Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
| Attempt | Result | Description |
|---|---|---|
| 1 | 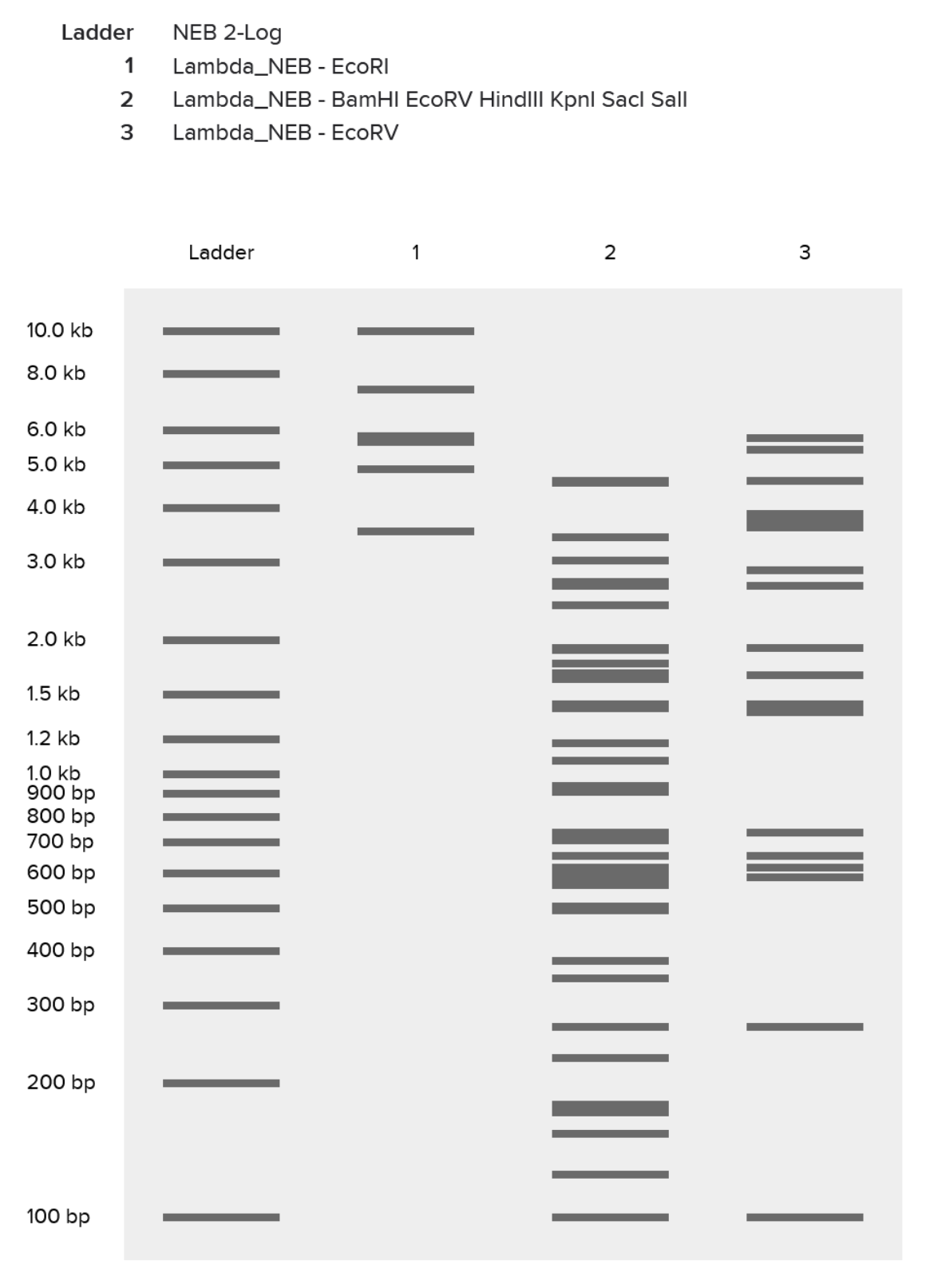
| Had an initial mess-up where I tried to “speedrun” the process and ended up with a ladder packed with the effects of multiple restriction enzymes. |
| 2 | 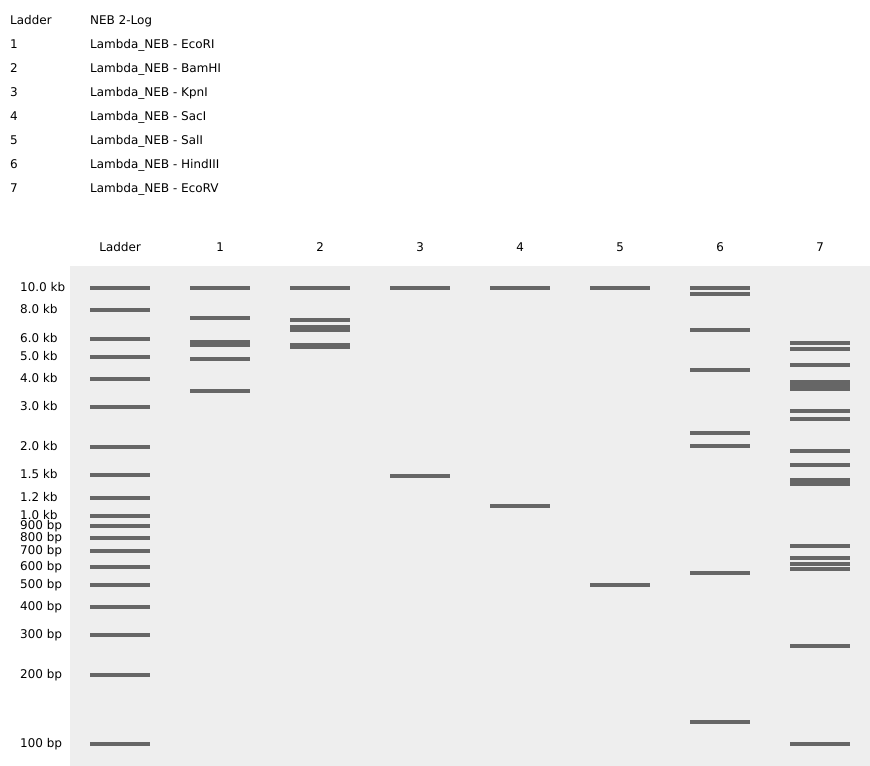
