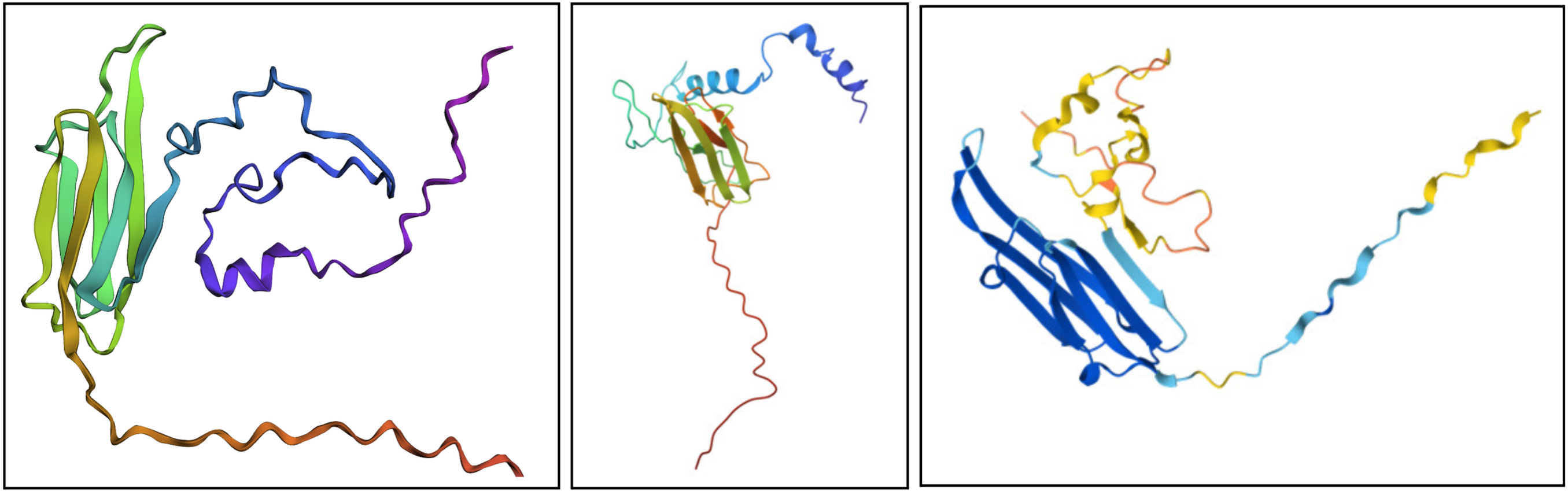
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I would say it looks pretty similar. On the left is the ESMFold result, the middle is a structure derived experimentally (on PDB), and on the right is a structure derived computationally (on PDB). You can see ESMFold has a similar sheet structure with the other two, but the placement of the loops (especially the bottom one) is identical to the right structure (both theoretical) while not necessarily resembling how the structure looks in the middle photo.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
It seems somewhat resiliant to mutations but not entirely. It’s possible to lose its shape with enough fiddling.
