Week 5 HW: Protein Design Part II
Part A SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Human SOD1 Sequence (UniProt)
A4V Mutant Sequence
Position 4: Ala → Val (A4V)
| Position | Wild-type | Mutant |
|---|---|---|
| 4 | A (Ala) | V (Val) |
PepMLM-Generated Peptides
4 candidate binders generated against the A4V mutant sequence + the known reference peptide.
Lower perplexity scores indicate sequences more confidently predicted by the model.
| # | Sequence | Perplexity | Note |
|---|---|---|---|
| PepMLM 1 | WHYPAAAAAWKK | 8.611 | — |
| PepMLM 2 | WRSPAVAAAHKE | 7.866 | Lowest perplexity |
| PepMLM 3 | WRYPAVALEWKK | 16.562 | Highest perplexity |
| PepMLM 4 | WHSYVVGARWWK | 13.338 | — |
| Known | FLYRWLPSRRGG | — | Reference binder |
Note on Perplexity: In PepMLM, perplexity reflects how confidently the masked language model predicts each residue in context. Lower perplexity suggests the sequence is more consistent with the model’s learned distribution of binders; however, higher perplexity sequences may still yield productive binding if their physicochemical and structural properties are favourable.
Part 2: Evaluate Binders with AlphaFold 3
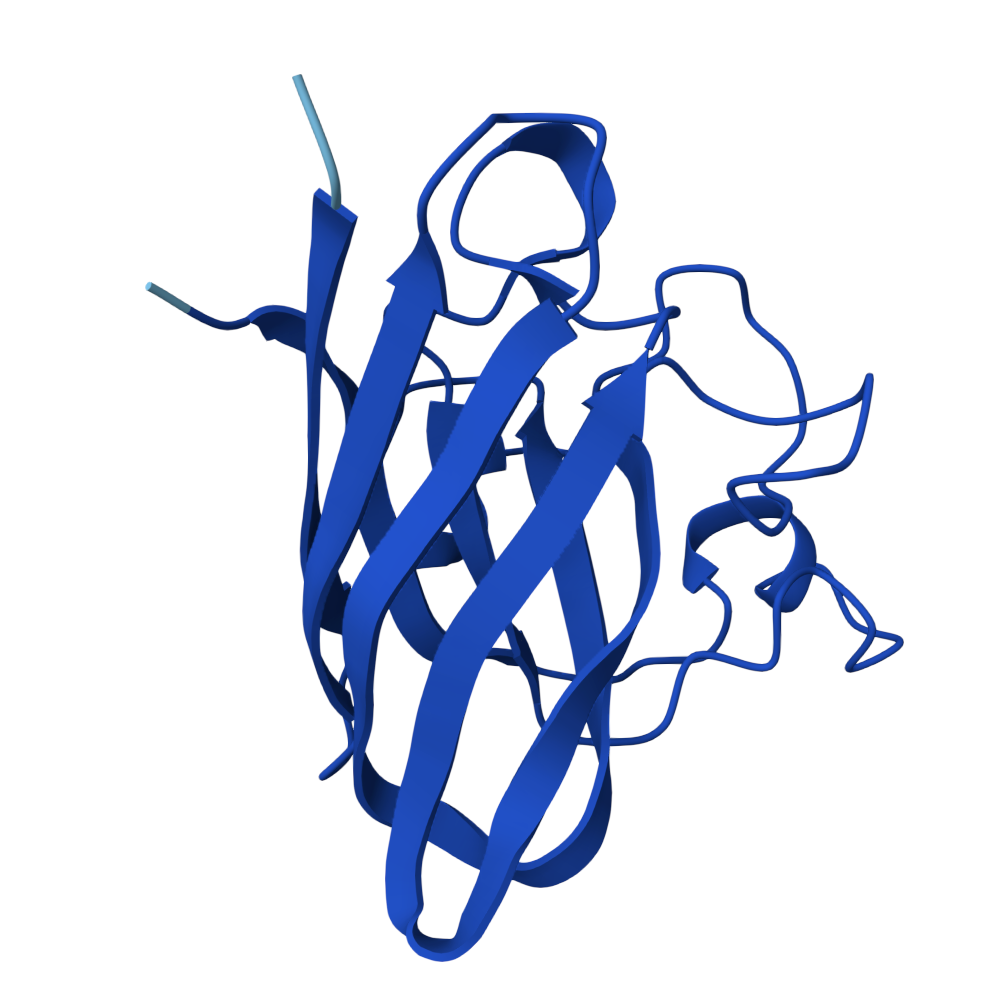 For the sake of my OCD or else with only 5 pics will look ugly | 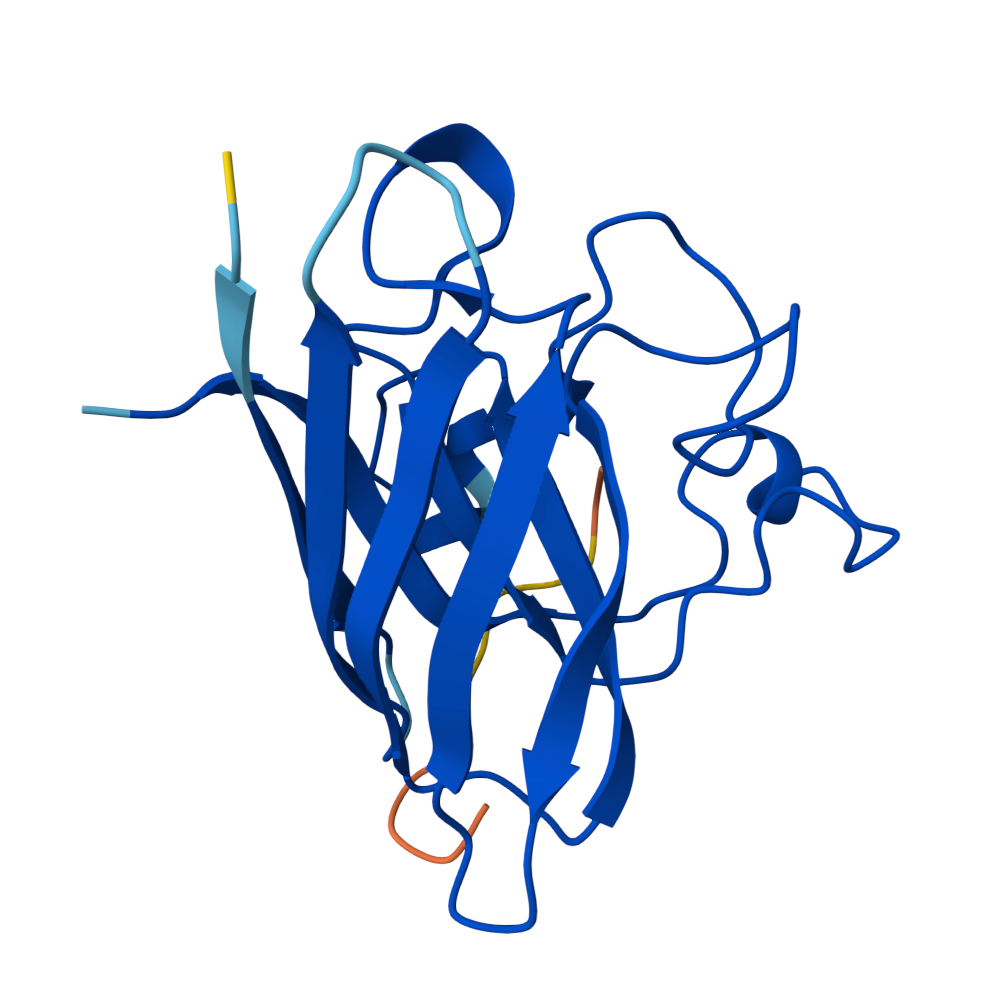 Known Peptide ipTM = 0.36 | 