Week 6 Lab: Gibson Assembly
Week 6 Lab: Gibson Assembly Lab
Overview
In this experiment we engineer color variants of the purple Acropora millepora chromoprotein (amilCP) by introducing targeted mutations at the chromophore (CP) site: cagTGTCAGtac. Substituting the TGTCAG hexamer with variant codons shifts the expressed color to orange, pink, magenta, or blue, as described by Liljeruhm et al. (2018).
Part 1 covers the preparation of two PCR fragments — a Backbone fragment and a Color insert fragment — which will be joined by Gibson Assembly and transformed into E. coli in Part 2.
Progress: PCR Setup → Thermal Cycling → DpnI Digest → Purification → Gel Electrophoresis ✓ → Gibson Assembly) → Transformation)
Part 1 — PCR Reaction Setup
Time estimate: ~1.5 hours total
Two parallel PCR reactions were prepared on ice using the mUAV plasmid as template. The Backbone reaction amplifies the vector (ori + CmR + promoter + RBS), while the Color reaction amplifies the chromophore region with a mutant forward primer that introduces the desired codon substitution at the CP site.
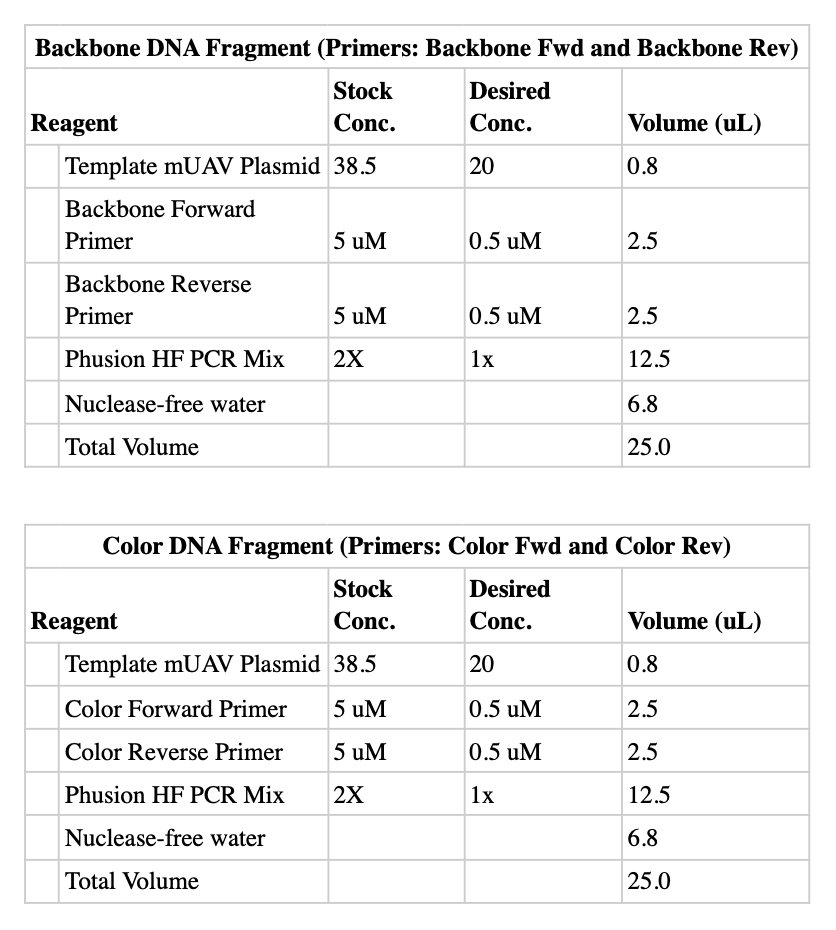
Reagent Tables
Backbone DNA Fragment (Primers: Backbone Fwd + Backbone Rev)
| Reagent | Stock Conc. | Desired Conc. | Volume (µL) |
|---|---|---|---|
| Template mUAV Plasmid | 38.5 ng/µL | 20 ng/µL | 0.8 |
| Backbone Forward Primer | 5 µM | 0.5 µM | 2.5 |
| Backbone Reverse Primer | 5 µM | 0.5 µM | 2.5 |
| Phusion HF PCR Mix | 2× | 1× | 12.5 |
| Nuclease-free water | — | — | 6.8 |
| Total Volume | 25.0 |
Color DNA Fragment (Primers: Color Fwd + Color Rev)
| Reagent | Stock Conc. | Desired Conc. | Volume (µL) |
|---|---|---|---|
| Template mUAV Plasmid | 38.5 ng/µL | 20 ng/µL | 0.8 |
| Color Forward Primer | 5 µM | 0.5 µM | 2.5 |
| Color Reverse Primer | 5 µM | 0.5 µM | 2.5 |
| Phusion HF PCR Mix | 2× | 1× | 12.5 |
| Nuclease-free water | — | — | 6.8 |
| Total Volume | 25.0 |
Thermocycler Programs
Backbone Fragment (BB_PCR) — run on Bio-Rad T100, 25 µL volume
Color Insert Fragment