C3. Protein Generation
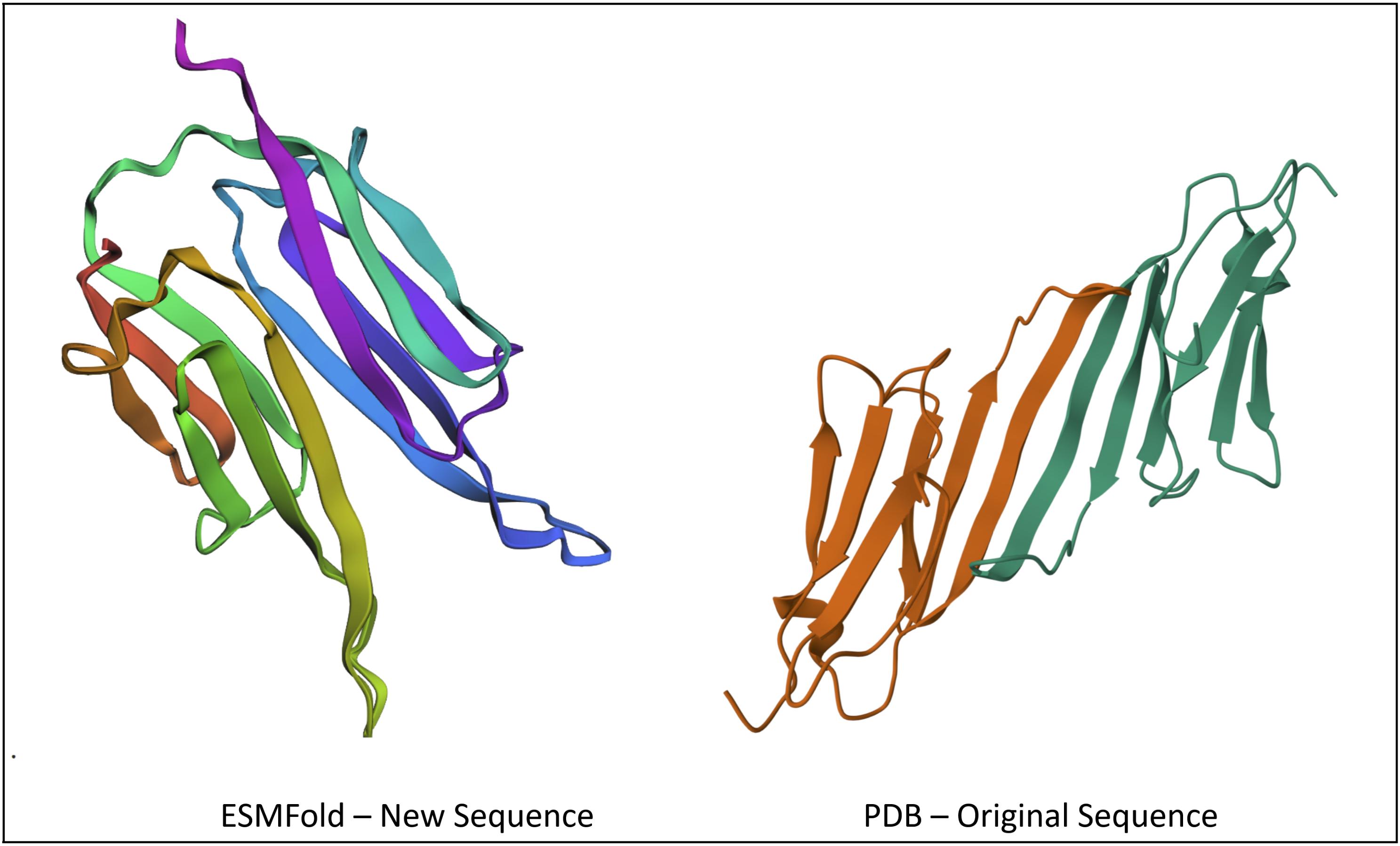
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
This part was pretty confusing to me, as the file I’d been referring to earlier (a 175 long sequence for Alpha-crystallin B, called P02511) wasn’t available on PDB. I ended up having to source 2N0K (pdb_00002n0k), a member within P02511 that ultimately had the same sequence, despite other members having different sequences.
I prompted the Colab to design for chain A and B. Both had a length of 82 AA (164 total) and had the following potential amino acid variations.
The new sequence ended up being
And I’ll swap MATKA → MATKV to get MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
See table below.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
See table below.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
See table below.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Binder
ipTM Score
Binding Spot
WHYGAVAAAHKE
0.31
Not near N-terminus, engages with β-barrel region. Surface-bound.
WRYGATGARHKE
0.33
Not near N-terminus, potentially engages with β-barrel region. Surface-bound.
WRYPVAALELWK
0.21
Not near N-terminus, potentially engages with β-barrel region. Surface-bound.
WRYPAVVLRLKE
0.3
Somewhat near N-terminus, does not engage with β-barrel region. Surface-bound.
FLYRWLPSRRGG (control)
0.36
On the complete opposite side of the N-terminus, with the β-barrel in between. Surface-bound.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
None of the ipTM values (max 0.33) exceed the known binder (0.36). However, all of the values are poor (< 0.6), which indicates the predictions might not be accurate. Some of the peptides do appear to bind better in their visualizations than the known (e.g. the first one WHYGAVAAAHKE and its interactions with the β-barrel).
