Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
The Dalton is an atomic mass unit that converts from mass using Avogadro’s number. So 500 grams = 500 * 6.022 * 1023 Daltons / 100 Daltons/amino acid = 5 * 6.022 * 1023 amino acids = 3.011 * 1024 amino acids.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Our bodies don’t interact with the meat that way. It’s a matter of absorbing the energy and nutrients from the cells, not carrying the cells over to reuse them. The protein also goes through our stomach and digestive track, which is a process more intended to break down what we eat and extract from it.
- Why are there only 20 natural amino acids?
The natural amino acids are determined by codons, which are determined by three nucleotides (of which can be adenine, uracil, guanine, cytosine). This gives 4 x 4 x 4 = 64 total codons, but redundancy among codons produces only 20 unique amino acids. More is definitely possible, but 20 seems to encapsulate all the amino acids we need.
- Can you make other non-natural amino acids? Design some new amino acids.
Natural amino acids come from the range of nucleotides across 3 spaces, 4 * 4 * 4 = 64. We can design new amino acids in a variety of ways, one notable way being reversing the chirality (from L-amino acids to D-amino acids), which could sort of be applied to any amino acid in existence for a “new” design.
- Where did amino acids come from before enzymes that make them, and before life started?
The 1953 experiment making “Primordial Soup” proved that amino acids (and other organic compounds) could come from natural reactions under ideal conditions with inorganic compounds (methane, ammonia, and hydrogen). It seems like there’s a couple of different ways organic compounds could be created abiotically.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If the helices are usually right handed, then this one will be left handed (opposite to what is normal).
- Can you discover additional helices in proteins?
Additional helices can always form in new proteins, though I don’t think you can “introduce” new helices to a protein whose folding pattern is known. But since new proteins are made all the time, yes, there will frequently be helices in their shapes, which can be figured out through protein modeling/prediction.
- Why are most molecular helices right-handed?
Not exactly sure why it became this way, but it definitely stays this way for ease of transcription and translation. One type of chirality tends to be dominant so that all templates and future proteins/molecules can be identical and interact with each other. Otherwise, left- and right- handed molecules have the exact same properties, but being able to piece together requires the same handedness.
- Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
Initially, you have multiple β-strands that can arrange laterally. These strands can sort of reach left and right and “link arms” with neighboring strands through strong hydrogen bonds. The repeated structure in each strand allows for repeated patterning.
- Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
β-sheets stem from a presence of strands in the protein with hydrogen backbones, so I’m guessing amyloid diseases often have that chemical makeup. Then it’ll tend to form the sheets just because it’s a very stable configuration.
- Design a β-sheet motif that forms a well-ordered structure.
Motifs appear to be ways to arrange the strands laterally against each other. This is easy to do in concept but might not reflect motifs that can occur naturally. I’m imagining a potential concept where you take a sheet and fold two ends to make a cylinder–that’s the current beta barrel design. If you imagine a long, rectangular sheet, though, there’s a way to fold it to make sort of a ribbon (like alpha helices’ shape). I wonder if it’s possible to get to that shape with very long but few beta strands.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
I picked crystallin, which is a protein in the eye responsible for the movement of your iris as you focus. It’s notably transparent, being part of the eye lens, and water-soluble, which was a callback to our lecture. I picked the protein because I was interested in how cataracts were formed.
The specific protein I went with for the following questions is P02511, or Alpha-crystallin B (in humans).
- Identify the amino acid sequence of your protein.
The AA sequence from UnitProt is
sp|P02511|CRYAB_HUMAN Alpha-crystallin B chain OS=Homo sapiens OX=9606 GN=CRYAB PE=1 SV=2 MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPTSTSLSPFYLRPPSFLRAPSW FDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEVHGKHEERQDEHGFISREFHR KYRIPADVDPLTITSSLSSDGVLTVNGPRKQVSGPERTIPITREEKPAVTAAPKK
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Using the Colab notebook, the protein is 175 amino acids long with the most common amino acid being P (and appearing 17 times).
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
According to UniProt, it’s part of the small heat shock protein (HSP20) family, along with all other Alpha-crystallin B proteins. However, according to the Transporter Classification Database, it’s part of the α-Crystallin Chaperone (CryA) family (where other Alpha-crystallin B proteins don’t appear).
- Does your protein belong to any protein family?
Homology refers to protein sequences that likely have a common ancestor (identified through having similarities in sequence/structure?). Using the BLAST software gives 250 results for similar proteins, with results primarily appearing to be Alpha-crystallin B in different animals.
- Identify the structure page of your protein in RCSB
This step was particularly difficult for me, as I didn’t always understand how to get to the answer based on what I had on the screen.
- When was the structure solved? Is it a good quality structure?
The structure seems to be initially solved in 2009 but has increased members up until 2025. Some particularly high resolution structures were identified in 2012 and 2014 through X-ray diffraction, with a resolution of 1.0 - 1.5 Å.
- Are there any other molecules in the solved structure apart from protein?
I’m not entirely sure how to identify this…at least visually, I didn’t identify any other components that that seemed to stick out as an entirely different molecule.
- Does your protein belong to any structure classification family?
