Week 4 HW: Protein Design Part I
Part A Conceptual Questions
Q1. How many molecules of amino acids do you take in with a piece of 500 g of meat?
Meat is approximately 25% protein by weight, so 500 g of meat contains about 125 g of protein. Using the given average molecular weight of ~100 Da (= 100 g/mol) per amino acid:
$500\text{ g} \times 0.25 = 125\text{ g of protein}$
Moles of amino acids = 125 g ÷ 100 g/mol = 1.25 mol
Number of molecules = 1.25 mol × 6.022 × 10²³ mol⁻¹ ≈ 7.5 × 10²³ amino acid molecules
Q2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Proteases break dietary proteins down into individual amino acids during digestion, which are chemically identical regardless of source. Once absorbed, your cells reassemble these amino acids into human proteins according to the instructions in your own DNA. No genetic information transfers from food to your genome; dietary DNA is degraded by nucleases in the gut. Food provides raw building blocks, but your genome provides the blueprint, so the output is always human protein.
Q3. Why are there only 20 natural amino acids?
The 20 canonical amino acids provide a near-optimal coverage of side-chain chemical properties — spanning small to large, polar to nonpolar, charged, aromatic, and nucleophilic — with minimal redundancy. The triplet genetic code can encode 64 codons, and after reserving stop signals and building in redundancy to buffer against mutation errors, 20 amino acids strikes a good balance between functional diversity and error tolerance. These 20 are also the ones that were biosynthetically accessible through early metabolic pathways derived from central metabolites. Once the translation machinery co-evolved around this set, changing it became prohibitively costly since it would affect every protein in every organism, so the system became frozen early in evolution.
Q4. Where did amino acids come from before enzymes that made them, and before life started?
Amino acids predate life and arise from chemistry. The Miller–Urey experiment demonstrated that electric discharges through a reducing atmosphere produce glycine, alanine, aspartate, and other amino acids. Life inherited these building blocks from prebiotic geochemistry and later evolved enzymatic pathways to produce them more efficiently.
Q5. If you make an α-helix using D-amino acids, what handedness would you expect?
A left-handed α-helix. The natural right-handed α-helix arises because L-amino acids position their side chains to minimize steric clashes with backbone carbonyls specifically in the right-handed conformation. D-amino acids are the mirror image of L-amino acids, so the favorable backbone dihedral angles flip sign — from (−57°, −47°) to (+57°, +47°) — producing a left-handed helix. This is confirmed experimentally: synthetic D-peptides give circular dichroism spectra that are exact mirror images of natural L-peptide helices.
Q6. Can you discover additional helices in proteins?
Yes. (according to google) Beyond the common α-helix, proteins contain 3₁₀-helices (3.0 residues/turn, i→i+3, common at helix termini), π-helices (4.4 residues/turn, i→i+5, rare single-turn insertions), and the collagen triple helix. In principle, any repeating set of backbone (φ, ψ) angles that permits regular hydrogen bonding defines a helix, and the main candidates have been systematically mapped from the Ramachandran plot.
Q7. Why are most molecular helices right-handed?
The dominance of right-handed helices stems from the universal use of L-amino acids–> the lowest-energy conformation due to favorable side-chain positioning.
Once L-amino acids became dominant, all downstream molecular machinery co-evolved around that chirality. If life had been founded on D-amino acids, left-handed helices would dominate and the biology would be equally functional.
Q8. Why do β-sheets tend to aggregate? What is the driving force?
β-sheets are inherently open-ended structures: unlike α-helices where all backbone hydrogen-bond donors and acceptors are satisfied internally, β-sheet edge strands have one face of exposed N–H and C=O groups available for hydrogen bonding with additional strands. This creates a thermodynamic driving force to recruit more strands and extend the sheet. The main forces driving aggregation are backbone hydrogen bonding between exposed edges, essentially intermolecular β-sheet extension, the hydrophobic effect from burying nonpolar side chains between stacked sheets, and van der Waals contacts in the cross-β arrangement.