| Finally got success with all of the listed enzymes, separately. |
| 3 | 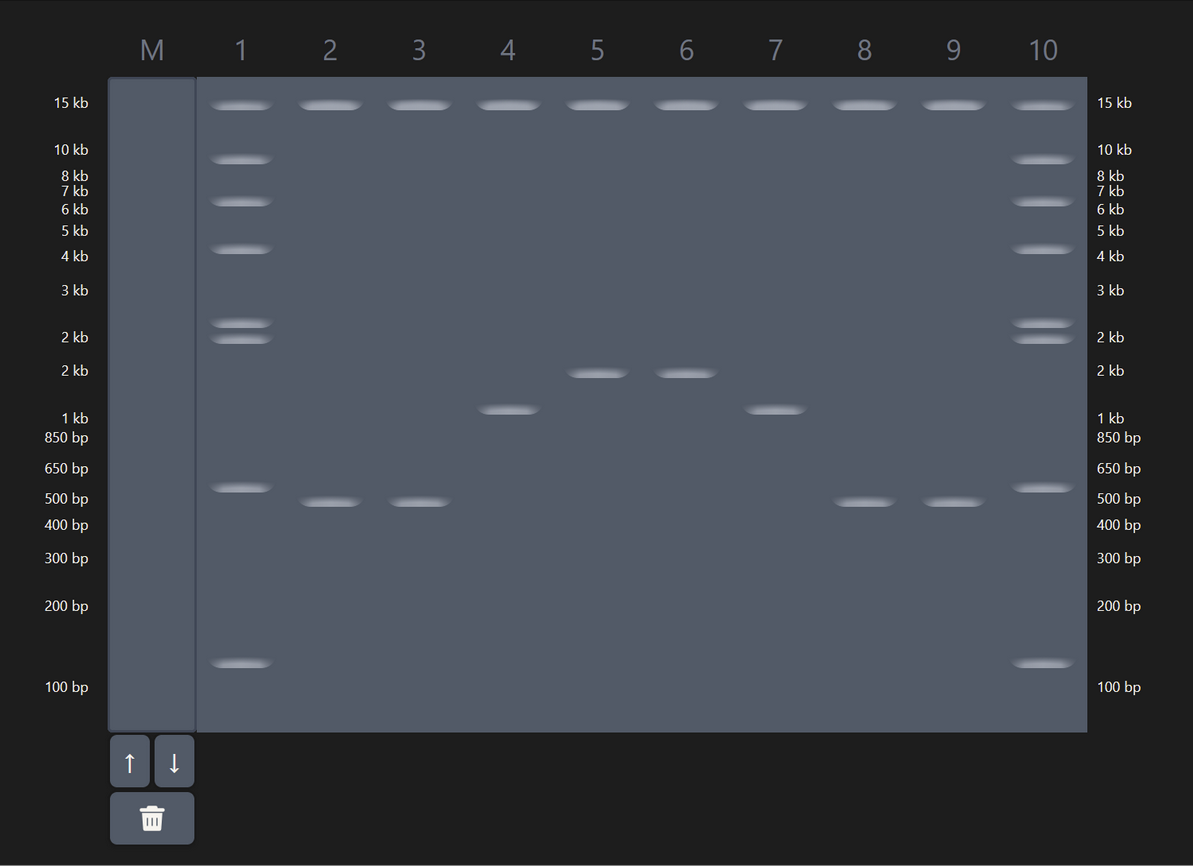
| Some experimentation on Ronan’s website got me this pattern that sort of looks like a pair of pants. In hindsight I should’ve definitely explored results from a combination of enzymes (e.g. EcoRI and HindIII together), which would’ve given me a bigger range of visual results. |