BB_PCR program (57°C anneal, 26 cycles, 25 µL volume).The Color forward primer carries an intentional mismatch in the 6-bp chromophore region (e.g. TGTCAG → GTTGGA for orange). Because the mismatch sits in the 5′ overhang, Phusion polymerase still extends efficiently from the matched 3′ binding region. The mutation is thus incorporated into every PCR copy and all downstream clones.
Part 1a — DpnI Digest
⏱ Time estimate: 45 min at 37°C
After PCR, 1 µL of DpnI was added directly to each 25 µL reaction and incubated at 37°C for 30–60 minutes. DpnI recognises methylated 5′-Gm6ATC-3′ sequences present on E. coli-propagated plasmid template, but absent from unmethylated PCR products. The enzyme therefore selectively digests the parental template while leaving new amplicons intact.
Residual un-digested template will generate wildtype (purple) background colonies that compete with and obscure your color-mutant transformants.
Part 1b — DNA Purification & Quantification
⏱ Time estimate: 30 min
PCR products were purified using the Zymo DNA Clean & Concentrator kit (silica-column adsorption) to remove primers, dNTPs, polymerase, and buffer salts before Gibson Assembly.
Equipment & Consumables
- Zymo DNA Clean & Concentrator kit (columns + buffers)
- Eppendorf Centrifuge 5415C (set to 13,000 rpm, ≈ 17,900 × g)
- 1.5 mL microcentrifuge tubes
- 50 mL Falcon tube (liquid waste)
- Nanodrop or Qubit spectrophotometer
- P20 and P200 pipettes with tips
- Nuclease-free water
Procedure
- Add 50 µL PCR product + 250 µL DNA Binding Buffer to a 1.5 mL tube. Vortex briefly.
- Transfer all 300 µL to a Zymo-Spin Column seated in a Collection Tube. Centrifuge 1 min at 13,000 rpm. Discard flow-through; keep the collection tube.
- Add 200 µL Wash Buffer. Centrifuge 1 min. Discard flow-through. Repeat once (2 washes total). Transfer column to a fresh 1.5 mL tube; discard the collection tube.
- Add 6 µL nuclease-free water directly to the column membrane. Rest at room temperature for 2 min. Centrifuge 1 min. Collect and save the elution.
- Measure concentration on Nanodrop: 2 µL per read. Target ≥ 30 ng/µL, A260/A280 ≈ 1.8–2.0.

Part 1c — Diagnostic Gel Electrophoresis
⏱ Time estimate: ~15 min at 100 V
Purified fragments were run on a 1% agarose E-Gel EX (Invitrogen) to confirm fragment sizes. Each lane received 3 µL sample + 3.3 µL 6× Loading Dye. DNA ladder loaded in lane M (leftmost).