Q9. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Proteins involved in amyloid diseases have aggregation-prone hydrophobic stretches or destabilizing mutations that lower the kinetic barrier to reaching this state, and once a nucleus forms it templates further conversion in a self-propagating manner.
Amyloid fibrils can be used as materials. They have tensile strength comparable to steel and Young’s moduli of 2–14 GPa, and they resist proteases, detergents, and heat. In bionanotechnology, amyloid fibrils serve as scaffolds for conductive nanowires, hydrogel matrices for tissue engineering and drug delivery, and membranes for heavy-metal water purification.
Part B — Protein Analysis and Visualization
Q1. Briefly describe the protein you selected and why you selected it.
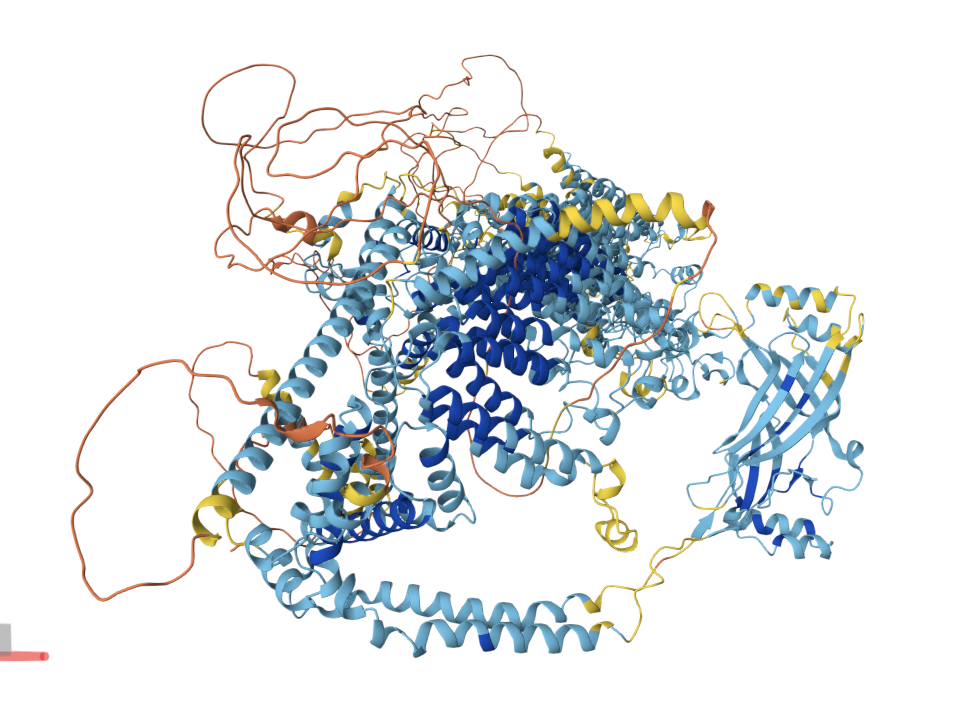
PIEZO1 is a homotrimeric mechanosensitive ion channel that converts physical forces — such as fluid shear stress, membrane stretch, and compressive pressure — into biochemical signals by allowing cation influx (primarily Ca²⁺) upon mechanical stimulation. Each subunit contains ~38 transmembrane helices that form a distinctive curved, propeller-like architecture with three peripheral “blades” and a central pore.
PIEZO1 is valuable because it serves as a fundamental mechanical switch for cellular programming: it governs processes including vascular development, red blood cell volume regulation, blood pressure sensing, and cell lineage determination in stem cells.
Q2. Identify the amino acid sequence of your protein.
Sequence length and composition
- Length: 2,521 amino acids (human PIEZO1, UniProt Q9H5I5).
- Most common amino acid: Leucine (L), appearing 367 times (~14.6% of the sequence). This is expected — leucine is the most abundant residue in transmembrane α-helices due to its hydrophobic character and favorable helix-forming propensity, and PIEZO1 is overwhelmingly α-helical with ~38 transmembrane passes per subunit.