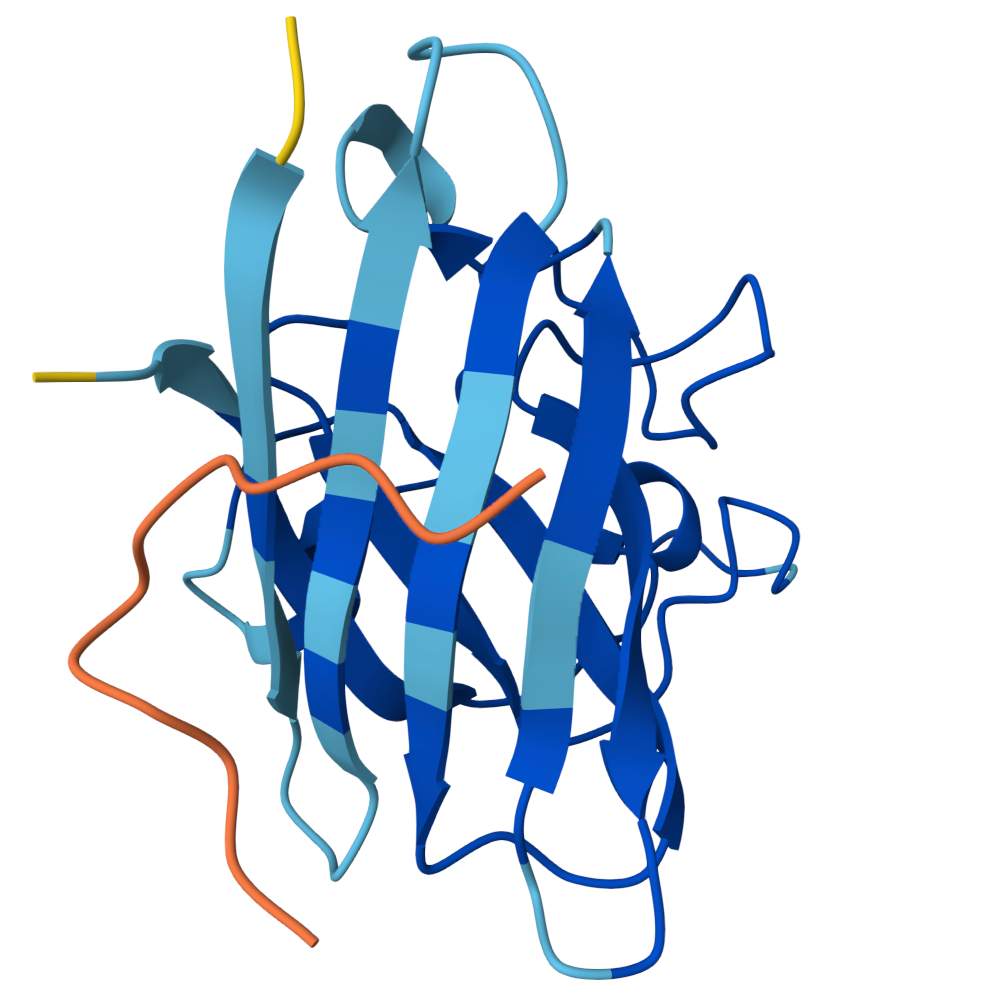 Peptide 1 ipTM = 0.27 |
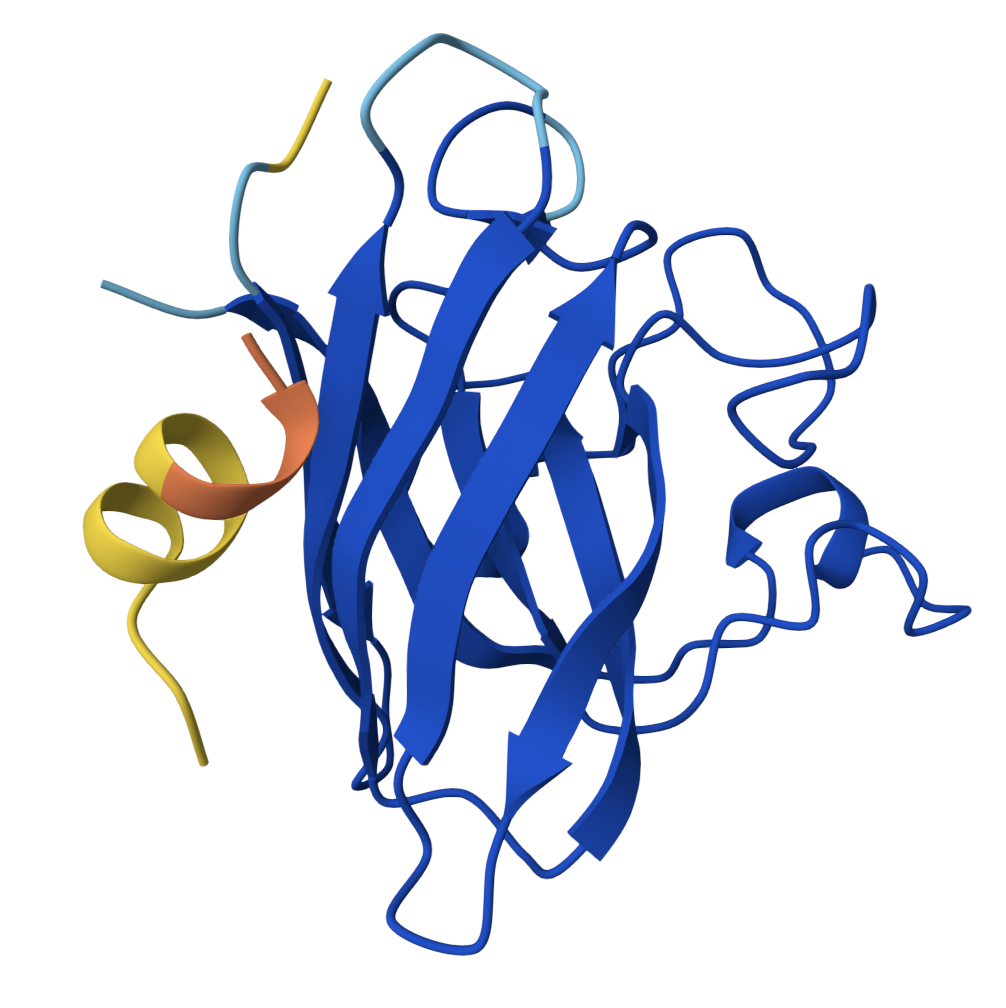 Peptide 2 ipTM = 0.40 | 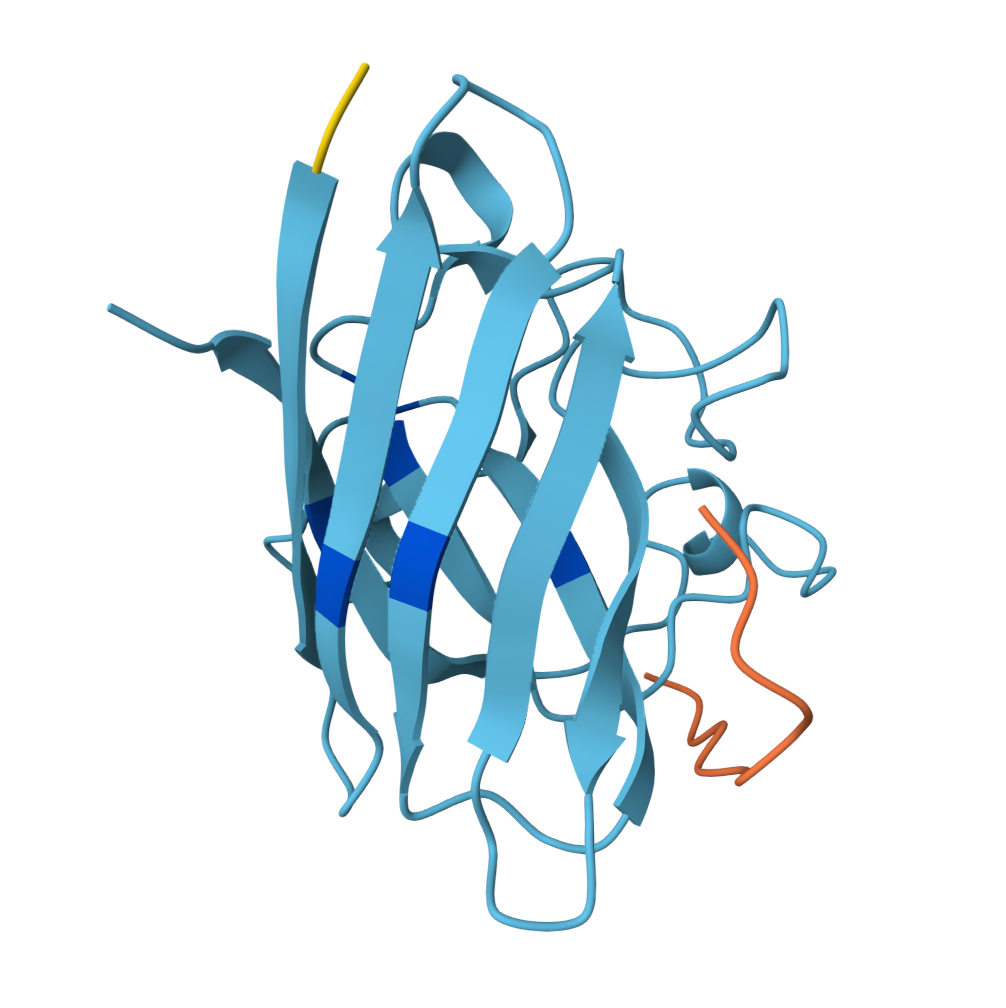 Peptide 3 ipTM = 0.19 | 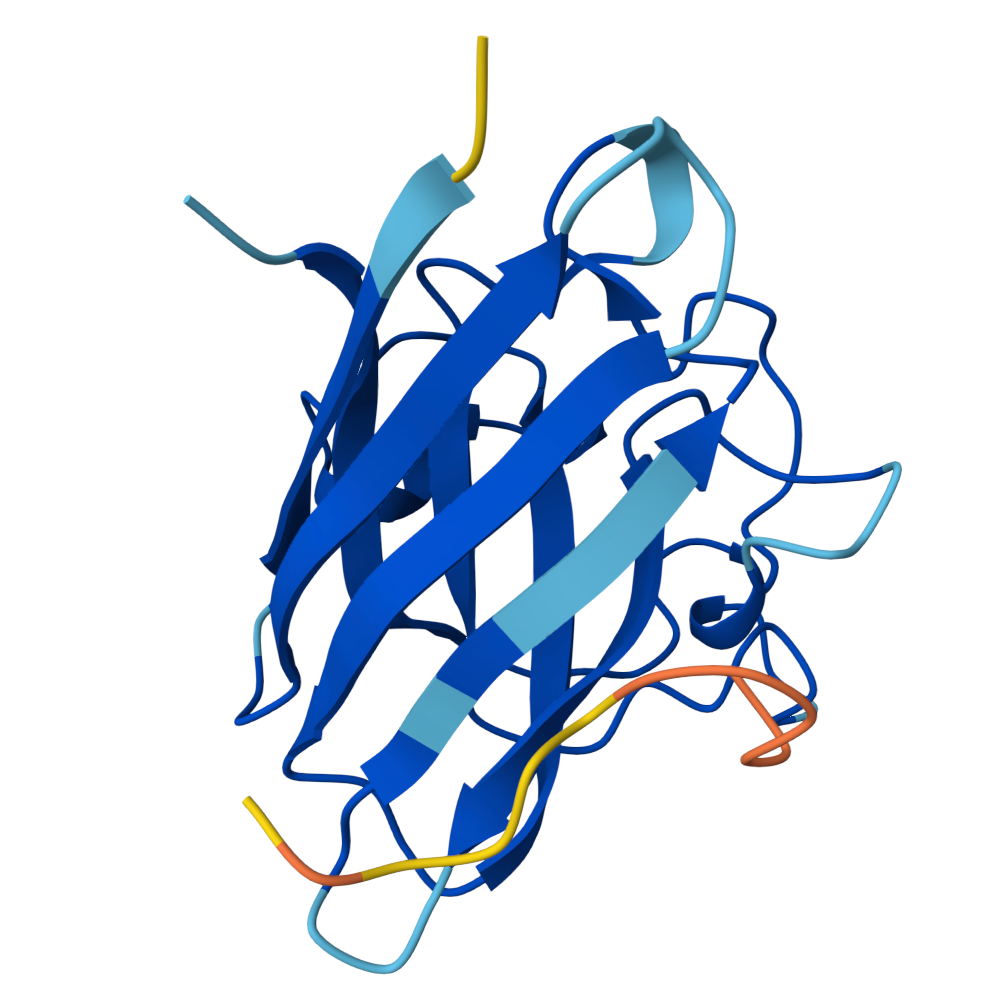 Peptide 4 ipTM = 0.39 |
ipTM (interface predicted TM-score) measures predicted interface accuracy.