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Binder
Predicted binding affinity (pKd/pKi)
Solubility (%)
Hemolysis probability (%)
Net charge (pH 7)
Molecular weight (Da)
WHYGAVAAAHKE
Weak binding, 5.893
Soluble, 1.000
Non-hemolytic, 0.024
-0.06
1339.5
WRYGATGARHKE
Weak binding, 6.087
Soluble, 1.000
Non-hemolytic, 0.028
1.85
1431.6
WRYPVAALELWK
Weak binding, 6.446
Soluble, 0.982
Non-hemolytic, 0.061
0.76
1531.8
WRYPAVVLRLKE
Weak binding, 6.506
Soluble, 0.816
Non-hemolytic, 0.050
1.77
1529.8
FLYRWLPSRRGG (control)
Weak binding, 6.361
Soluble, 0.608
Non-hemolytic, 0.047
2.76
1507.7
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
Actually, the extreme outlier is WRYPVAALELWK with an ipTM of 0.21, but it doesn’t vary as drastically with its other properties. It’s generally in the middle of the pack with binding affinity, solubility, and charge. One notable difference is its hemolysis probability is a bit higher, but not by much–still enough to be non-hemolytic.
The best performing two, WRYGATGARHKE with 0.33 and the control with 0.36, have solubilities on either extreme and very low to somewhat low hemolysis probability. They do have higher than average net charge, though–maybe that’s something that can be leveraged.
Choose one peptide you would advance and justify your decision briefly.
I don’t have an obvious contender, but WRYGATGARHKE is just behind the control in ipTM and is quite different in properties, so it’d be worth optimizing that to see if there’s a new direction that could produce good peptides.
Part 4: Generate Optimized Peptides with moPPIt
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
I used the following inputs for generation:
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Binder
Predicted binding affinity (pKd/pKi)
Solubility (%)
Hemolysis probability (%)
ipTM
CTAGSTVGVGVW
6.7832
0.9996
0.0618
0.36
ASATFEPPPVCH
5.8068
1
0.0223
0.39
VSEKYCVQFGKT
6.2623
1
0.0405
0.33
MSAGICNEFKQK
5.6404
1
0.0238
0.55
KNPCEAYCFNWV
6.7200
1
0.0346
0.28
I’d say there’s more variety. PepMLM repeated a lot of beginning and ending amino acids in the sequence, but all these sequences look completely unique. It doesn’t reflect as much in the properties, though. For evaluating, I’d run each sequence through the same software and compare properties/ipTM to see if there’s any improvement. E.g. MSAGICNEFKQK had a huge jump in ipTM to 0.55–that’s promising!
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Week 6 HW: Genetic Circuits Part I
Part 1: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The master mix can be found here and contains “Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2”. The polymerase is an essential part of PCR, nucleotides are the base materials needed to form new DNA sequences, and reaction buffer lowers the energy needed to start the process.
What are some factors that determine primer annealing temperature during PCR?
The master mix can be found here and contains “Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2”. The polymerase is an essential part of PCR, nucleotides are the base materials needed to form new DNA sequences, and reaction buffer lowers the energy needed to start the process.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Method
PCR
Restriction Enzyme
Throughput
High (2^n)
Low (n)
Protocol
Requires 3 step process of varying temperature, 1 pot reaction
Requires plasmid preparation to form recognition sites in DNA
Protein Involved
Primer, Polymerase
Endonuclease
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
You can verify experimentally how well the DNA is assembled. Plasmids are usually built with an antibiotic resistance gene, and in the test you put an antibiotic on your plasmids to see which survives. The ones that survive should be fully assembled.
How does the plasmid DNA enter the E. coli cells during transformation?
The heat shock method puts the bacteria and plasmid at a high temperature for about a minute, which “shocks” the bacteria into forming pores that the plasmid can enter through.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is also a form of molecular cloning, but uses restriction enzymes unlike Gibson Assembly. It uses recognition sites instead of overlapping sequences, and the enzymes are responsible for stripping the DNA (in place of exonuclease) and removing recognition sites to create the final construct (in place of ligase). This allows Golden Gate Assembly to accommodate multiple fragments in a one-pot reaction to create a single long strand of DNA. On the other hand, it needs more specific design and choosing of enzymes, including making sure recognition sites are unique within the sequence and do not show up accidentally.
Model this assembly method with Benchling or Asimov Kernel!
Further modeling in Asimov in link below.