| 4 | 
| Replicated “sort-of pants” on Benchling, and my final result. |
Part 3: DNA Design Challenge
3.1. Choose your protein.
I recently read about snake venom and how its majority composition of proteins/enzymes make it (theoretically) edible, since it can be digested in the stomach. That was a pretty fun fact. For this assignment, I picked irditoxin, a three-finger toxin that is selectively neurotoxic towards birds and lizards (but not mammals).
I found two subunits on UniProt and went with A.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I located irditoxin subunit A in the European Nucleotide Archive, with the following DNA sequence:
3.3. Codon optimization.
Codon optimization can be useful in controlling gene expression within a sequence (both increasing and decreasing it). It can also make mRNA production more efficient and impact translation speed, which can in turn affect things like folding speed (fast-translated sequences fold while waiting for slow-translated sequences).
I tried out IDT’s Codon Optimization Tool. IDT recognized a couple different stop and start codons, so I picked the sequence between the first codon (ATG, a start) and the first stop codon (TGA at position 330). The sequence was optimized for E.coli to go for a standard, well understood, commonly used organism.
Shortened old sequence:
Optimized sequence:
Another mess-up: IDT denied the optimized sequence due to its complexity, which means this sequence isn’t currently manufacturable and needs to be further redesigned. Seems some of my enzyme recognition sites weren’t ideal.
3.4. You have a sequence! Now what? What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
As we’re working with a neurotoxin, and not for high-volume production, I’d probably turn to a cell-free method.
Part 4: Prepare a Twist DNA Synthesis Order
4.1-2. Create a Twist account and a Benchling account, Build Your DNA Insert Sequence
From the downloaded FASTA file:
My expression casset can be accessed here.
4.3-6. Select The “Genes” Option, “Clonal Genes”, Import your sequence, and Choose Your Vector
Here’s my plasmid. She’s beautiful!
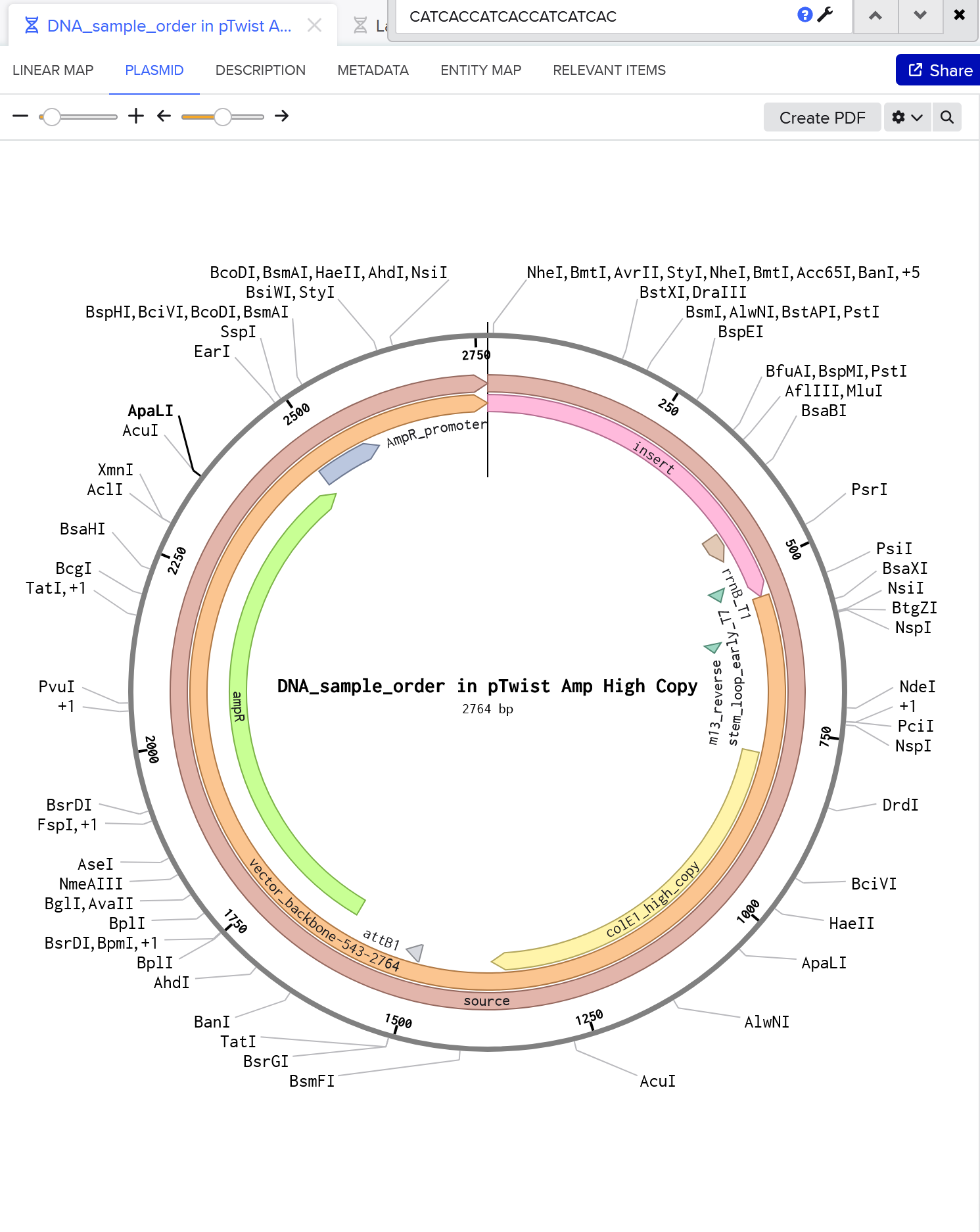
Part 5: DNA Read/Write/Edit
5.1 DNA Read
- What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I think bioindicators are an interesting group of organisms, and sequencing them could help us isolate genes that react to surroundings and get used for a more standardized, widespread environment monitoring tool. E.g. microalgae can detect wide range of water quality issues, from heavy metals to nanoparticles, yet “only a few species have been fully sequenced without any gaps”1.
- In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