Using the Structural Classification website, it belongs to the “Alpha crystallin-like” family, further within the “Hsp20 chaperone-like” family.
- Open the structure of your protein in any 3D molecule visualization software:
I chose to use PyMol to open my structure, getting the structure below.
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
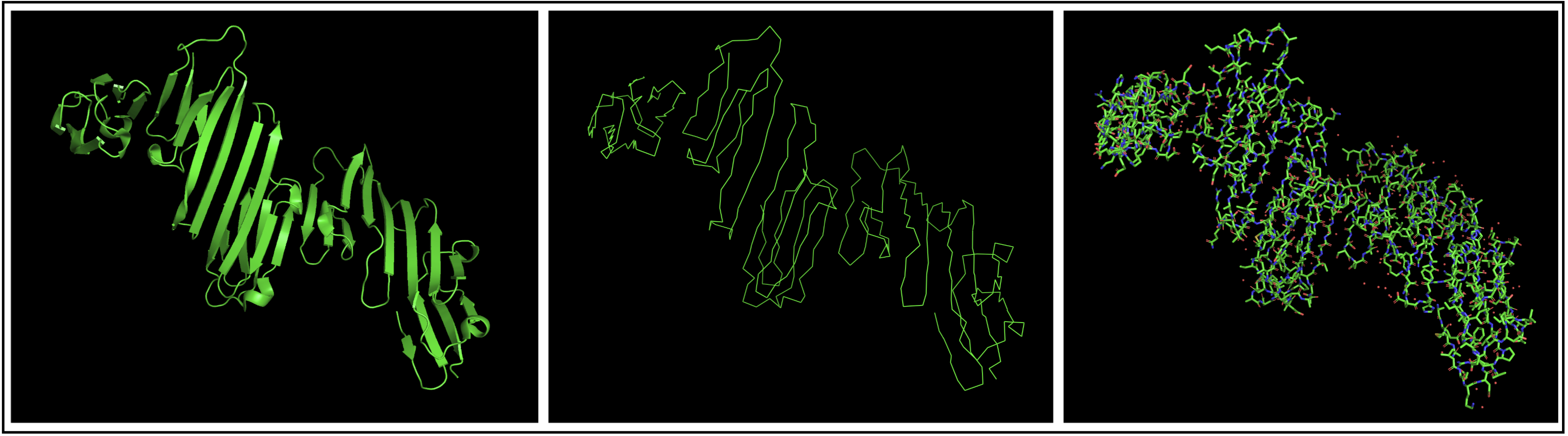
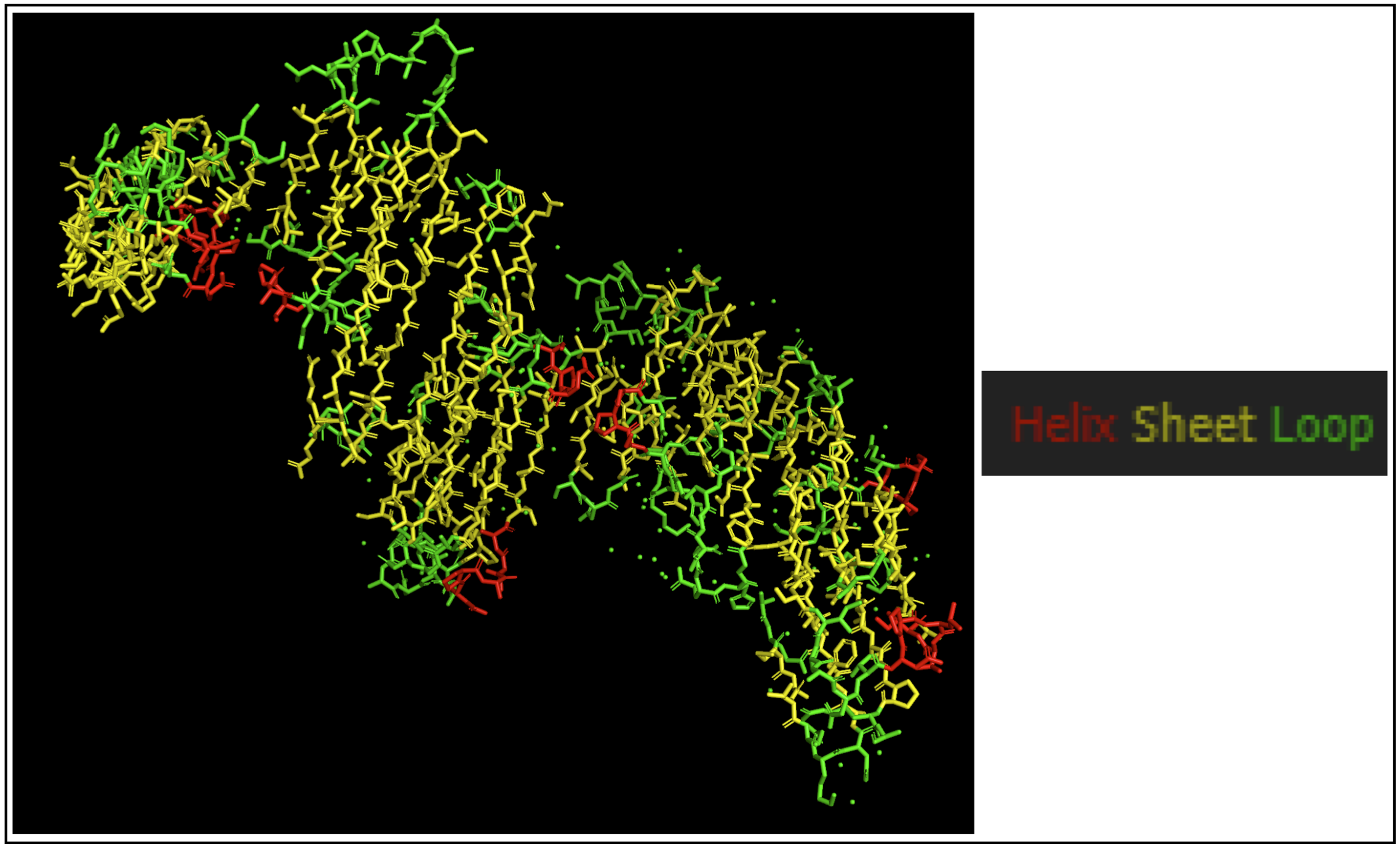
- Color the protein by secondary structure. Does it have more helices or sheets?
