Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
P00441 can be found on the UnitProt site here. It has the following sequence:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
And I’ll swap MATKA → MATKV to get MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
See table below.
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
See table below.
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
See table below.
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
| Binder | Pseudo Perplexity |
|---|---|
| WHYGAVAAAHKE | 7.5475612006564115 |
| WRYGATGARHKE | 11.178195011097767 |
| WRYPVAALELWK | 21.190836127543506 |
| WRYPAVVLRLKE | 13.790132945872145 |
| FLYRWLPSRRGG (control) |
Part 2: Evaluate Binders with AlphaFold3
- Navigate to the AlphaFold Server
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
| Binder | ipTM Score | Binding Spot |
|---|---|---|
| WHYGAVAAAHKE | 0.31 | Not near N-terminus, engages with β-barrel region. Surface-bound. |
| WRYGATGARHKE | 0.33 | Not near N-terminus, potentially engages with β-barrel region. Surface-bound. |
| WRYPVAALELWK | 0.21 | Not near N-terminus, potentially engages with β-barrel region. Surface-bound. |
| WRYPAVVLRLKE | 0.3 | Somewhat near N-terminus, does not engage with β-barrel region. Surface-bound. |
| FLYRWLPSRRGG (control) | 0.36 | On the complete opposite side of the N-terminus, with the β-barrel in between. Surface-bound. |
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
None of the ipTM values (max 0.33) exceed the known binder (0.36). However, all of the values are poor (< 0.6), which indicates the predictions might not be accurate. Some of the peptides do appear to bind better in their visualizations than the known (e.g. the first one WHYGAVAAAHKE and its interactions with the β-barrel).
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
| Binder | Predicted binding affinity (pKd/pKi) | Solubility (%) | Hemolysis probability (%) | Net charge (pH 7) | Molecular weight (Da) |
|---|---|---|---|---|---|
| WHYGAVAAAHKE | Weak binding, 5.893 | Soluble, 1.000 | Non-hemolytic, 0.024 | -0.06 | 1339.5 |
| WRYGATGARHKE | Weak binding, 6.087 | Soluble, 1.000 | Non-hemolytic, 0.028 | 1.85 | 1431.6 |
| WRYPVAALELWK | Weak binding, 6.446 | Soluble, 0.982 | Non-hemolytic, 0.061 | 0.76 | 1531.8 |
| WRYPAVVLRLKE | Weak binding, 6.506 | Soluble, 0.816 | Non-hemolytic, 0.050 | 1.77 | 1529.8 |
| FLYRWLPSRRGG (control) | Weak binding, 6.361 | Soluble, 0.608 | Non-hemolytic, 0.047 | 2.76 | 1507.7 |
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
Actually, the extreme outlier is WRYPVAALELWK with an ipTM of 0.21, but it doesn’t vary as drastically with its other properties. It’s generally in the middle of the pack with binding affinity, solubility, and charge. One notable difference is its hemolysis probability is a bit higher, but not by much–still enough to be non-hemolytic.
The best performing two, WRYGATGARHKE with 0.33 and the control with 0.36, have solubilities on either extreme and very low to somewhat low hemolysis probability. They do have higher than average net charge, though–maybe that’s something that can be leveraged.
Choose one peptide you would advance and justify your decision briefly.
I don’t have an obvious contender, but WRYGATGARHKE is just behind the control in ipTM and is quite different in properties, so it’d be worth optimizing that to see if there’s a new direction that could produce good peptides.
Part 4: Generate Optimized Peptides with moPPIt
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
I used the following inputs for generation:
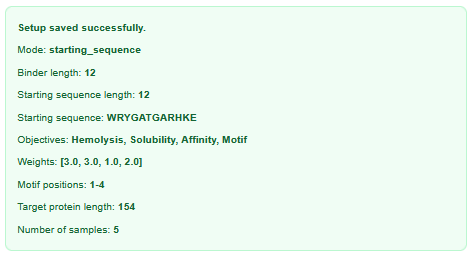
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
| Binder | Predicted binding affinity (pKd/pKi) | Solubility (%) | Hemolysis probability (%) | ipTM |
|---|---|---|---|---|
| CTAGSTVGVGVW | 6.7832 | 0.9996 | 0.0618 | 0.36 |
| ASATFEPPPVCH | 5.8068 | 1 | 0.0223 | 0.39 |
| VSEKYCVQFGKT | 6.2623 | 1 | 0.0405 | 0.33 |
| MSAGICNEFKQK | 5.6404 | 1 | 0.0238 | 0.55 |
| KNPCEAYCFNWV | 6.7200 | 1 | 0.0346 | 0.28 |
I’d say there’s more variety. PepMLM repeated a lot of beginning and ending amino acids in the sequence, but all these sequences look completely unique. It doesn’t reflect as much in the properties, though. For evaluating, I’d run each sequence through the same software and compare properties/ipTM to see if there’s any improvement. E.g. MSAGICNEFKQK had a huge jump in ipTM to 0.55–that’s promising!
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.