Lab
Weekly lab sessions:
- Introduction to lab safety and basic synthetic biology techniques.
- Gel Electrophoresis

- Opentrons and cloud laboratory tools for automated protocols.

- Chromophore Color Cloning Quest

- NeuroMorphic Wizard

- Collective Art

By the end of this lab, you will be able to:
A micropipette (commonly just called a “pipette” in the lab) is a precision instrument used to measure and transfer very small volumes of liquid — typically between 0.1 µL and 1000 µL (1 mL). Accurate pipetting is one of the most fundamental skills in biology, chemistry, and biomedical research. Even small errors in volume measurement can ruin an experiment, skew results, or waste expensive reagents.
Volumes in the lab are measured in:
| Unit | Abbreviation | Equivalent |
|---|---|---|
| Milliliter | mL | 1/1000 of a liter |
| Microliter | µL | 1/1000 of a mL = 1/1,000,000 of a liter |
| Nanoliter | nL | 1/1000 of a µL (specialized instruments only) |
💡 Quick reference: 1 mL = 1,000 µL. A typical raindrop is ~50 µL. A grain of salt is ~60 nL.
There are three standard micropipettes you will use in this course. Each is color-coded and designed for a specific volume range. Never exceed the maximum or go below the minimum volume — this damages the internal piston mechanism.
| Pipette Name | Common Color | Volume Range | Typical Use |
|---|---|---|---|
| P20 | Yellow | 0.5 µL – 20 µL | Small volumes: enzymes, DNA samples |
| P200 | Yellow | 20 µL – 200 µL | Medium volumes: PCR reactions, buffers |
| P1000 | Blue | 200 µL – 1000 µL | Large volumes: media, stock solutions |
⚠️ Rule: Always choose the pipette whose range most closely fits your target volume. Using a P1000 to measure 5 µL will be wildly inaccurate.
The volume display has three digits read from top to bottom. How you interpret those digits depends on which pipette you are using.
| Display | Volume |
|---|---|
| 2 / 0 / 0 | 20.0 µL (maximum) |
| 1 / 0 / 0 | 10.0 µL |
| 0 / 5 / 0 | 5.0 µL |
| 0 / 1 / 0 | 1.0 µL |
| Display | Volume |
|---|---|
| 2 / 0 / 0 | 200 µL (maximum) |
| 1 / 0 / 0 | 100 µL |
| 0 / 5 / 0 | 50 µL |
| 0 / 2 / 5 | 25 µL |
| Display | Volume |
|---|---|
| 1 / 0 / 0 | 1000 µL = 1.0 mL (maximum) |
| 0 / 5 / 0 | 500 µL = 0.5 mL |
| 0 / 2 / 5 | 250 µL |
| 0 / 2 / 0 | 200 µL (minimum) |
Turn the volume adjustment dial to your desired volume.
Press the tip cone firmly into a fresh pipette tip in the tip box. Give it a slight twist or firm push to create an airtight seal. Never touch the tip with your fingers after attachment — this contaminates your sample.
Before aspirating your actual sample, aspirate and expel the liquid 2–3 times to wet the inner walls of the tip. This reduces evaporation error, especially important for small volumes (<10 µL).
⚠️ Releasing too fast creates bubbles and inaccurate volumes. Speed matters!
Hold the tip up to the light. If you see a bubble, expel the liquid and re-aspirate. Bubbles displace liquid and reduce your actual volume.
Press the tip ejector button firmly over a waste container. Never remove used tips by hand — tips may be contaminated with biological or chemical material.