Homologs
 Using UniProt BLAST on the human PIEZO1 sequence returns homologs across a broad range of eukaryotes — vertebrates, insects, plants, and even single-celled eukaryotes — reflecting the ancient evolutionary origin of mechanosensation. The closest homolog is PIEZO2 (human, ~42% sequence identity), which mediates light touch and proprioception. Beyond PIEZO2, orthologs of PIEZO1 are found in most metazoan genomes (mouse, zebrafish, Drosophila, C. elegans), with more distant homologs in plants (Arabidopsis) and protists. A typical BLAST search returns several hundred significant hits (E-value < 0.05), though the number depends on the database and threshold used.
Using UniProt BLAST on the human PIEZO1 sequence returns homologs across a broad range of eukaryotes — vertebrates, insects, plants, and even single-celled eukaryotes — reflecting the ancient evolutionary origin of mechanosensation. The closest homolog is PIEZO2 (human, ~42% sequence identity), which mediates light touch and proprioception. Beyond PIEZO2, orthologs of PIEZO1 are found in most metazoan genomes (mouse, zebrafish, Drosophila, C. elegans), with more distant homologs in plants (Arabidopsis) and protists. A typical BLAST search returns several hundred significant hits (E-value < 0.05), though the number depends on the database and threshold used.
Protein family
PIEZO1 belongs to the Piezo family , a eukaryote-specific family of mechanosensitive channels with no significant homology to any other known ion channel family (e.g., TRP channels, Degenerin/ENaC, or MscL/MscS bacterial mechanosensitive channels). This makes the Piezo family an evolutionarily independent solution to mechanotransduction.
Q3. Identify the structure page of your protein in RCSB.
 Structure and resolution
Structure and resolution
The primary full-length structure is PDB: 5Z10 . The human PIEZO1 also has related entries (e.g., PDB 7WLT).
- Resolution: 3.97 Å. For a cryo-EM structure of a ~900 kDa trimeric membrane protein, this is a reasonable resolution — sufficient to trace the backbone, assign secondary structure, and identify transmembrane helix positions. However, it is not high resolution by crystallographic standards; individual side-chain conformations and water molecules are generally not resolvable at this resolution.
Other molecules in the structure
The solved structure contains:
- Lipid molecules — Phospholipids are resolved in the transmembrane domain, consistent with PIEZO1’s curved membrane-embedded architecture and its sensitivity to membrane composition and tension.
- Detergent molecules — from the purification process (typically digitonin or LMNG).
- Ions — depending on the specific entry, Ca²⁺ or other cations may be modeled in or near the pore region.
Structure classification
In the RCSB classification, it falls under membrane proteins → ion channels → mechanosensitive channels. Its unique propeller-blade topology does not closely resemble any other structurally characterized ion channel family, making it a distinct structural class.