The protein seems to mostly be composed of sheets with some helices.
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I colored hydrophobic residues in red and hydrophillic residues in green.
- Hydrophobic (in red): glycine (Gly), alanine (Ala), valine (Val), leucine (Leu), isoleucine (Ile), proline (Pro), phenylalanine (Phe), methionine (Met), tryptophan (Trp)
- PyMol code:
select hydrophobic, resn Gly resn Ala resn Val resn Leu resn Ile resn Pro resn Phe resn Met resn Trp
- PyMol code:
- Hydrophilic (in green): serine (Ser), threonine (Thr), asparagine (Asn), glutamine (Gln), cysteine (Cys), glycine (Gly)
- PyMol code:
select hydrophillic, resn Ser resn Thr resn Asn resn Gln resn Cys resn Gly
- PyMol code:
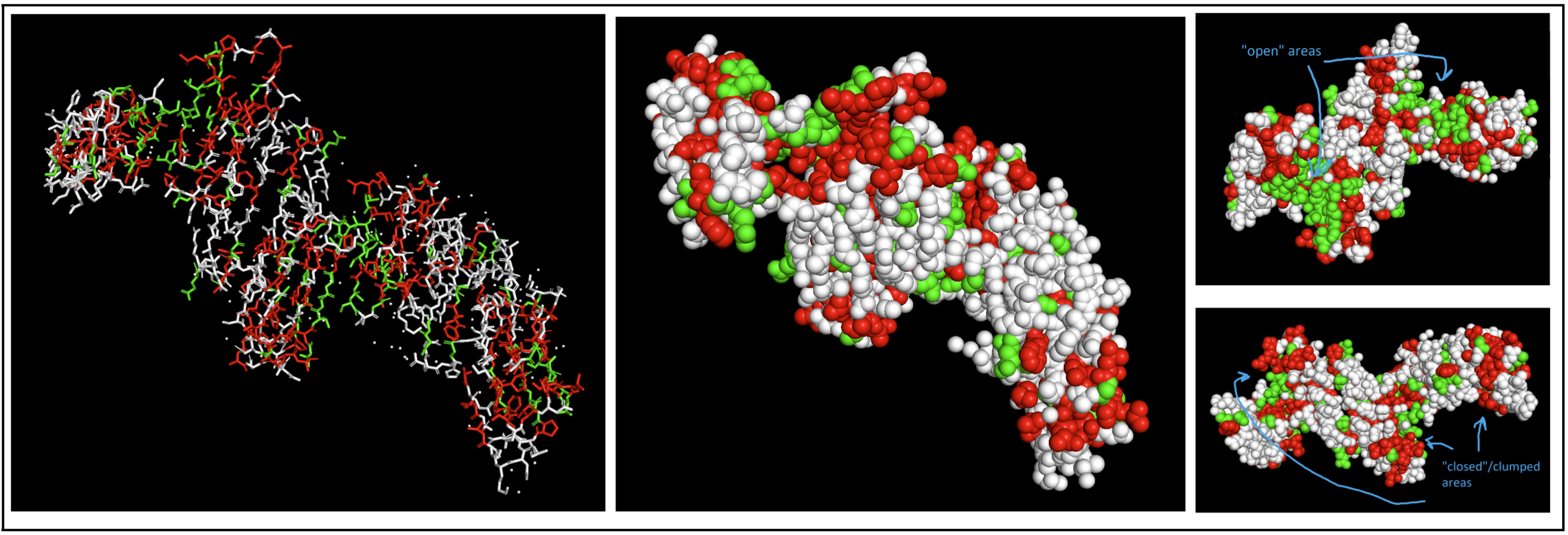 I had to switch to a spheres visualization to better see how molecules were interacting. It was a little hard for me to see a significant pattern, but I do feel like the hydrophilic residues have more “open” facing areas, whereas the hydrophobic residues were more clumped (both together and with neighboring residues).