| Mistake | What Goes Wrong | How to Fix It |
|---|---|---|
| Releasing the plunger too fast | Creates air bubbles; aspirates incorrect volume | Always release slowly and steadily |
| Angling the pipette too far | Liquid runs back into the barrel, damaging the piston | Keep pipette vertical (±20°) |
| Inserting tip too deep | Liquid coats the outside of the tip and is carried over | Insert only 2–6 mm depending on pipette |
| Not pre-wetting the tip | First aspiration is inaccurate (surface tension effect) | Aspirate and expel 2–3 times before sampling |
| Pushing to second stop during aspiration | Aspirates too much volume / introduces air | Only push to first stop when aspirating |
| Reusing tips between samples | Cross-contamination of reagents | Change tip between every new sample |
| Setting volume outside the range | Inaccurate measurement; piston damage | Always choose the right pipette for the volume |
| Touching the tip with fingers | Introduces skin oils, DNA, and microbes | Handle only the pipette body; use tip boxes |
These two terms are distinct and both matter in pipetting:
A pipette can be precise but inaccurate if it is mis-calibrated. Always check calibration before critical experiments.
You can test your pipetting accuracy by weighing water. Since water has a density of 1.00 g/mL, 100 µL of water should weigh exactly 0.100 g.
| Trial | Expected Mass (g) | Measured Mass (g) | Error (g) | % Error |
|---|---|---|---|---|
| 1 | 0.100 | |||
| 2 | 0.100 | |||
| 3 | 0.100 | |||
| 4 | 0.100 | |||
| 5 | 0.100 | |||
| Mean | 0.100 |
% Error formula:
✅ Good pipetting: % error < 2% for P200 at 100 µL
⚠️ Acceptable: % error 2–5%
❌ Needs improvement: % error > 5%
Image 1 (Mid-run photograph): The photograph taken during electrophoresis shows the gel submerged in TAE within the gel box. Two colored dye fronts are faintly visible — a blue band and a dark purple band — but they appear localized to only one or two lanes. The majority of the gel appears empty, with no visible dye migration in the other wells. This is already an early indicator that most wells were either not loaded successfully or contained insufficient DNA.
Image 2 (GeneSnap image): The final imaging result is largely dark. Only a single lane shows any detectable fluorescence — a faint, somewhat smeared signal concentrated in what appears to be one lane, with no clearly resolved discrete bands. The remaining lanes are entirely blank. This represents an unsuccessful gel run in terms of the intended gel art pattern.
Analysis of What Went Wrong Based on the observations made during lab sessions and the photographic evidence, several compounding factors likely contributed to the result:
This figure represents the end-to-end automated pipeline: from DNA library preparation through robotic CFPS assembly, incubation, fluorescence readout, and computational analysis.
Each row represents a unique biosensor construct variant; each column a different inducer concentration. Construct C5 shows the steepest dose-response (ideal switch-like behavior) with minimal background — identified as the top hit for downstream validation.
References: Hossain et al. (2020) ACS Synth. Biol.; Pardee et al. (2014) Cell; Opentrons Protocol API v2 Documentation.
In this experiment we engineer color variants of the purple Acropora millepora chromoprotein (amilCP) by introducing targeted mutations at the chromophore (CP) site: cagTGTCAGtac. Substituting the TGTCAG hexamer with variant codons shifts the expressed color to orange, pink, magenta, or blue, as described by Liljeruhm et al. (2018).
Part 1 covers the preparation of two PCR fragments — a Backbone fragment and a Color insert fragment — which will be joined by Gibson Assembly and transformed into E. coli in Part 2.
Progress: PCR Setup → Thermal Cycling → DpnI Digest → Purification → Gel Electrophoresis ✓ → Gibson Assembly) → Transformation)
Time estimate: ~1.5 hours total
Two parallel PCR reactions were prepared on ice using the mUAV plasmid as template. The Backbone reaction amplifies the vector (ori + CmR + promoter + RBS), while the Color reaction amplifies the chromophore region with a mutant forward primer that introduces the desired codon substitution at the CP site.