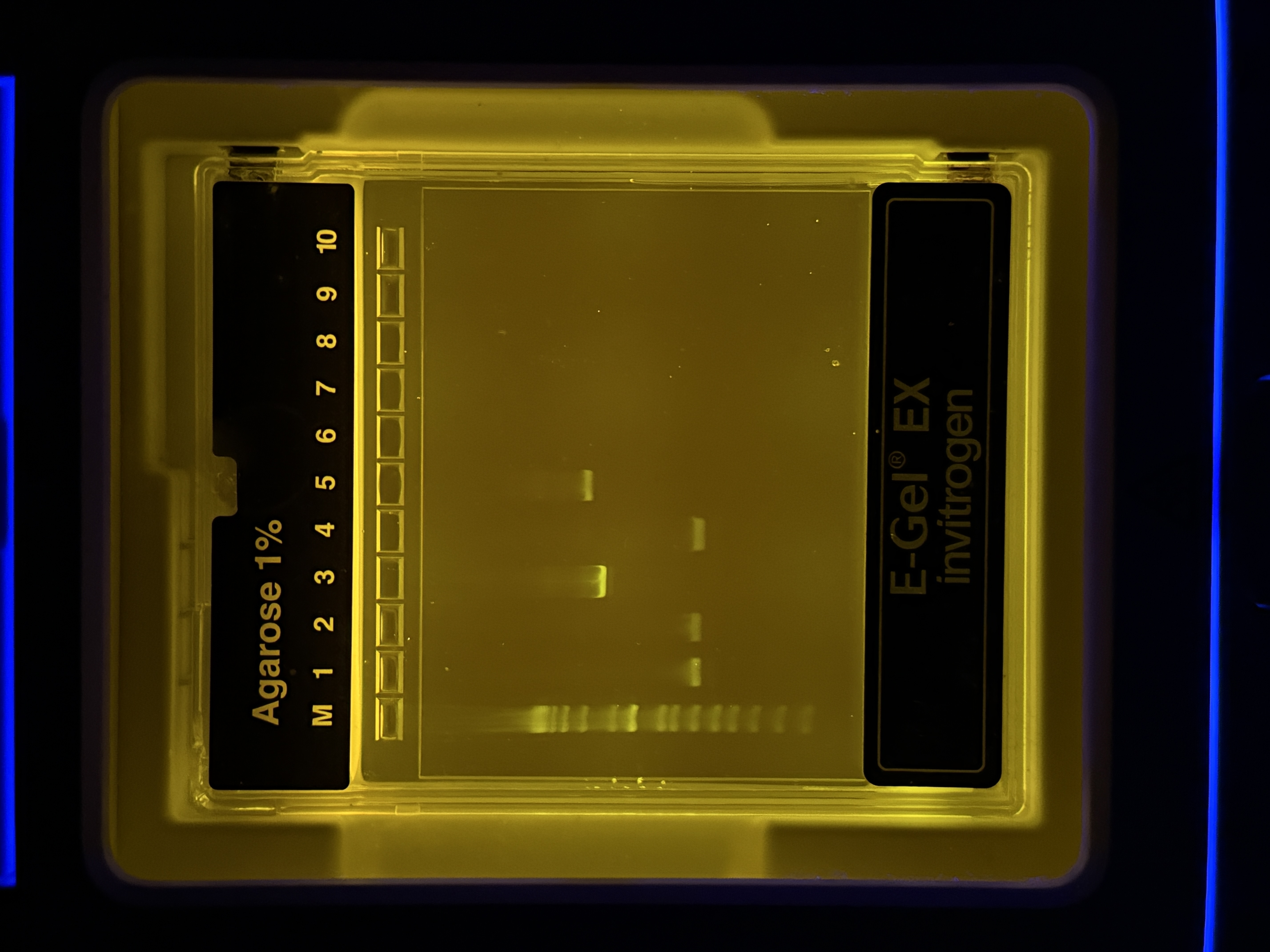
Band Interpretation
| Lane | Observation | Interpretation |
|---|---|---|
| M | Ladder bands across full range | Reference marker |
| 1 | Faint band ~400–500 bp | Likely primer-dimer or low-yield non-specific product |
| 2 | Faint band, similar to lane 1 | Same as above; low amplification |
| 3 | Bright band ~600–750 bp | Color insert fragment (~700 bp) — strong, clean yield |
| 4 | Faint lower band | Minor non-specific; likely negligible for downstream steps |
| 5 | Bright band ~2.7–2.9 kb | Backbone fragment (~2800 bp) — strong, clean yield |
| 6–10 | Empty | — |
Expected fragment sizes:
- Backbone: ~2800 bp (ori + CmR + promoter + RBS)
- Color insert: ~700 bp (24 bp upstream of CP site + chromophore + terminator)
Lanes 3 and 5 show bright, clean bands at the expected sizes for Color insert and Backbone respectively. Faint bands in lanes 1, 2, and 4 represent minor non-specific products that will be diluted out during Gibson Assembly and will not affect the outcome. Both fragments are confirmed — proceed to Gibson Assembly.