Values range from 0 to 1 — higher is better. Scores ≥ 0.5 generally indicate confident predictions.
Binding Analysis
| Structure | ipTM | Near A4V / N-term? | β-barrel engagement | Surface character |
|---|---|---|---|---|
| Known (Reference) | 0.36 | Yes | Lateral strand edge | Surface-bound, extended |
| PepMLM Peptide 1 | 0.27 | No | Minimal | Surface, poorly engaged |
| PepMLM Peptide 2 | 0.40 | Partial — dimer face | Lateral interface cleft | Surface docked |
| PepMLM Peptide 3 | 0.19 | No | None | Peripheral, non-specific |
| PepMLM Peptide 4 | 0.39 | Distal (C-term base) | Bottom loop region | Surface-bound |
Notes
- PepMLM Peptide 2 is the strongest candidate: highest ipTM, adopts α-helical secondary structure upon binding, and docks into the concave groove at the lateral β-barrel interface — the region destabilised by the A4V mutation. One face of the helix contacts SOD1 while the other remains solvent-exposed. This binding mode is consistent with therapeutic peptides that stabilise misfolding-prone interfaces.
- PepMLM Peptide 4 has a comparable ipTM (0.39) but localises to the base of the barrel near C-terminal loops, distal from the A4V site, limiting its therapeutic relevance.
- PepMLM Peptides 1 and 3 show poor interface engagement and are unlikely to be productive binders.
ipTM (interface predicted TM-score) measures predicted interface accuracy.
Values range from 0–1; scores ≥ 0.5 generally indicate confident predictions. All values here are modest, consistent with flexible peptide–protein interfaces typical in AlphaFold-Multimer assessments.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
swipe left for more
PepMLM 1WHYPAAAAAWKK | PepMLM 2WRSPAVAAAHKE | PepMLM 3WRYPAVALEWKK | PepMLM 4WHSYVVGARWWK | Known (Reference)FLYRWLPSRRGG | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
All peptides are predicted soluble and non-hemolytic. Binding affinity (pKd/pKi): higher = stronger predicted affinity. Negative GRAVY scores reflect hydrophilic character across all sequences. Across the five peptides, there is no clear correlation between ipTM and predicted binding affinity.
The peptide I selected is PepMLM Peptide 2 (WRYPAVALEWKK). While its predicted affinity is modest, it has the highest ipTM, adopts stable α-helical secondary structure upon docking — a hallmark of productive peptide–protein interfaces — and engages the lateral cleft of the β-barrel at precisely the region destabilised by A4V. It is the only candidate where the structural, physicochemical, and site-specificity evidence converge.
Part C: Final Project: L-Protein Mutants
The MS2 bacteriophage lysis protein (L-protein) is a 74 amino acid protein responsible for killing E. coli host cells by perforating the bacterial membrane. A critical vulnerability of this system is that a single point mutation in the host chaperone protein DnaJ can prevent the lysis protein from functioning, allowing E. coli to acquire resistance to MS2.
The L-protein has two structurally and functionally distinct regions:
- Soluble N-terminal domain (positions 1–38): responsible for interaction with DnaJ
- Transmembrane domain (positions 39–73): responsible for membrane insertion and lysis
At least 2 in the transmembrane region and at least 2 in the soluble region.