I had to switch to a spheres visualization to better see how molecules were interacting. It was a little hard for me to see a significant pattern, but I do feel like the hydrophilic residues have more “open” facing areas, whereas the hydrophobic residues were more clumped (both together and with neighboring residues).
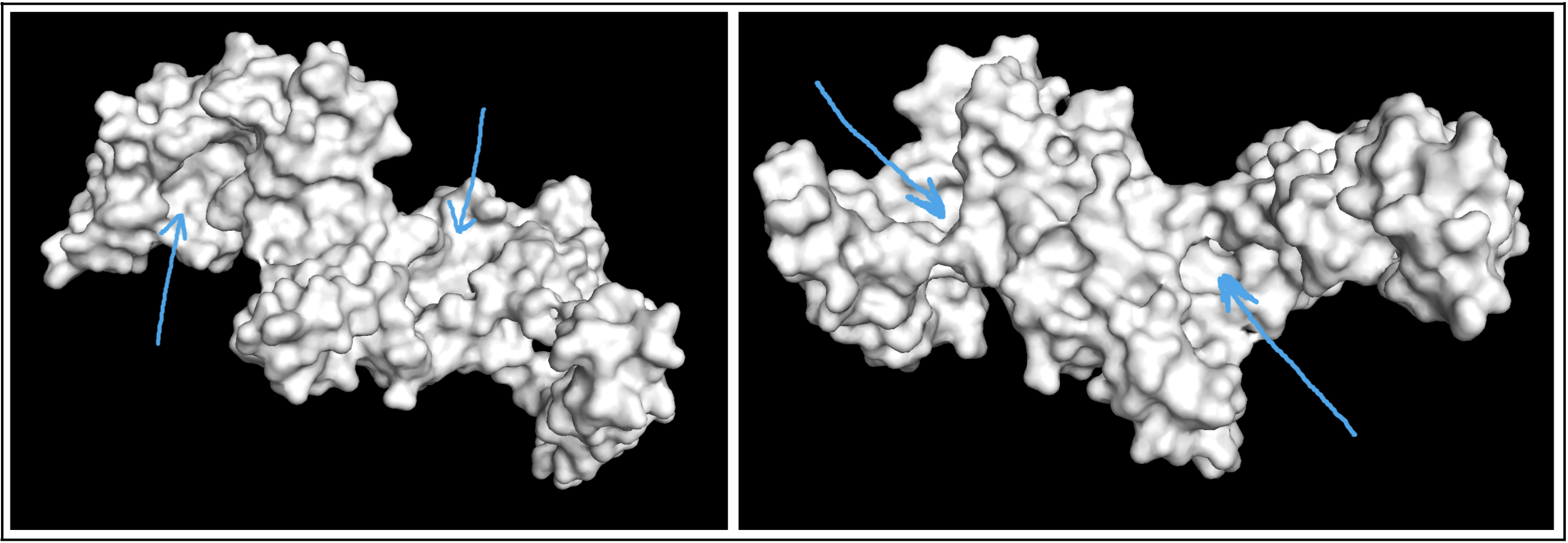
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Visualizing the protein as a surface was really helpful! I could easily find a couple areas that could be binding pockets. It’s a little difficult to show it accurately in a photo, but I indicated potential areas below:
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
I had to pivot here as I originally chose vicilin from lentil beans (Lens culinaris), which was a type of globulin storage protein. It has a rather long sequence (shown below) and as a result made reading some results pretty challenging. I ended up switching back to crystallin from Part B.
C1. Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
- (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
sp|P02511|CRYAB_HUMAN Alpha-crystallin B chain OS=Homo sapiens OX=9606 GN=CRYAB PE=1 SV=2 MDIAIHHPWIRRPFFPFHSPSRLFDQFFGEHLLESDLFPTSTSLSPFYLRPPSFLRAPSW FDTGLSEMRLEKDRFSVNLDVKHFSPEELKVKVLGDVIEVHGKHEERQDEHGFISREFHR KYRIPADVDPLTITSSLSSDGVLTVNGPRKQVSGPERTIPITREEKPAVTAAPKK
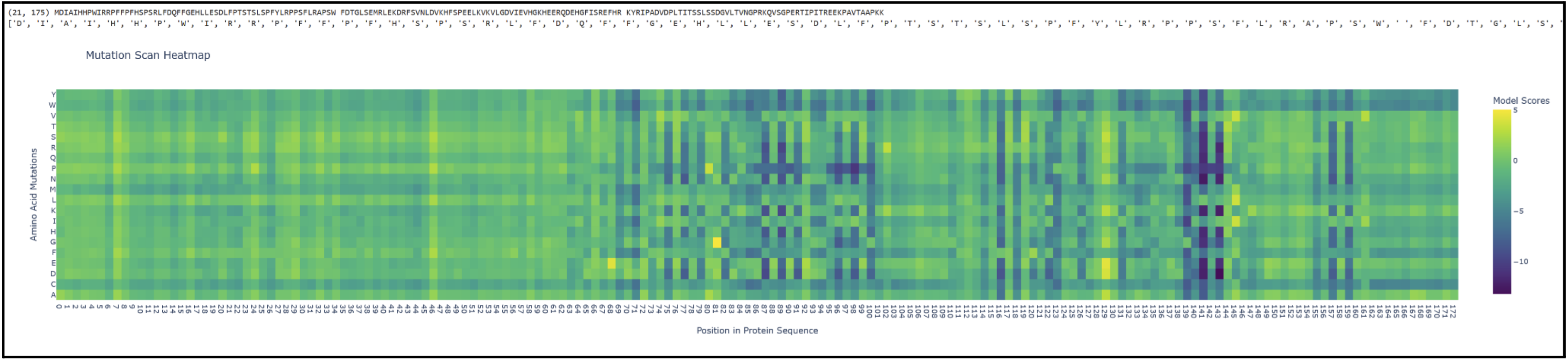
Proteins in the latter ⅔ of the sequence, particularly 87 (L), 89 (V), 96 (I), 98 (V), 139 (G), 141 (L), and 143 (V), seem subject to severely detrimental mutations (looking at the dark blue streaks). This indicates to me that Leucine and Valine are rather important residues that should avoid mutation. Meanwhile, the first ⅓ of the sequence seems pretty tolerant to any changes. Notably, protein 129 seems like it can be mutated with mostly beneficial outcomes.
- Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
3D t-SNE Visualization (done with Plotly) in the Colab.
 Shoutout to Nourelden Rihan for his helpful guide on the forum! I was able to plot my protein pretty easily thanks to him.
Shoutout to Nourelden Rihan for his helpful guide on the forum! I was able to plot my protein pretty easily thanks to him.
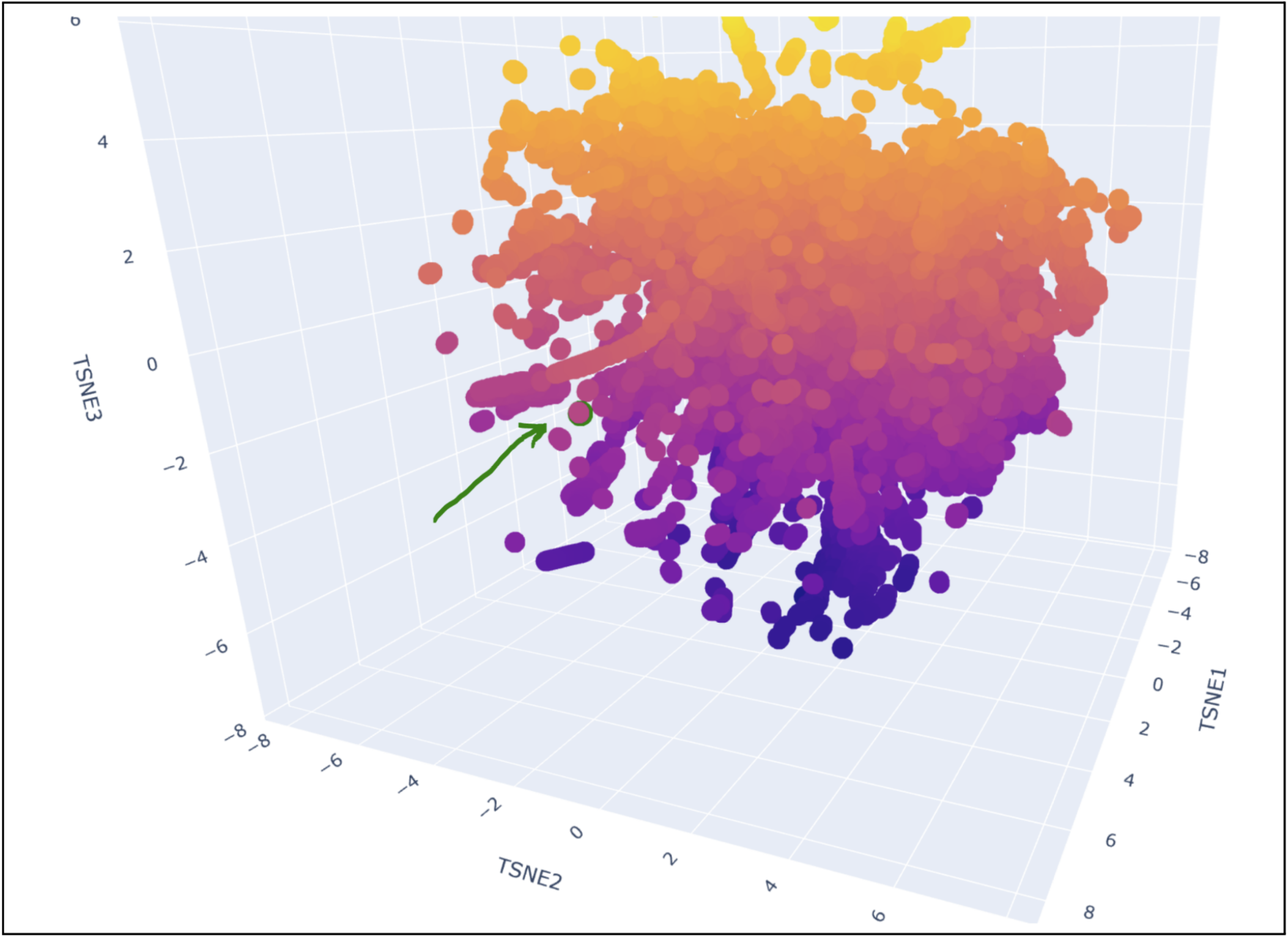 My protein was pretty close to a cluster of other proteins, seen in the photo below.