Part 2: Asimov Kernel
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
My folder, along with the constructs and notebook documentation, can be found here.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Increased complexity in what it can process. Genetic circuits are capable of digital logic, but neural networks can fine tune connections and weights to represent a nuanced system with a set of many inputs and outputs.
This is in particular in a cell’s ability to contain a “weighted summation” dependent on the situation it gets trained on.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs can be used to represent complex systems in biology. An example would be cancer, as cancer cells rapidly mutate and provide continuous data. Input would be DNA, while the output would be a tag like GFP or any other form of fluorescence. IANN is good at making predictions for these systems, but has limitations like increased processing time (as with all neural networks).
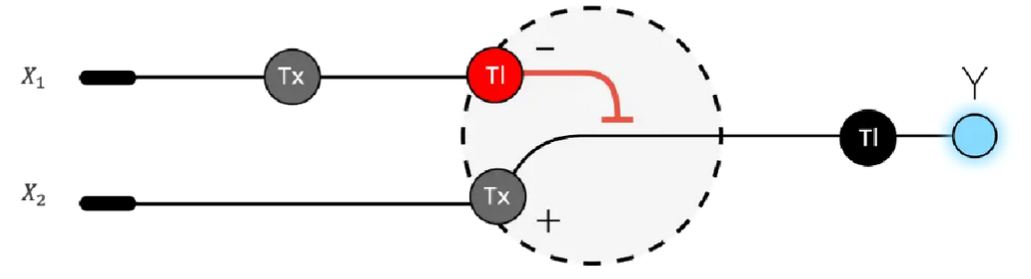
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
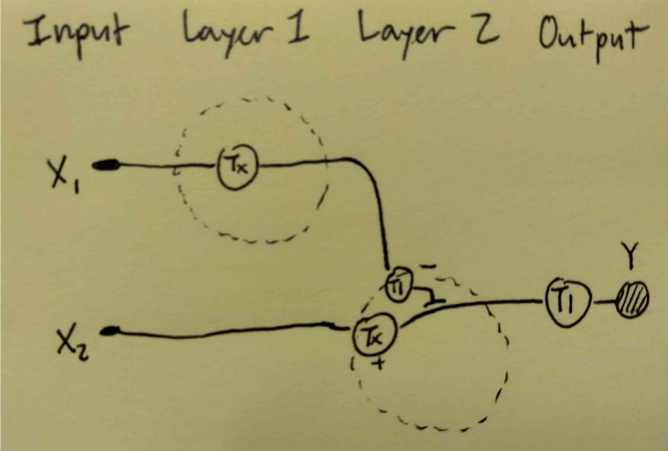
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
This diagram is neither entirely correct nor represents the multilayer perception, will be updated.
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium is an example that has structural integrity. You see it in architecture concepts as a naturally grown building material. Fungi can bring a biodegradable quality to traditional materials while keeping performance.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi already have natural properties that can be further optimized with genetic engineering. E.g. mycelium is fire retardant and insulatory after it’s dried, but can be engineered to be as strong as the currently used materials. You can also introduce a new aspect like color.
Bacteria-produced material is currently more fragile than fungi-produced. Stuff like mycelium has inherent structure and rigidity, which occupies a different niche in material applications.
Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW: Cell-Free Systems
Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free systems have an advantage in “extreme” situations, while in-vivo occurs within cells that have to be kept alive. In taking the process outside of the cell, you can handle it more roughly i.e. freeze drying the system for long-distance transport, or making a system that can be kickstarted just by adding water (in remote locations). Cell-free systems also have more control because they’re synthetic, so you can determine how big the cell is and what exactly goes in it.
Describe the main components of a cell-free expression system and explain the role of each component.
The components of a cell-free system are:
Cell Extract/Lysate – the internal components of the cell that contains molecular machinery/cofactors such as ribosomes, RNA polymerase, etc
tRNA – an RNA molecule needed to transcribe the genetic code
Membrane channels – for communication within/out of the cell
Plasmids/linear DNAs – containing the DNA sequence needed for the reaction
Nucleotides – the building blocks of RNA, aka ATP, GTP, CTP, UTP
Salts and buffer – needed to maintain pH (e.g. HEPES buffer)
Other enzymes/cofactors that aid in part of the process → Coenzyme A, 3-PGA, Spermidine, NAD etc
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy is needed to run the reactions, and having a constant supply is crucial the longer the reaction goes. One way of continuously providing ATP is picking chemical processes that can replenish resources. This is part of why some cell-free reactions (like ribose NMP) are more efficient, because they can produce ATP which is used for both RNA synthesis and an energy source, allowing the reaction to continue for longer.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cells differ from eukaryotic cells in components; notably, they lack membrane-bound organelles. For cell-free, they are the standard and are very low cost/high throughput. They’re a simple iteration of cell-free systems.