Option 1: Mutagenesis
Running the ESM-2 protein language model
(facebook/esm2_t6_8M_UR50D) on the full wild-type L-protein sequence:
The model scores every possible single amino acid substitution at every position using a Log Likelihood Ratio (LLR):
- Positive score → the substitution looks evolutionarily natural and compatible
- Negative score → the substitution disrupts what the model expects at that position
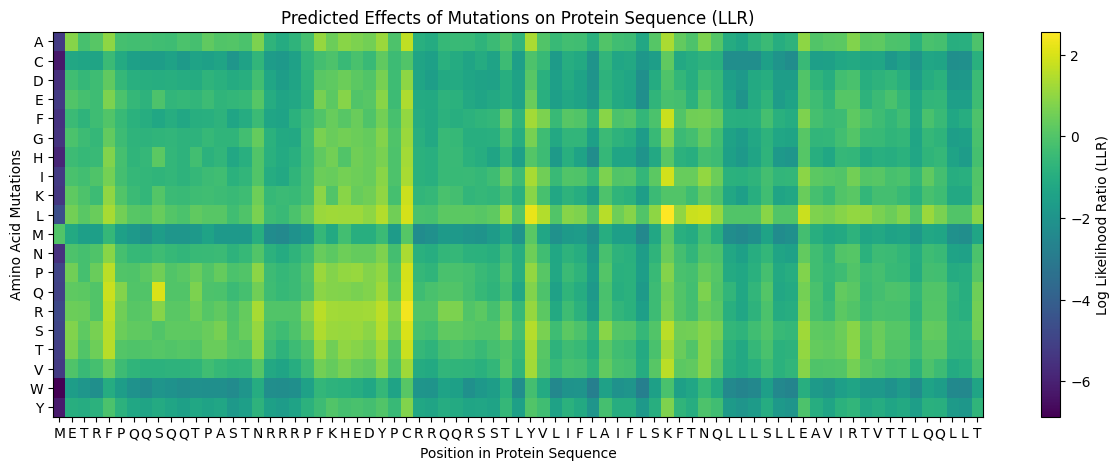
- Position 1 (M) showed almost entirely dark purple scores, confirming the start methionine is essential and should not be mutated
- Rows M, W, Y were dark across most positions — large/bulky amino acids are generally disruptive substitutions
- The transmembrane region (~positions 39–73) showed brighter yellow/green scores for hydrophobic substitutions (L, I, V, F) — consistent with the hydrophobic nature of membrane-spanning helices
- Bright yellow hotspots at positions 29, 39, and 50 stood out as positions where specific mutations are strongly predicted
The notebook was first run with a focused query on the transmembrane region (positions 38–60), producing the following top-scored mutations:
Three positions dominate the top scores: 50, 39, and 45. The model strongly favors leucine (L) substitutions at positions 50 and 39, and also at position 45. This is the first signal pointing toward K50L, Y39L, and A45(L or P) as strong TM candidates. Notably, multiple substitutions at position 50 rank highly (L, I, F, V, S, A),suggesting this position is generally flexible — but leucine scores the highest of all.
The notebook was then run on the full protein sequence to get a global ranking across all 74 positions:
The top 10 globally are dominated by three positions: 50 (K→L), 29 (C→R/S/Q/P/L), and 39 (Y→L). This globally confirms what the TM scan already suggested, and additionally highlights C29 in the soluble region as a computationally interesting mutation site.
The full ranking also produced a second merged output combining both score datasets:
The computational shortlist from the ESM model was:
- K50L (score: +2.56) — highest in entire protein
- C29R (score: +2.40) — highest in soluble region
- Y39L (score: +2.24) — strong TM candidate
- A45L (score: +1.54) — noted in TM scan
The L-Protein Mutants CSV was uploaded into the notebook, which displayed the first rows of the experimental dataset:
This dataset contains experimentally measured lysis outcomes (0 = no lysis, 1 = lysis) for mutations that have already been tested in the lab. Cross-referencing this with the ESM scores revealed which computational predictions align with real biology.
Merging both datasets exposed a critical finding: the ESM model only partially agrees with experimental lysis outcomes.
| Mutation | ESM Score | Lysis (Lab) | Agreement? |
|---|---|---|---|
| P13L | +0.10 | Yes | ✅ |
| S15A | +0.04 | Yes | ✅ |
| K23E | +0.18 | Yes | ✅ |
| E25G | +0.45 | Yes | ✅ |
| A45P | +0.04 | Yes | ✅ |
| I46F | -0.10 | Yes | ❌ |
| R18G | -0.85 | Yes | ❌ |
| R31I | -0.93 | Yes | ❌ |
| L44P | -1.59 | Yes | ❌ |
| R20W | -2.18 | Yes | ❌ |
The disagreements (especially R18G, I46F, L44P) suggest that the ESM model scores general protein structural fitness (the ability to fold into a stable, functional, three-dimensional shape (conformation) that is energeticaly favorable), not functional lysis activity (the process of breaking open cell membranes).