My protein was pretty close to a cluster of other proteins, seen in the photo below.
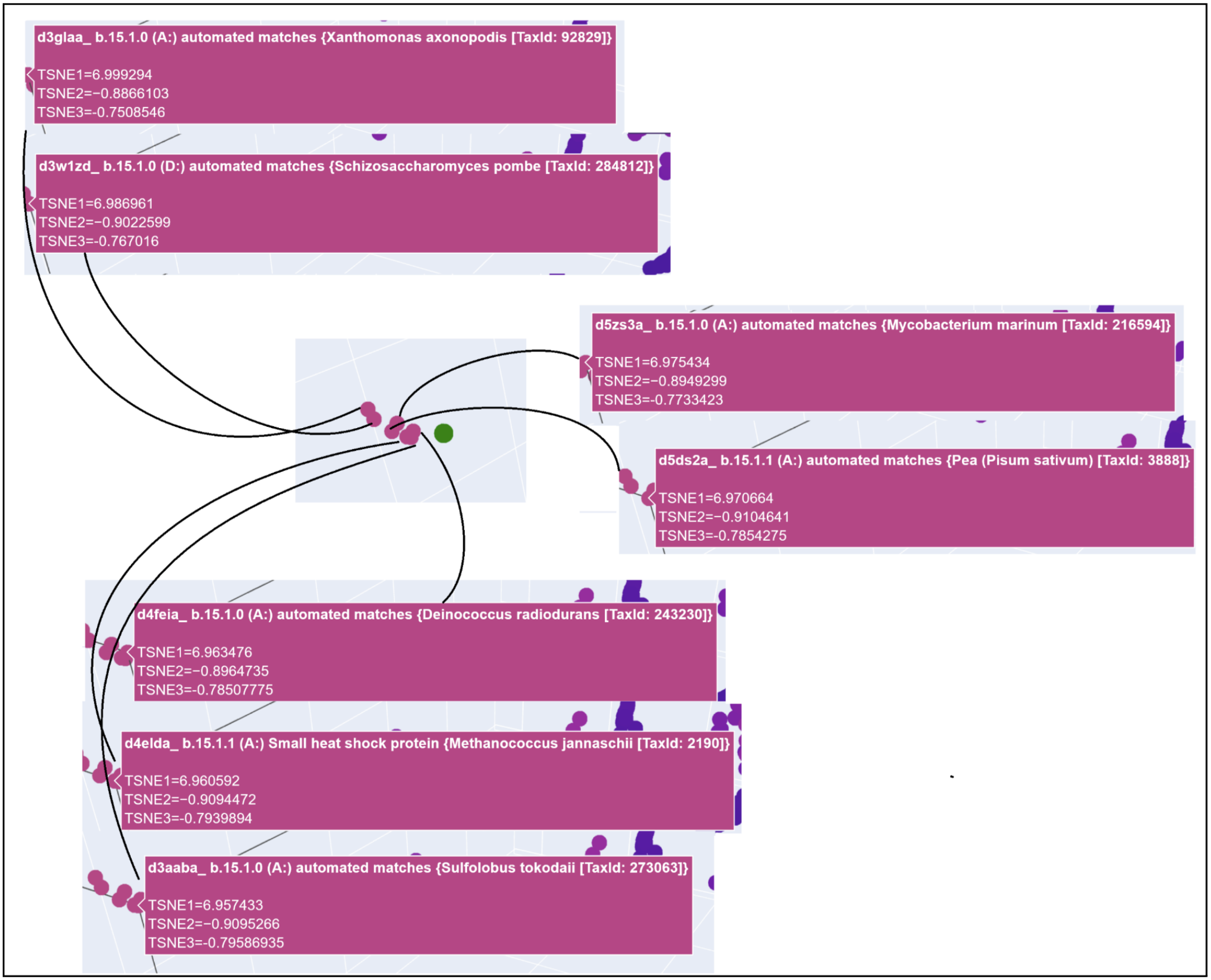
C2. Protein Folding
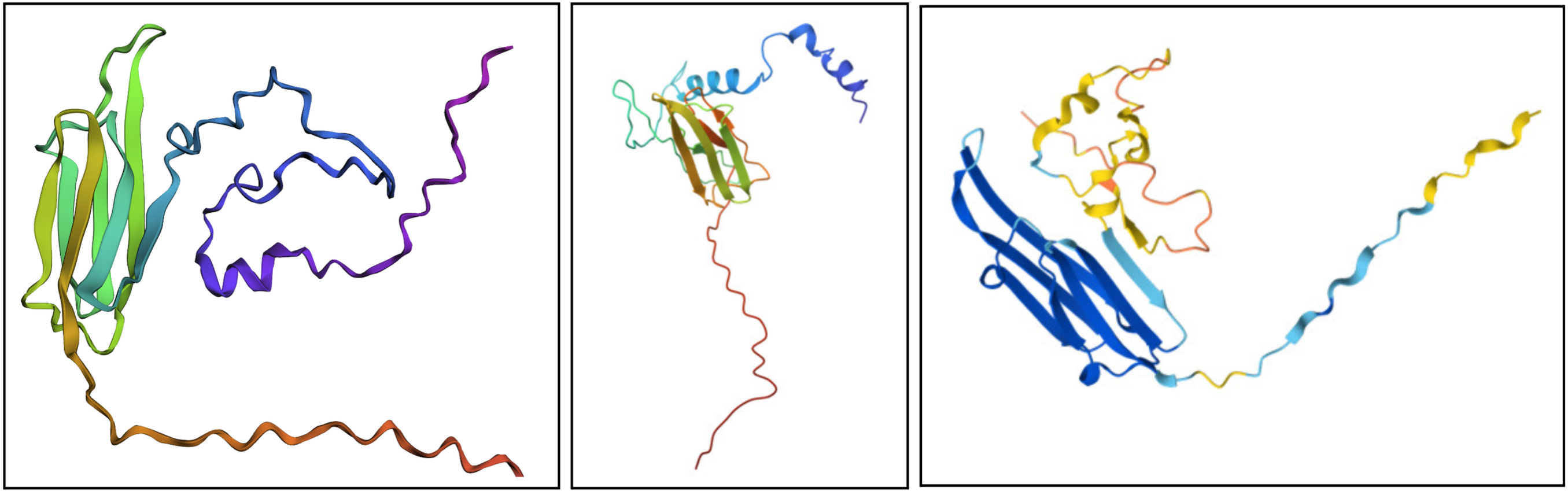
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I would say it looks pretty similar. On the left is the ESMFold result, the middle is a structure derived experimentally (on PDB), and on the right is a structure derived computationally (on PDB). You can see ESMFold has a similar sheet structure with the other two, but the placement of the loops (especially the bottom one) is identical to the right structure (both theoretical) while not necessarily resembling how the structure looks in the middle photo.
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
It seems somewhat resiliant to mutations but not entirely. It’s possible to lose its shape with enough fiddling.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