Q4. Visualize the structure of your protein.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon
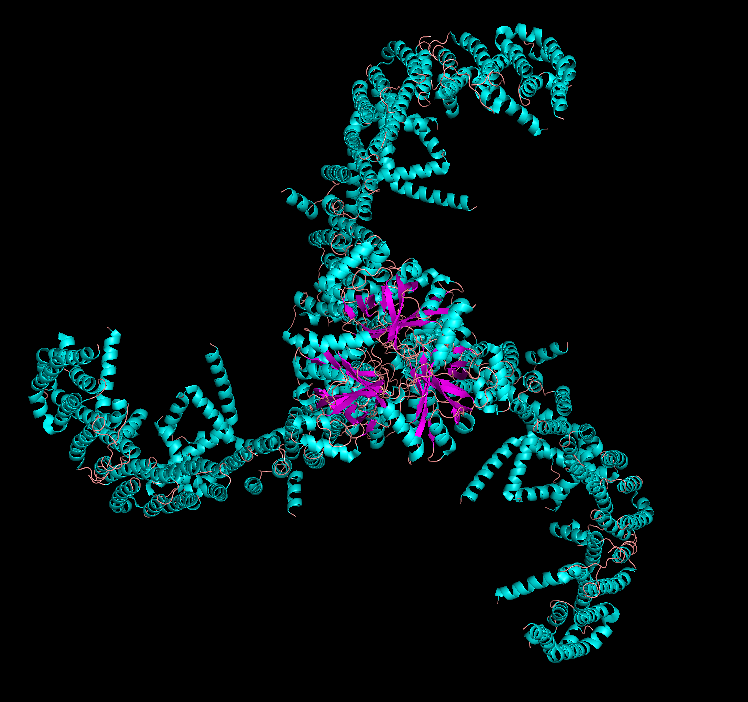
Ribbon

Ball and Stick

Secondary structure
PIEZO1 is overwhelmingly α-helical. Each subunit contains ~38 transmembrane helices organized into repeated structural units called “Piezo repeats” (or “transmembrane helical units”), which form the curved blades of the propeller. The central pore region includes an inner helix (TM37), outer helix (TM38), and the C-terminal extracellular domain (CED). There are virtually no β-sheets in the structure — only short loops and turns connect the helices. This extreme α-helical bias is consistent with its identity as a multi-pass transmembrane protein.
Residue type distribution (hydrophobic vs. hydrophilic)
When colored by residue type:
- The transmembrane blade regions are dominated by hydrophobic residues (Leu, Ile, Val, Phe, Ala) — these face the lipid bilayer and form the core of helix-helix packing within the membrane. This explains why leucine is the most frequent amino acid.
- Hydrophilic and charged residues (Arg, Lys, Glu, Asp) are concentrated at the intracellular and extracellular surfaces, at helix termini (anchoring the protein at the membrane-water interface), and lining the central ion conduction pore (where they contribute to ion selectivity and gating).
- The CED (C-terminal extracellular domain), which protrudes above the membrane at the trimer center, has a higher proportion of polar and charged residues, consistent with its aqueous environment.
This distribution follows the classic “positive-inside rule” — positively charged residues (Arg, Lys) are enriched on the cytoplasmic side of the membrane.
Surface and binding pockets
The surface of PIEZO1 reveals several notable features:
- Central pore. The most prominent “hole” is the ion conduction pathway at the trimer axis. This is the functional pore through which cations flow upon channel activation.
- Lateral fenestrations. Between the blade domains near the membrane plane, there are openings (fenestrations) that may allow lateral lipid access to the pore — a feature shared with some other ion channels and potentially important for lipid-mediated gating.
- Intracellular “cap” cavity. On the cytoplasmic face, the converging beam-like structures create an enclosed cavity that has been proposed as a binding site for intracellular modulators.
- Yoda1 binding site. The small-molecule agonist Yoda1 binds in a pocket between the blade and pore module (identified in structures like PDB 7WLT), confirming a druggable pocket in the structure.
Overall, the surface is not smooth — the curved, dome-shaped architecture creates multiple grooves and pockets that are functionally relevant for lipid interaction, mechanical force transduction, and pharmacological targeting.
Part C - Using ML-Based Protein Design Tools
1. Deep Mutational Scans
1.1 Method
ESM2 was used to generate an unsupervised deep mutational scan of human PIEZO1 (UniProt Q9H5I5, 2,521 amino acids). For every position in the sequence, the model scores the log-likelihood of substituting the wild-type residue with each of the 20 amino acids. The resulting heatmap displays Model Scores across all positions (x-axis) and all possible amino acid substitutions (y-axis), where green/yellow indicates neutral or favorable substitutions and dark blue/purple indicates substitutions the model predicts to be strongly deleterious.