Mutations that disrupt DnaJ binding (like R18G) are penalised by the model because the arginine is evolutionarily conserved — but conserved because it binds DnaJ.
This insight shaped the final selection strategy:
Use ESM scores to identify novel untested candidates with high computational confidence, and use experimental data to validate or override those scores based on known biology.
With all evidence assembled, five mutations were selected spanning both protein regions:
Soluble Region Mutations (Positions 1–38)
P13L — Position 13, Proline → Leucine
- ESM Score: +0.10 | Lysis: Confirmed | Protein Level: Confirmed
- Proline at position 13 creates a rigid backbone kink within the DnaJ-binding domain. Replacing it with leucine (flexible, hydrophobic) removes this constraint, potentially allowing the soluble domain to fold independently of DnaJ. Supported by both model and lab.
S15A — Position 15, Serine → Alanine
- ESM Score: +0.04 | Lysis: Confirmed | Protein Level: Confirmed
- Serine at position 15 sits within the NRRRP arginine-rich DnaJ-binding motif. Its hydroxyl side chain is a candidate hydrogen-bonding contact point with DnaJ. Replacing it with alanine (no side chain beyond a methyl group) directly removes a potential DnaJ interaction site. Both ESM and lab confirm this is tolerated. Selected alongside P13L because the two mechanisms are complementary — P13L addresses backbone rigidity, S15A addresses the interaction surface.
Transmembrane Region Mutations (Positions 39–73)
Y39L — Position 39, Tyrosine → Leucine
- ESM Score: +2.24 | Lysis: Not yet tested
- Position 39 is the first residue of the transmembrane domain — the boundary point where the protein transitions from soluble to membrane-spanning. Tyrosine is large and polar (hydroxyl group), which is chemically unusual at the start of a hydrophobic TM helix. Leucine is hydrophobic and small, making for a cleaner, sharper TM helix start. The ESM model strongly favors this change, and it ranked 3rd globally across the entire protein. The only tested mutation at this position (Y39H) failed — but histidine is charged and polar, making it incomparable to leucine. Selected as the highest-confidence novel TM candidate.
A45P — Position 45, Alanine → Proline
- ESM Score: +0.04 | Lysis: Confirmed | Protein Level: Confirmed
- Introducing proline into a transmembrane helix creates a structural kink — a feature found in many natural pore-forming proteins and ion channels. This kink at position 45 (sitting centrally in the TM helix) may promote the conformational change needed to open the transmembrane pore. Supported by both the ESM model and direct experimental confirmation.
K50L — Position 50, Lysine → Leucine
- ESM Score: +2.56 (highest in entire protein) | Lysis: Not yet tested
- Lysine (K) is a charged, hydrophilic amino acid — unusual to find it buried deep in a hydrophobic transmembrane helix. The ESM model assigns the highest score in the entire protein to replacing it with leucine (hydrophobic), which is thermodynamically much more compatible with a membrane environment. This substitution could improve membrane insertion efficiency, increase protein expression, or stabilize the TM assembly. It is acknowledged that four other K50 variants (K50E, K50N, K50I, K50Q) have failed in the lab, suggesting this position may be sensitive. However, K50L is specifically a hydrophobic substitution — chemically distinct from the charged/polar variants that failed — and its extremely high ESM score justifies testing it as a novel candidate.
Final Mutations
| # | Mutation | Region | ESM Score | Lysis | Protein | Rationale |
|---|---|---|---|---|---|---|
| 1 | P13L | Soluble | +0.10 | ✅ | ✅ | Removes proline kink; enables DnaJ-independent folding |
| 2 | S15A | Soluble | +0.04 | ✅ | ✅ | Removes DnaJ contact site within NRRRP motif |
| 3 | Y39L | TM | +2.24 | ❓ | — | Sharpens TM helix entry; 3rd highest ESM score globally |
| 4 | A45P | TM | +0.04 | ✅ | ✅ | Proline kink promotes pore-forming conformation |
| 5 | K50L | TM | +2.56 | ❓ | — | Highest ESM score in protein; removes charged residue from TM core |
AI Prompt used in this section for mutation selection: Given the provided mutations, could you explain the rationale behind each and why would each serve as potentially candidates?