- Input this sequence into ESMFold and compare the predicted structure to your original.
This part was pretty confusing to me, as the file I’d been referring to earlier (a 175 long sequence for Alpha-crystallin B, called P02511) wasn’t available on PDB. I ended up having to source 2N0K (pdb_00002n0k), a member within P02511 that ultimately had the same sequence, despite other members having different sequences.
I prompted the Colab to design for chain A and B. Both had a length of 82 AA (164 total) and had the following potential amino acid variations.
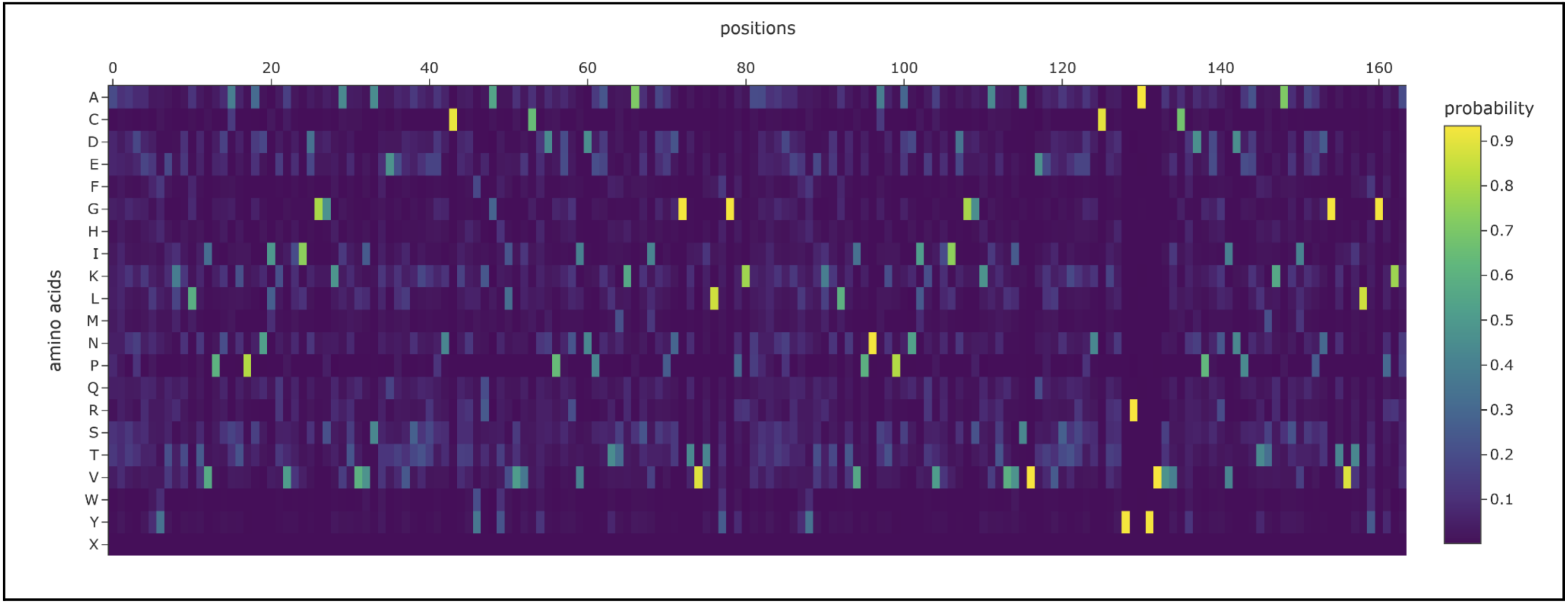 The new sequence ended up being
The new sequence ended up being
PATPEERTIELKVPNAKPENIEVIIDGGRITVKAKELVEKRENCDYYKGYLVECDDPERVDPETMKAEIDEDGTVTIYGPGAPATPEERTIELKVPNAKPENIEVIIDGGRITVKAKELVEKRENCDYYKGYLVECDDPERVDPETMKAEIDEDGTVTIYGPGA
After plugging into ESMFold, I got a structure that looks pretty similar to the original, at least in the way the sheets fold on each other.