Backbone DNA Fragment (Primers: Backbone Fwd + Backbone Rev)
| Reagent | Stock Conc. | Desired Conc. | Volume (µL) |
|---|---|---|---|
| Template mUAV Plasmid | 38.5 ng/µL | 20 ng/µL | 0.8 |
| Backbone Forward Primer | 5 µM | 0.5 µM | 2.5 |
| Backbone Reverse Primer | 5 µM | 0.5 µM | 2.5 |
| Phusion HF PCR Mix | 2× | 1× | 12.5 |
| Nuclease-free water | — | — | 6.8 |
| Total Volume | 25.0 |
Color DNA Fragment (Primers: Color Fwd + Color Rev)
| Reagent | Stock Conc. | Desired Conc. | Volume (µL) |
|---|---|---|---|
| Template mUAV Plasmid | 38.5 ng/µL | 20 ng/µL | 0.8 |
| Color Forward Primer | 5 µM | 0.5 µM | 2.5 |
| Color Reverse Primer | 5 µM | 0.5 µM | 2.5 |
| Phusion HF PCR Mix | 2× | 1× | 12.5 |
| Nuclease-free water | — | — | 6.8 |
| Total Volume | 25.0 |
Backbone Fragment (BB_PCR) — run on Bio-Rad T100, 25 µL volume
Color Insert Fragment


BB_PCR program (57°C anneal, 26 cycles, 25 µL volume).The Color forward primer carries an intentional mismatch in the 6-bp chromophore region (e.g. TGTCAG → GTTGGA for orange). Because the mismatch sits in the 5′ overhang, Phusion polymerase still extends efficiently from the matched 3′ binding region. The mutation is thus incorporated into every PCR copy and all downstream clones.
⏱ Time estimate: 45 min at 37°C
After PCR, 1 µL of DpnI was added directly to each 25 µL reaction and incubated at 37°C for 30–60 minutes. DpnI recognises methylated 5′-Gm6ATC-3′ sequences present on E. coli-propagated plasmid template, but absent from unmethylated PCR products. The enzyme therefore selectively digests the parental template while leaving new amplicons intact.
Residual un-digested template will generate wildtype (purple) background colonies that compete with and obscure your color-mutant transformants.
⏱ Time estimate: 30 min
PCR products were purified using the Zymo DNA Clean & Concentrator kit (silica-column adsorption) to remove primers, dNTPs, polymerase, and buffer salts before Gibson Assembly.

⏱ Time estimate: ~15 min at 100 V
Purified fragments were run on a 1% agarose E-Gel EX (Invitrogen) to confirm fragment sizes. Each lane received 3 µL sample + 3.3 µL 6× Loading Dye. DNA ladder loaded in lane M (leftmost).

| Lane | Observation | Interpretation |
|---|---|---|
| M | Ladder bands across full range | Reference marker |
| 1 | Faint band ~400–500 bp | Likely primer-dimer or low-yield non-specific product |
| 2 | Faint band, similar to lane 1 | Same as above; low amplification |
| 3 | Bright band ~600–750 bp | Color insert fragment (~700 bp) — strong, clean yield |
| 4 | Faint lower band | Minor non-specific; likely negligible for downstream steps |
| 5 | Bright band ~2.7–2.9 kb | Backbone fragment (~2800 bp) — strong, clean yield |
| 6–10 | Empty | — |
Expected fragment sizes:
Lanes 3 and 5 show bright, clean bands at the expected sizes for Color insert and Backbone respectively. Faint bands in lanes 1, 2, and 4 represent minor non-specific products that will be diluted out during Gibson Assembly and will not affect the outcome. Both fragments are confirmed — proceed to Gibson Assembly.
⏱ Incubation: 72 hours at 37°C | Selection: LB-Agar + Chloramphenicol 25 µg/mL








All colonies across every plate — regardless of intended color variant (blue, pink, light pink) — express a uniform blue-purple color consistent with wildtype amilCP. The intended color shifts to pink or blue did not appear. One notable exception is the red-circled colony in the final plate (Fig. 5h), which is transparent/colorless.
Gibson Assembly outcome is highly sensitive to the molar ratio of insert to backbone, not just the volumes used. The protocol specifies 0.5 µL backbone and 1.0 µL insert — but those volumes assume both fragments are at exactly the stated concentrations after purification.
When backbone is in excess, the probability of the two backbone ends annealing to each other increases sharply — rather than each end finding the insert:
Because the ratio imbalance originates in the Gibson reaction before transformation, it would affect all three volume groups (2µL, 4µL, 7µL) equally — explaining the consistency of the wildtype purple outcome across all plates.