I’d likely use a NGS method to handle the sequencing of the entire genome, and of the multiple species that we come across at that. So potentially something like PacBio, which is third-generation. Both the sample used and the library need to be prepared, with the sample needing to be purified and the DNA needing to be fragmented to length and end-capped. The DNA is decoded through a polymerase that runs along the sequence. As it interacts with each nucleotide, it emits light, which is recorded live and appended onto the current sequence. The end result is a straightforward sequence of DNA nucleotides, given in a file that can be read on any notepad app.
5.2 DNA Write
- What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I’m continuously interested in biomaterials as an alternative to things like plastic. There’s been a lot of work on getting plastic that is biodegradable, and I’m wondering if there’s a way to go about it from the opposite direction, like fortifying kombucha leather to last longer.
- What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Solid-phase synthesis using the phosphoramidite method seems to be the go-to method, so I’d stick with that. The steps are coupling the base with phosphoramidite, capping unreacted sites, oxidating the phosphate, deblocking, and then repeating as needed. The limitation to this process is that it decreases in efficiency past a certain number of bp (~200 as discussed last week) so could potentially be difficult as the needed sequence becomes longer.
5.3 DNA Edit
- What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
This feels like a touchy subject in line with our ethical considerations from last week. The responsibility of human genome sequencing seems enormous, so I’ll consider other organisms. I’m thinking of how filter feeders play an important role in the water ecosystem, essentially “purifying” the water. Could something like that be intentionally edited into plants (or other microorganisms) to boost its “purifying” effect on the air?
- What technology or technologies would you use to perform these DNA edits and why?
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I’m unsure of this part as I’m not too familiar with the work needed to introduce this behavior into land organisms. I believe CRISPR-Cas9 is also a go-to for gene editing, so would probably be a popular approach regardless. The process begins with a guide RNA finding the target sequence in the DNA, at which Cas-9 “cleaves” the segment. Then either new DNA can be added (replacing the segment), or the DNA strands repair (deleting the segment). For preparation, the appropriate guide RNA needs to be sourced. A limitation of CRISPR-Cas9 is in its effectiveness; it has the potential to insert DNA incorrectly, which can lead to mutations when applied to human genome sequencing.
Citations
[1] Evangelia Stavridou, Lefkothea Karapetsi, Georgia Maria Nteve, Georgia Tsintzou, Marianna Chatzikonstantinou, Meropi Tsaousi, Angel Martinez, Pablo Flores, Marián Merino, Luka Dobrovic, José Luis Mullor, Stefan Martens, Leonardo Cerasino, Nico Salmaso, Maslin Osathanunkul, Nikolaos E. Labrou, Panagiotis Madesis, Landscape of microalgae omics and metabolic engineering research for strain improvement: An overview, Aquaculture, Volume 587, 2024, https://doi.org/10.1016/j.aquaculture.2024.740803.