Part 2 — Transformation Results & Analysis
⏱ Incubation: 72 hours at 37°C | Selection: LB-Agar + Chloramphenicol 25 µg/mL
Colony Plates
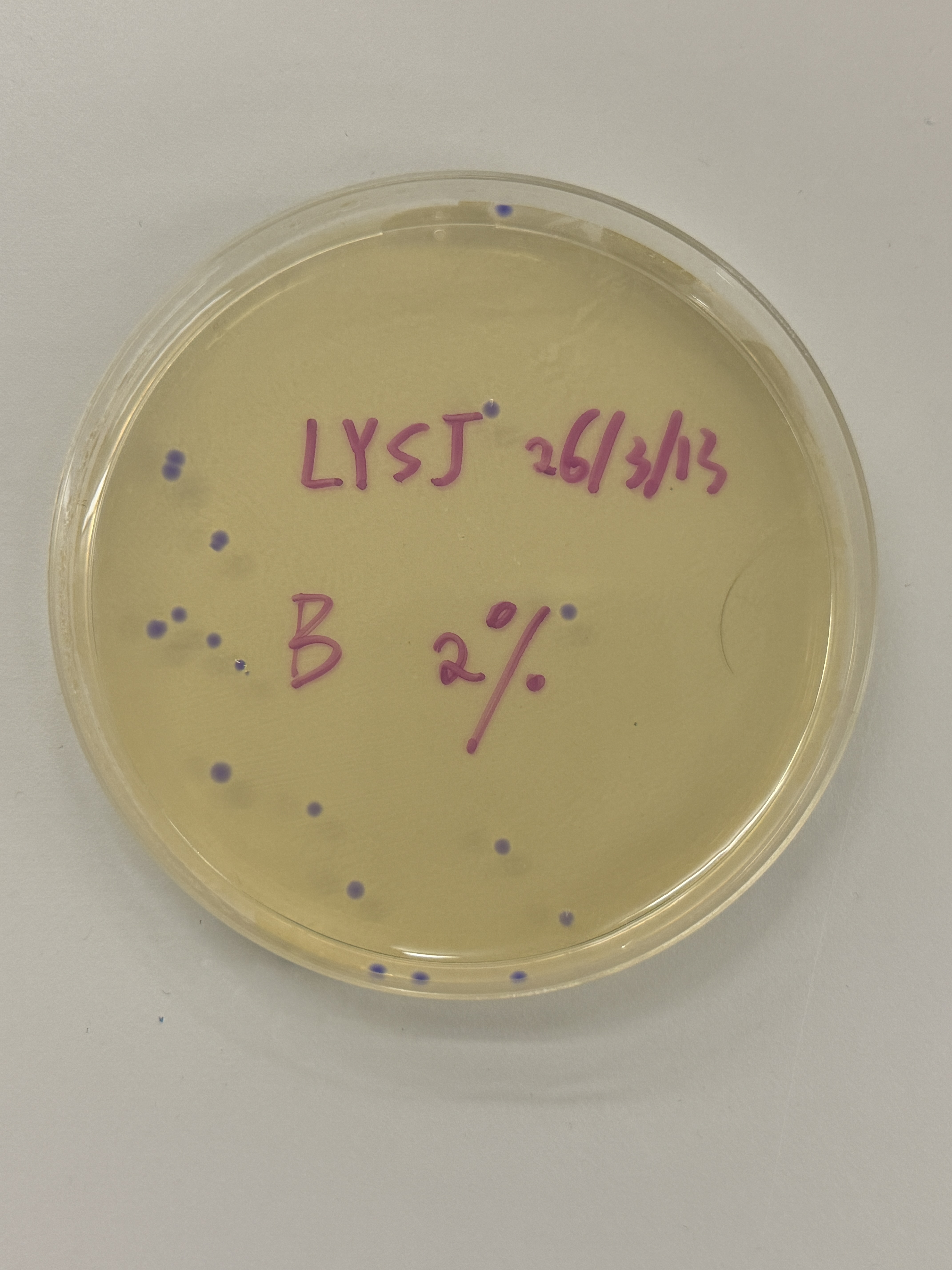
~15 colonies