Eukaryotic cells are more complex and high cost, but can work with certain proteins and antibodies that are toxic to prokaryotic cells. All eukaryotic cells have an endoplasmic reticulum, which lets them represent functions like protein folding and post-translational modifications in cell-free systems.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
I’d set it up similar to the lab process in Homework 11–with a standardized list of all components that go in (salt/buffer, enzymes, nucleotides, cofactors, etc) and vary input volume to observe how expression of the protein changes. A potential challenge could be in measuring the expression of the membrane protein (something I’m not as familiar with myself). This could be addressed by linking production of the protein to a more easily measurable protein (like GFP) to be produced as well.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Possible reasons and their solutions are
The pH of the system is off – vary the concentration of buffer to adjust the pH across different trials
Maturation time is not within the window I’m observing – observe protein expression across the entire range of time and look at relative difference in expression at different times
Protein expression does not match up to the range I’m looking at (ie a fluorescent protein with low brightness) – recalibrating equipment to a known control? Or attempt a trial with a similar protein to see if results change (ie with a different fluorescent protein)
Homework question from Kate Adamala – Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell can be designed for medical applications, such as in drug production. The input would be the template for the molecule, e.g. inserted as a plasmid, with the output being assembly of the target drug and potentially a light indicator for success.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
The process needs to be isolated, so a membrane (and therefore synthetic cell) is necessary.
Could this function be realized by genetically modified natural cell?
It could potentially, but the process of biomanufacturing generally needs to happen in a cleanroom or at least a clean space. A synthetic cell has more understood components and a more controlled environment within itself compared to a natural cell, and the process could be better standardized.
Describe the desired outcome of your synthetic cell operation.
Ideally, given a plasmid containing the sequence of the drug, they are able to produce it and a fluorescent protein.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of? Phospholipids + cholesterol.
What would you encapsulate inside? Enzymes, small molecules.
Cell-free Tx/Tl system, plasmid/DNA encoding drug, fluorescent protein, amino acids, nucleotides, other enzymes and coenzymes TBD
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial (and probably E. coli) should be fine. I’m not currently aware of any further modifications that need to occur after translation.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
I would likely need membrane channel pores to allow for molecules to cross over, especially for the output proteins.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC (phospholipid), cholesterol
Genes: fluorescent protein (e.g. GFP), E. coli, gene for channel pores
How will you measure the function of your system?
The results should be measured in fluorescence output.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
I propose that cell-free biosensors for pollutants found in acid rain could be integrated into rain jackets as a concept of wearable environmental sensors.
How will the idea work, in more detail? Write 3-4 sentences or more.
This idea builds upon detection of acid rain through pH testing, by incorporating the response of test strips into textiles. Rain jackets are already designed for a rainy environment. If additional cell-free layers were engineered on top of the waterproof layer, similar to what was discussed in class, we could design a rain jacket that could change color in response to low pH in rainwater. This could be done by joining the cell-free system with a fluorescent protein or other colorimetric protein that operates well in low pH. The end result would be a more “live” response to the individual’s local environmental contaminants, allowing the user to constantly evaluate their surroundings while also acting as a functional article of clothing.
What societal challenge or market need will this address?