Partially succeeded, as this is consistent with a scenario where the backbone reassembled without the color insert — the Gibson exonuclease chewed back both ends of the backbone, they annealed to each other rather than to the insert, and ligase sealed the nick. The result is a backbone-only plasmid that carries CmR but lacks the amilCP CDS entirely, hence no color.
Alternatively, the insert was incorporated but with a frameshift or premature stop codon introduced during the Gibson join, knocking out chromoprotein expression without replacing it with a new color.
Either way, this colony is evidence that the Gibson Assembly chemistry was active and processing DNA correctly. The colorless result is not a failure — it is a partial success where the backbone was modified but the color swap did not complete as intended.
the wildtype blue-purple across all other colonies most likely reflects surviving template from incomplete DpnI digestion, while the single transparent colony shows that at least one genuine assembly event occurred.
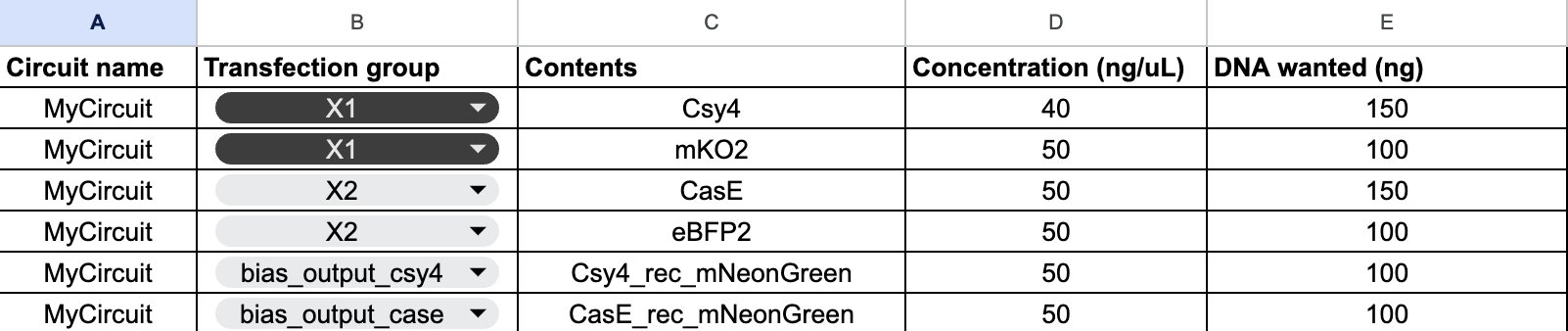
For the neuromorphic circuit, our group aimed to design a “L” shaped heatmap. We added two bias corresponding to X1 and X2 ERNs.
Looked perfect
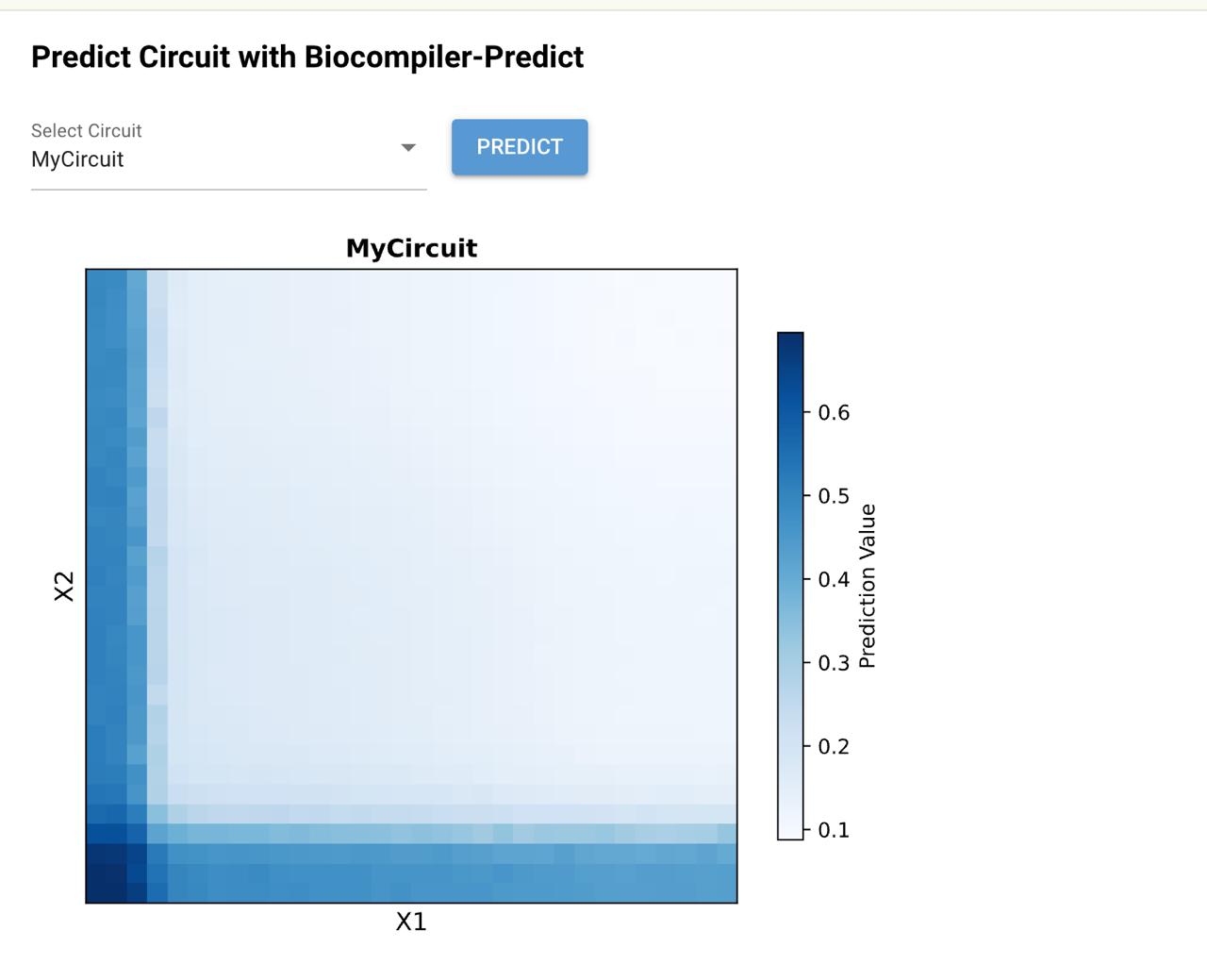