~2 colonies
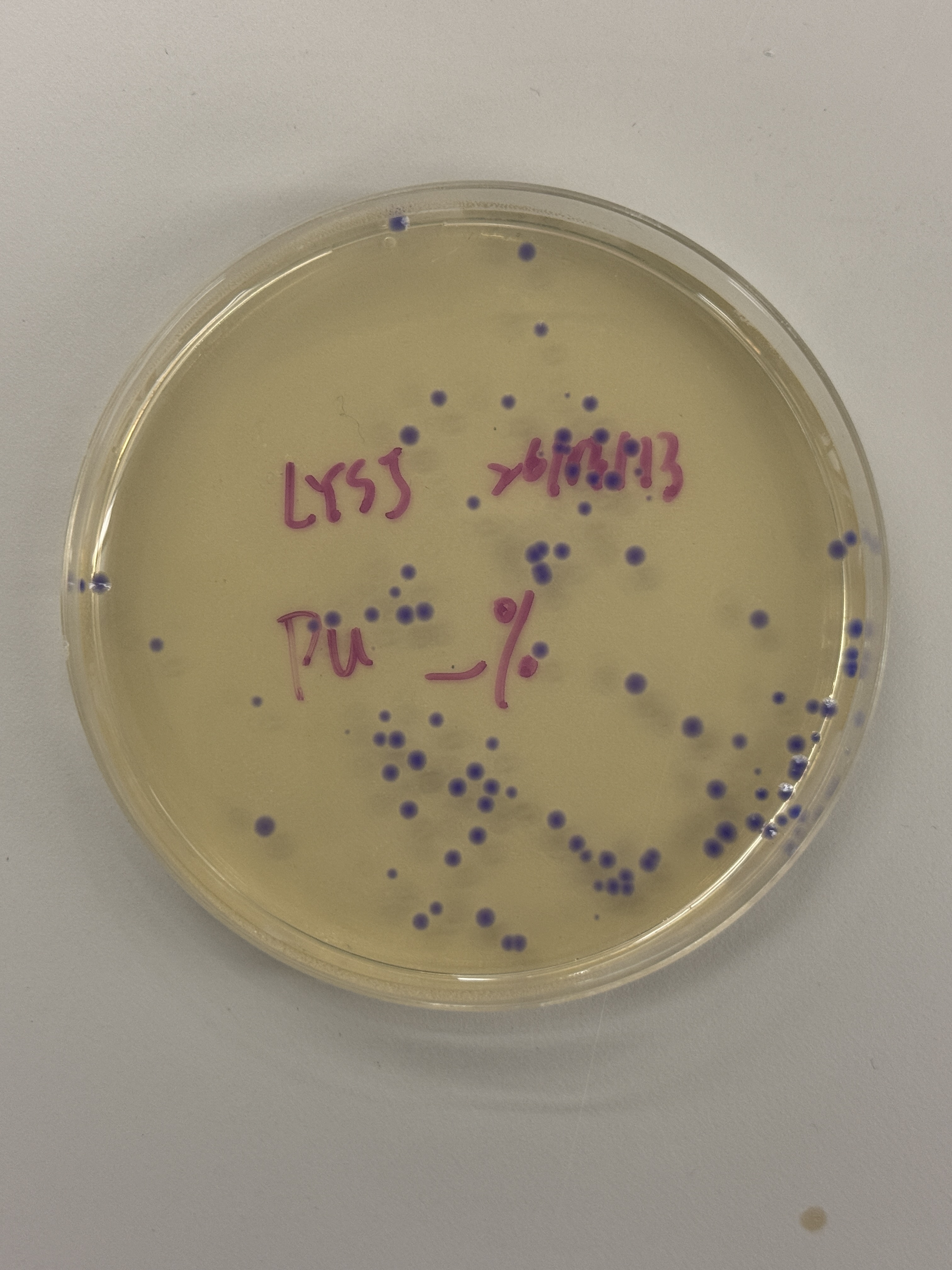
~100+ colonies
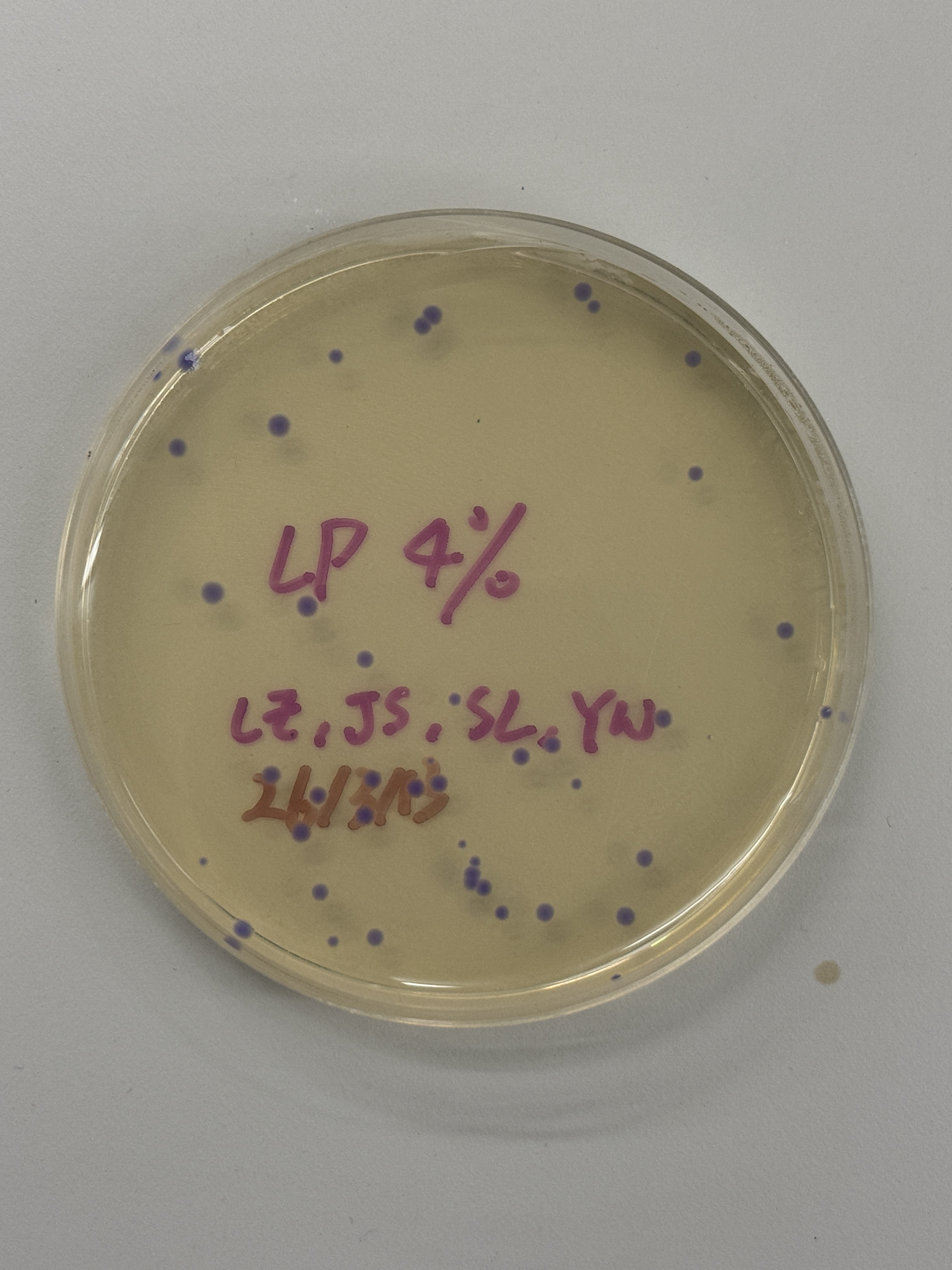