This is intended to be a first step in responding to local environmental injustice, giving individuals more agency to observe and detect the extent of pollution in their backyard.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
One concern is the possibility of overwhelming the cell-free systems with water, since rain jackets are generally worn longterm (whenever it rains). Additionally, the jacket might be a one-time use, since the detection of acid rain once would render it ineffective for the second rainfall. This could be addressed by intentional design, allowing only a limited amount of water to reach the cell-free system (i.e. smaller access holes for water droplets) and having a swappable cell-free system layer on the jacket.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Quorum sensing is a method for bacteria to communicate on a large, colony-wide scale. This occurs through autoinducers, chemical molecules that diffuse or are transported between the cells. I think this is an interesting topic to apply to space, as testing these cells can reveal how the process of diffusion/osmosis is impacted by microgravity as well as opening a path up to explore different mediums of communication for biology in space.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Aliivibrio fischeri would be interesting as it’s a bacteria that provides bioluminescence within a squid once its population grows enough. The proteins involved are luciferase and LuxY.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Lots of prior proposals tackle the effects of microgravity, so this is sort of similar. The luciferase protein will get produced as a result of coordination across a bacteria colony. In getting to that result of fluorescence, we want to see how successfully the bacteria cells communicate with each other in microgravity.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
I hypothesize that the proteins will have a faster response time and increased quorum sensing in spaceflight, as this seems to be the pattern with prior quorum sensing experiments. This might show itself in the form of fluorescence appearing much faster than it would on Earth, but might also be a false positive where fluorescence appears before cells have reached an appropriate density. This is actually a bit different than what I would have thought by intuition–I imagined that microgravity would make it harder for chemical molecules to travel across cells, which seems true, but in this case the settled molecules would be wrongfully interpreted as “positive” signals sent by other cells, resulting in a denser perceived network of cells.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
The experiment is outlined below, though I’m a little skeptical of how easy it would be to implement:
To represent cell density, I would have a cell-free system be composed of the internals of Aliivibrio fischeri, including a way to generate and maintain autoinducers. The reaction could be carried out in several samples with different cell densities each.
The cell-free system would also include a plasmid that codes for luciferase, for when the switch to produce fluorescent protein is “flipped”.
Controls would require samples that are above and below the density needed for the bacteria to glow, based on experimental data gathered on Earth
Data would be detected value of fluorescence, which would need specific measurement tools.
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z for each adjacent pair of peaks (n, n+1) using:
Let’s take the two largest peaks on the graph, labelled at m/zn = 903.7148 and m/zn+1 = 875.4421. z = 875.4421 / (903.7148 - 875.4421) = 30.96
Determine the MW of the protein using the relationship between m/zn , MW, and z
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with $\frac{m}{z}$ values.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, I would need a second peak for the adjacent charge state approach. The second noticeable peak is not in the zoomed in photo, and I am skeptical that the small rise I see in the photo is considered a peak.
Waters Part II — Secondary/Tertiary structure (OPTIONAL)
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $\frac{m}{z}$ on a mass spectrometer with 30,000 resolution.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
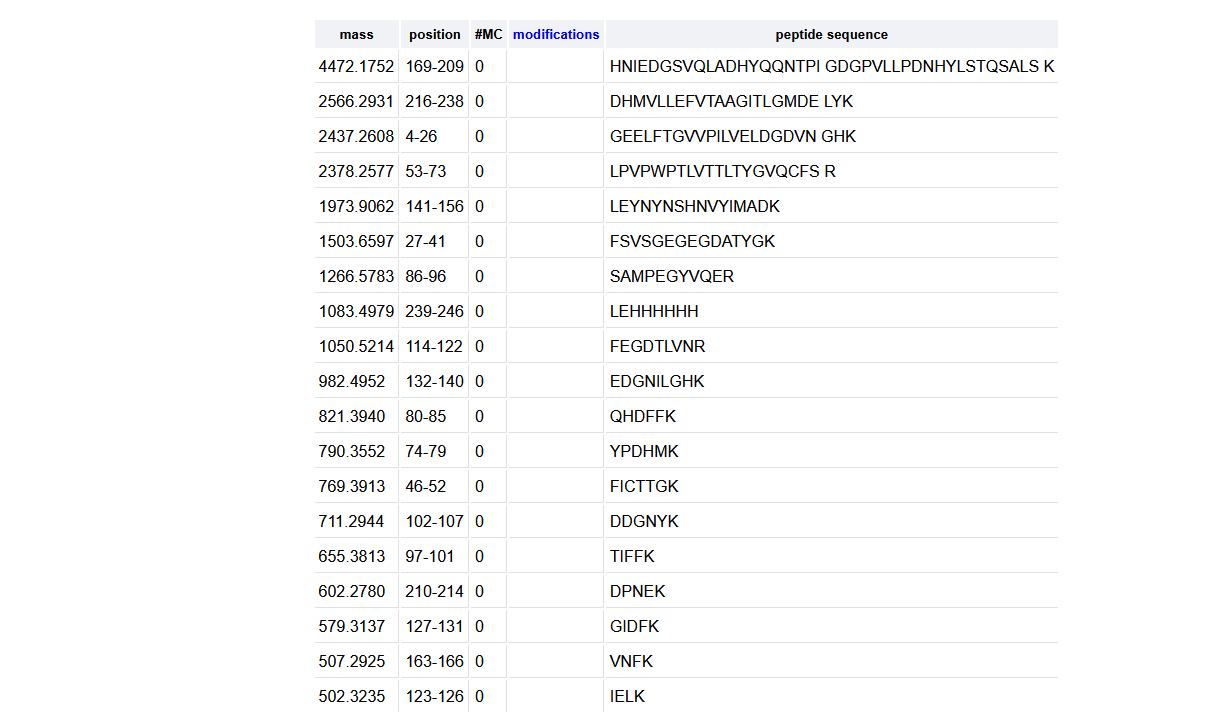
How many peptides will be generated from tryptic digestion of eGFP?