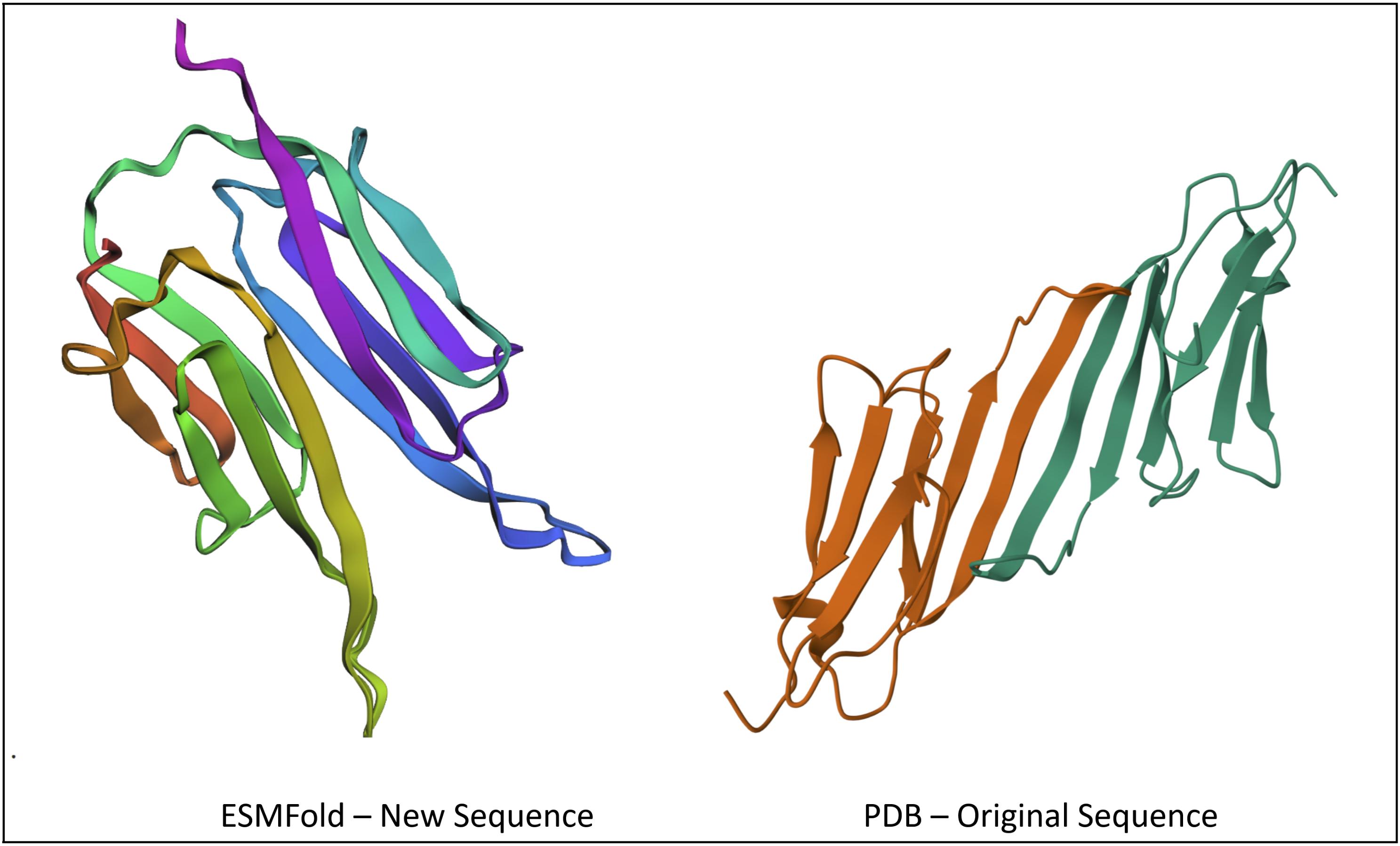
Part D. Group Brainstorm on Bacteriophage Engineering
Our proposal (and research notes) for this assignment can be accessed here.