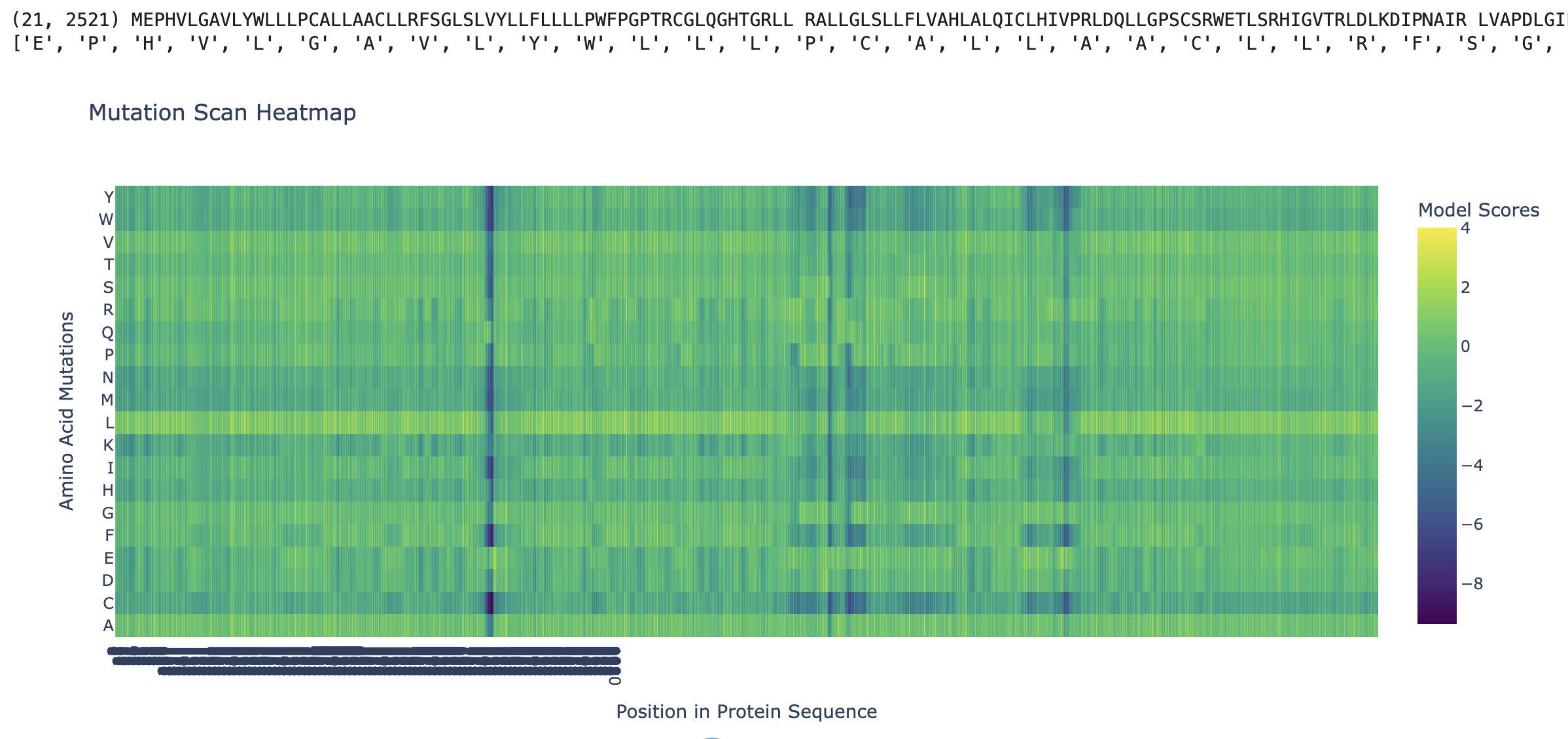
1.2 Observed Patterns
Conserved positions appear as dark vertical columns. Several positions show strongly negative scores across nearly all 20 substitutions, indicating that the model considers any change at those positions highly unlikely based on evolutionary sequence patterns. These columns correspond to residues that are critical for PIEZO1’s structure or function — they map primarily to the pore-lining region and the C-terminal anchor domain, where even conservative substitutions would disrupt ion conduction or mechanical gating.