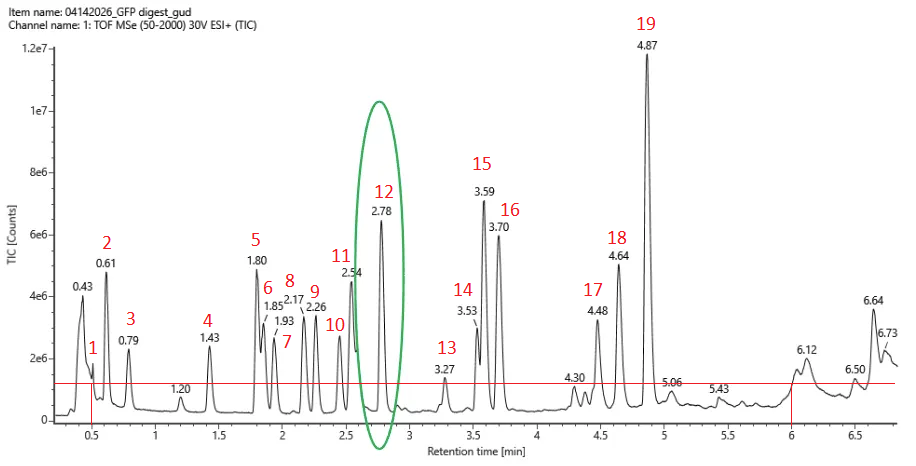
19 peptides were generated as seen in the photo below.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
I counted 19 and used roughly 10% * 1.2e7 = 0.12e7 as a threshold (photo below).
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
It matches exactly. I’m pleasantly surprised!
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H}+]) based on its m/z and z.
The m/z of the most abundant charge state is 525.76712, with the charge being z = 1/(Δm/z) = 1/(525.76712 - 526.25918) = 2.0322 ≈ 2
As a result, the mass is [M+H]+ = (m/z * z) - (z - 1) * H = 1051.53424 - 1.00727 = 1050.527
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
It’s closest to FEGDTLVNR, which has a mass of 1050.5214.
AccuracyFEGDTLVNR = |1050.527 - 1050.5214| / 1050.5214 = 5.30 ppm
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6. Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
91.1% of my sequence is covered, not excluding peptides less than 500 Da.
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). Identify where the following oligomeric species are on the spectrum shown below from the CDMS:
For CDMS, calculation of mass is just a function of m/z * z. So for the following:
7FU Decamer → 340 kDa * 10 = 3.4 MDa
8FU Didecamer → 400 kDa * 20 = 8 MDa
8FU 3-Decamer → 400 kDa * 30 = 12 MDa
8FU 4-Decamer → 400 kDa * 40 = 16 MDa
Waters Part V — Did I make GFP?
This is from data given in the homework, not lab work.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
27875.41
27951.85
2742.545 ppm
My error was unusually high for observed mass.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
Unfortunately, I did not get to contribute this year, but I did discuss the project with friends in the class. I really like the concept of a collaborative (and also competitive, occasionally) project with an end result that is artistic (while also leading to the lesson next week). I think the process was very lovely, with people’s ideas growing and shifting until it reaches a fully developed design.