I think we might’ve submitted the wrong file ahahaha, so the final output only displayed the bottom part of the “L”
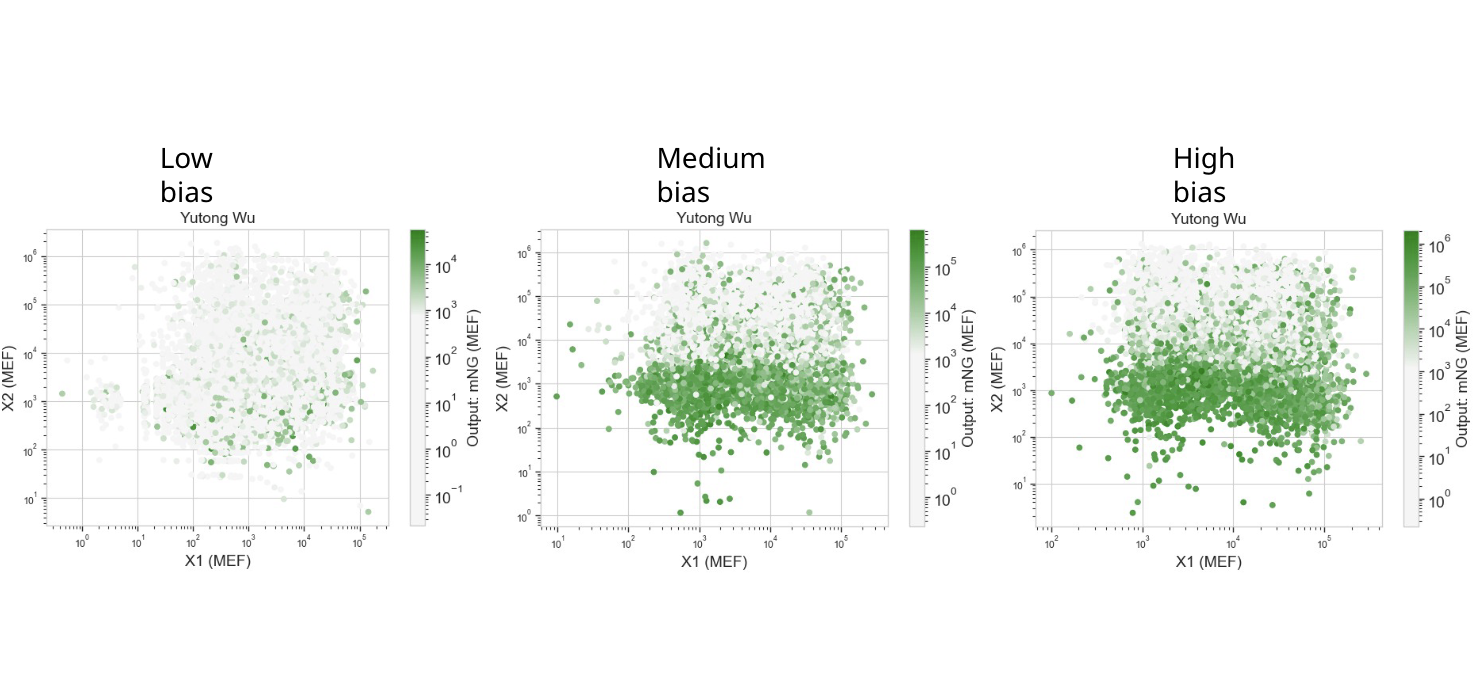
Each dot in these scatterplots represents a single human cell. The color shows the level of output (mNeonGreen) as a function of X1 and X2 and, optionally, varying levels of bias.
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
sfGFP: primary advantage is robust folding kinetics; it is engineered to fold correctly even when fused to insoluble proteins, making it highly resistant to aggregation in the crowded environment of a cell-free extract.
mRFP1: characterized by slow maturation kinetics and a tendency for photobleaching; the delay between peptide synthesis and chromophore formation can lead to an underestimation of protein yield in short-term reactions.
mKO2: features fast maturation and oxygen dependence; while it reaches peak fluorescence quickly, the final oxidative step of chromophore formation requires sufficient O2 levels, which may become limiting in deep-well plates.
mTurquoise2: known for high quantum yield and acid stability; its low pKa makes it less sensitive to the $pH$ drops that naturally occur as metabolic byproducts (like organic acids) accumulate during long-term cell-free incubation.
mScarlet_I: a high-brightness variant with accelerated maturation compared to earlier red FPs; however, it remains sensitive to the oxidative environment, as oxygen is required to complete the cyclization of its chromophore.
Electra2: optimized for ultra-fast maturation; its rapid “time-to-bright” makes it the ideal candidate for real-time monitoring of transcription-translation (TX-TL) kinetics where immediate feedback is required.
Protein: mScarlet_I
Reagent Adjustment: Increase Glucose and Nicotinamide concentrations while utilizing a semi-permeable reaction seal.
Expected Effect:In a 36-hour run, the primary bottleneck for a bright red FP like mScarlet_I is the depletion of energy and the requirement for oxygen for chromophore maturation. By increasing Glucose and Nicotinamide, we extend the metabolic “runway” for $ATP$ regeneration via the NMP-Ribose-Glucose pathway; combining this with a semi-permeable seal ensures a constant influx of O2 to drive the oxidative maturation of the chromophore, thereby maximizing the total fluorescent signal over the extended incubation period.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by 4/24). Make sure that your final project slide is in the slide deck below to be included!
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (TBD!).