The Leucine (L) row is notably bright across most positions. Mutations to leucine are generally well-tolerated, which is consistent with PIEZO1’s identity as a multi-pass transmembrane protein (~38 TM helices per subunit). Leucine is the most common residue in α-helical transmembrane domains due to its hydrophobic character and favorable helix-forming propensity, so substituting to leucine is a “safe” change at most positions.
The Glycine (G) row shows scattered deep blue spots. Positions where the wild-type is glycine tend to show dark columns across other substitutions. Glycines in transmembrane helices are critical for helix packing and flexibility — they allow tight inter-helix contacts that bulkier residues would sterically prevent. Mutating these glycines is therefore strongly disfavored.
A specific example: One of the most prominent dark vertical bands appears in the region corresponding to the inner pore helix of PIEZO1. Conserved charged residues in this region (e.g., glutamate or arginine residues lining the pore) score very negatively when mutated to hydrophobic residues like leucine, isoleucine, or valine. This is biologically expected — charged residues in the pore domain are essential for cation selectivity and gating, and replacing them with hydrophobic side chains would destroy channel function.
2. Latent Space Analysis
2.1 Method
15,177 structurally classified protein domains from the SCOPe/ASTRAL database were embedded using ESM2-8M (hidden dimension = 320) into 320-dimensional vectors. t-SNE then projected these into 3D for visualization. The color scale represents TSNE3 (yellow = high, purple = low), providing visual depth. Despite using the smallest ESM2 model, the projection recovers meaningful structural groupings, demonstrating that protein language models encode structural information implicitly from sequence alone.
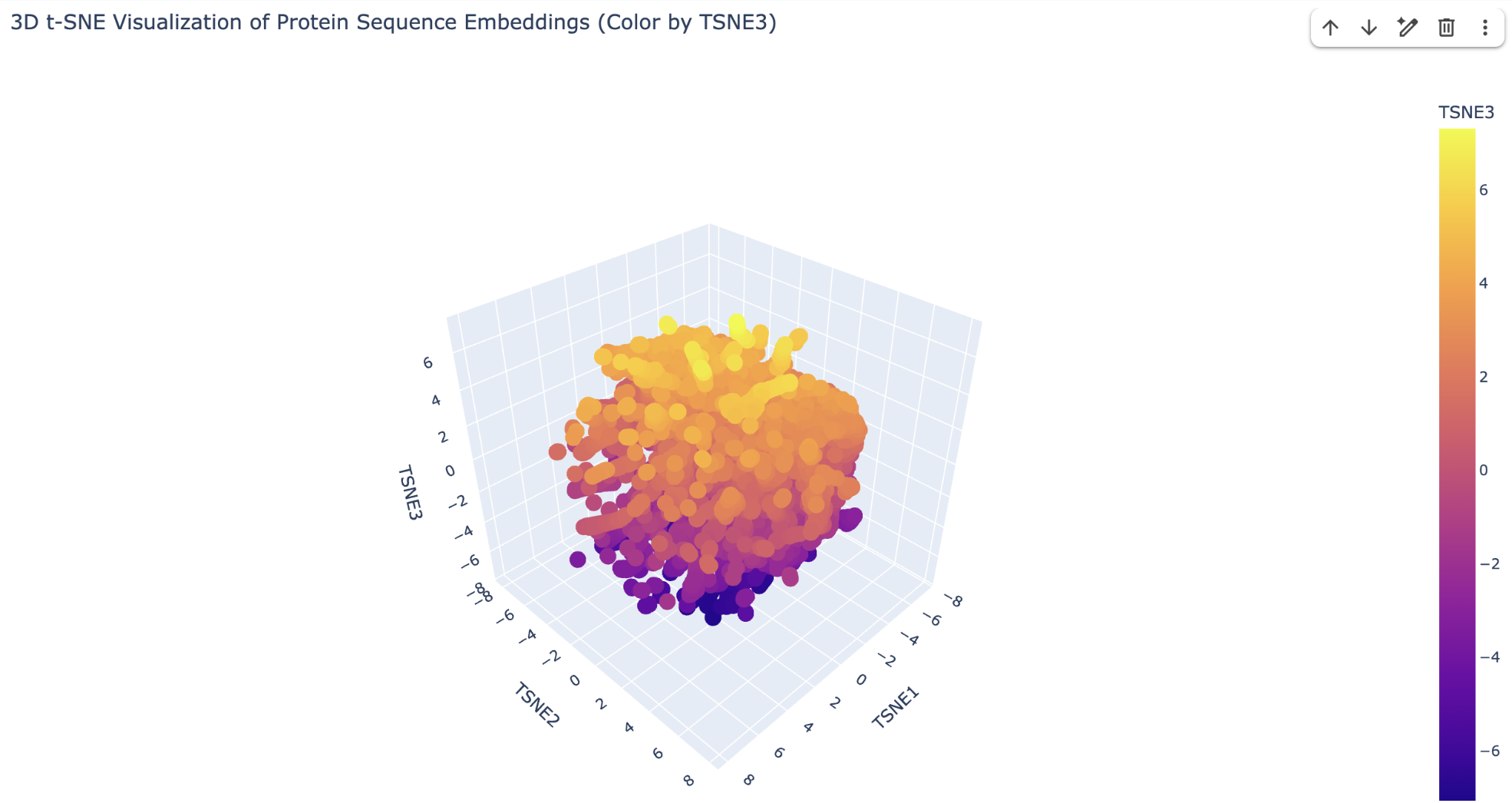
2.2 Neighborhood Analysis
I took three corresponding coordinates for analysis:
Upper yellow region (high TSNE3) — β-sheet-rich proteins.