I’m not too sure if this would lose the spirit of the assignment, but coordinating within nodes or between people might better guide us towards a final design. I feel like the end result, with the four different designs on each well, was a little lucky.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) – Lysate contains cell internals that are necessary in cell-free systems, notable RNA, ribosomes, and other small molecules. In this case, it provides the T7 RNA polymerase needed for transcription.
Salts/Buffer
Potassium Glutamate – This provides an ideal condition for the reaction by providing salts to create a buffer, which can maintain pH.
HEPES-KOH pH 7.5 – This is a buffer that maintains pH within the system (around 7.5).
Magnesium Glutamate – This provides the Mg2+ ions necessary for maintaining pH and ion concentration.
Potassium phosphate monobasic – The process of turning a _MP → _TP (monophosphate to triphosphate) requires additional phosphates, which this component provides.
Potassium phosphate dibasic – Same as above, but monobasic is acidic while dibasic is…basic. They tackle two ranges of pH in the reaction.
Energy / Nucleotide System
Ribose – Ribose is part of the process of turning NMPs to NTPs.
Glucose – Glucose provides energy used in the system.
AMP – AMP turns into ATP, which is the nucleotide for adenine necessary for RNA synthesis. ATP is also used as an energy source.
CMP – CMP turns into CTP, which is the nucleotide for cytosine necessary for RNA synthesis.
GMP – GMP turns into GTP, which is the nucleotide for guanine necessary for RNA synthesis. GTP is also used as an energy source.
UMP – UMP turns into UTP, which is the nucleotide for uracil necessary for RNA synthesis.
Guanine – Guanine turns into GMP, which turns into GTP. It’s a more “raw” material and would require more time/complexity to assemble for better efficiency.
Translation Mix (Amino Acids)
17 Amino Acid Mix – These amino acids are the building blocks of our target proteins, and will be used in translation after the RNA has been made.
Tyrosine – Same as above, but with tyrosine only. Perhaps it is implemented at a higher rate than other amino acids.
Cysteine – Same as above, but with cysteine only. Perhaps it is implemented at a higher rate than other amino acids.
Additives
Nicotinamide – This additive turns into NAD, which is an important cofactor responsible for cellular metabolism.
Backfill
Nuclease Free Water – The water is needed to suspend all these components in a solution, allowing them to mix together.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The difference lies in nucleotides. PEP-NTP has nearly ready-to-use ATP, GTP, CTP, UTP, which is the “final form” before translation. However, the process is inefficient and peters out quickly. Ribose uses AMP, GMP, CMP, UMP, which requires an additional process to convert to ATP, GTP, CTP, UTP, but the process is more sustainable and generates more energy/less byproducts for long term reacting.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine is just a tertiary layer removed from GTP. Guanine would have to undergo a process to turn into GMP, and then another process to turn into GTP. Transcription would only occur after those two steps.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP – This protein takes the structure of a β-barrel, indicating a hydrophilic outside with a hydrophobic inside. It’d likely be readily receptive to water and a good solution for “just-add-water” cell-free systems.
mRFP1 – This protein has high brightness, so would not need as many proteins expressed for detection. This could help the cell-free system optimize volume.
mKO2 – This protein has a pKA of 5.5, which means it’s pretty sensitive to acidity and would drop in fluorescence intensity when exposed to liquid of that pH or lower.
mTurquoise2 – This protein is known to be rapidly maturing. That means it’ll be very responsive in cell-free systems that are time-sensitive (e.g. an on-the-spot diagnosis).
mScarlet_I – This protein has moderate acid sensitivity, so is also pretty sentitive to acidity similar to mKO2.
Electra2 – This protein has high brightness similar to mRFP1, so would behave similarly.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Most of these fluorescent proteins are less effective in acidic conditions, so we want to shift the pH in the other direction. We can adjust the concentration of salts/buffer that maintain that pH, as well as increase the concentration of potassium phosphate dibasic, which operates at a higher pH than monobasic.
So from the initial concentrations, we can adjust for mKO2, which has a pKa of 5.5. We can increase the volume of HEPES-KOH and potassium phosphate dibasic (or potentially both dibasic and monobasic if they need to be at the same volume).
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Was unable to complete the lab.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!