- d2g5da1 (TSNE: −2.29, −1.13, 4.05) is Membrane-bound lytic murein transglycosylase A (MLTA) from Neisseria gonorrhoeae. Its neighbors in this yellow cluster are predominantly other β-barrel and β-sheet-rich domains, including outer membrane proteins from gram-negative bacteria that share the β-barrel architecture.
Dense central orange region (intermediate TSNE3) — common α/β folds.
- d3cwna_ (TSNE: −0.82, 0.88, 0.34) is an E. coli protein matching SCOP class c.1.10.1 (α/β, TIM barrel fold). The TIM barrel is the most common enzyme fold in nature (found in glycolysis enzymes, aldolases, tryptophan synthase, etc.), and its position in the densest part of the plot reflects both its abundance in protein databases.
Lower purple region (low TSNE3) — unusual/transmembrane proteins.
- d1x2ma1 (TSNE: −0.79, 0.85, −6.20) is Lag1 longevity assurance homolog 6 (LASS6/CerS6) from mouse. LASS6 is a multi-pass transmembrane ceramide synthase with ~5–6 TM helices and a unique Lag1p motif. Its position far from the soluble enzyme core reflects ESM2’s recognition that its hydrophobic, membrane-spanning sequence features are fundamentally distinct from typical soluble proteins.
2.3 Placing PIEZO1
PIEZO1 would be expected to sit in the purple periphery or as an isolated outlier given that
It is an extremely large multi-pass transmembrane protein, so its sequence composition is heavily biased toward hydrophobic residues. This transmembrane character would push it away from the soluble-protein-dominated central core, similar to how LASS6 sits in the purple region.
PIEZO1 has no sequence homology to any other known ion channel family. Its “Piezo repeat” domains and propeller-blade architecture are structurally unique. ESM2 would therefore embed it far from other channel proteins.
The only protein expected to sit nearby is PIEZO2 (~42% sequence identity), the sole close homolog. If PIEZO2 is absent from the dataset, PIEZO1 would sit alone — reflecting the evolutionary isolation of the Piezo family as a structurally novel, independent solution to mechanosensation.