~40–50 colonies
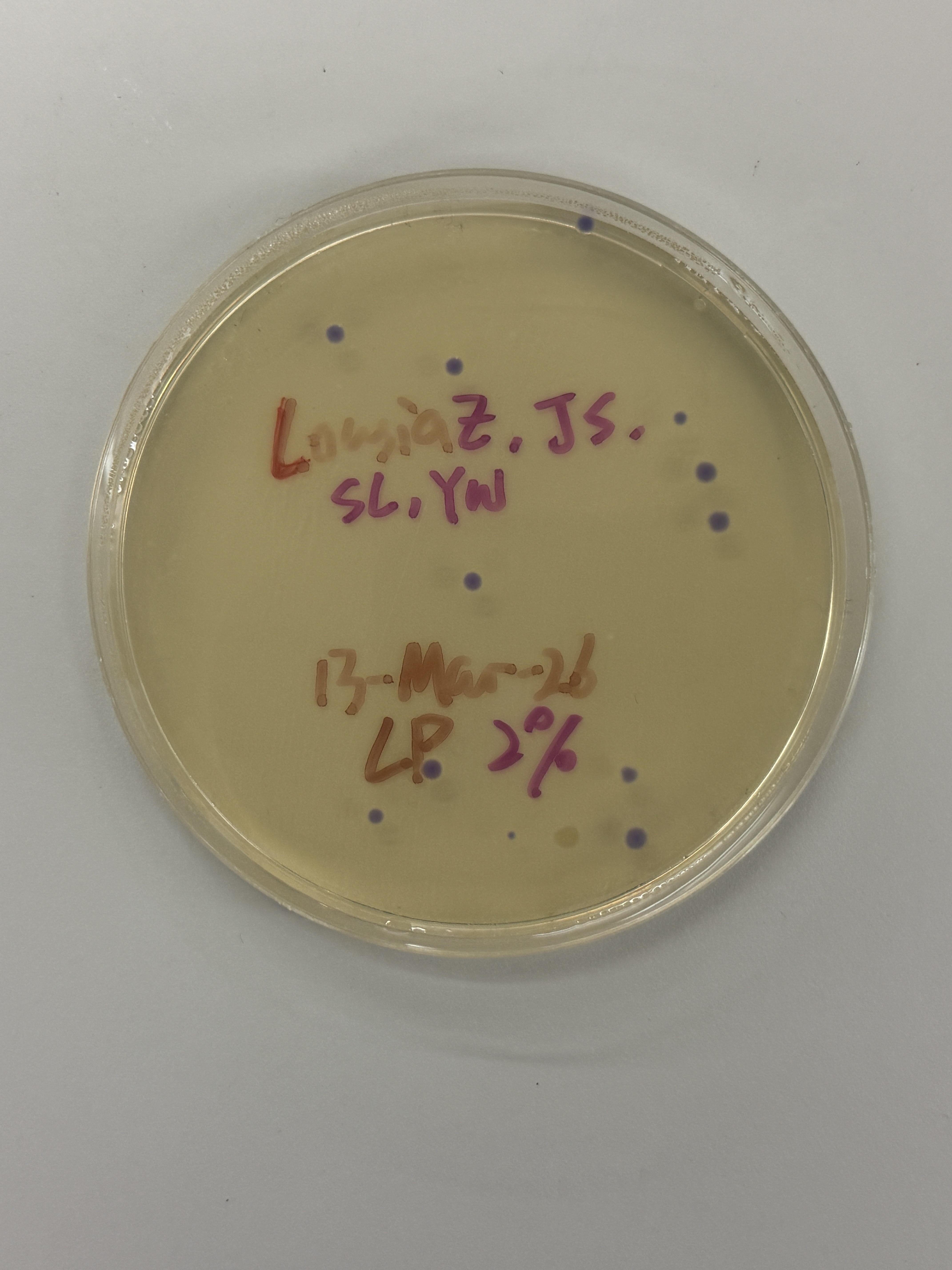
~15–20 colonies
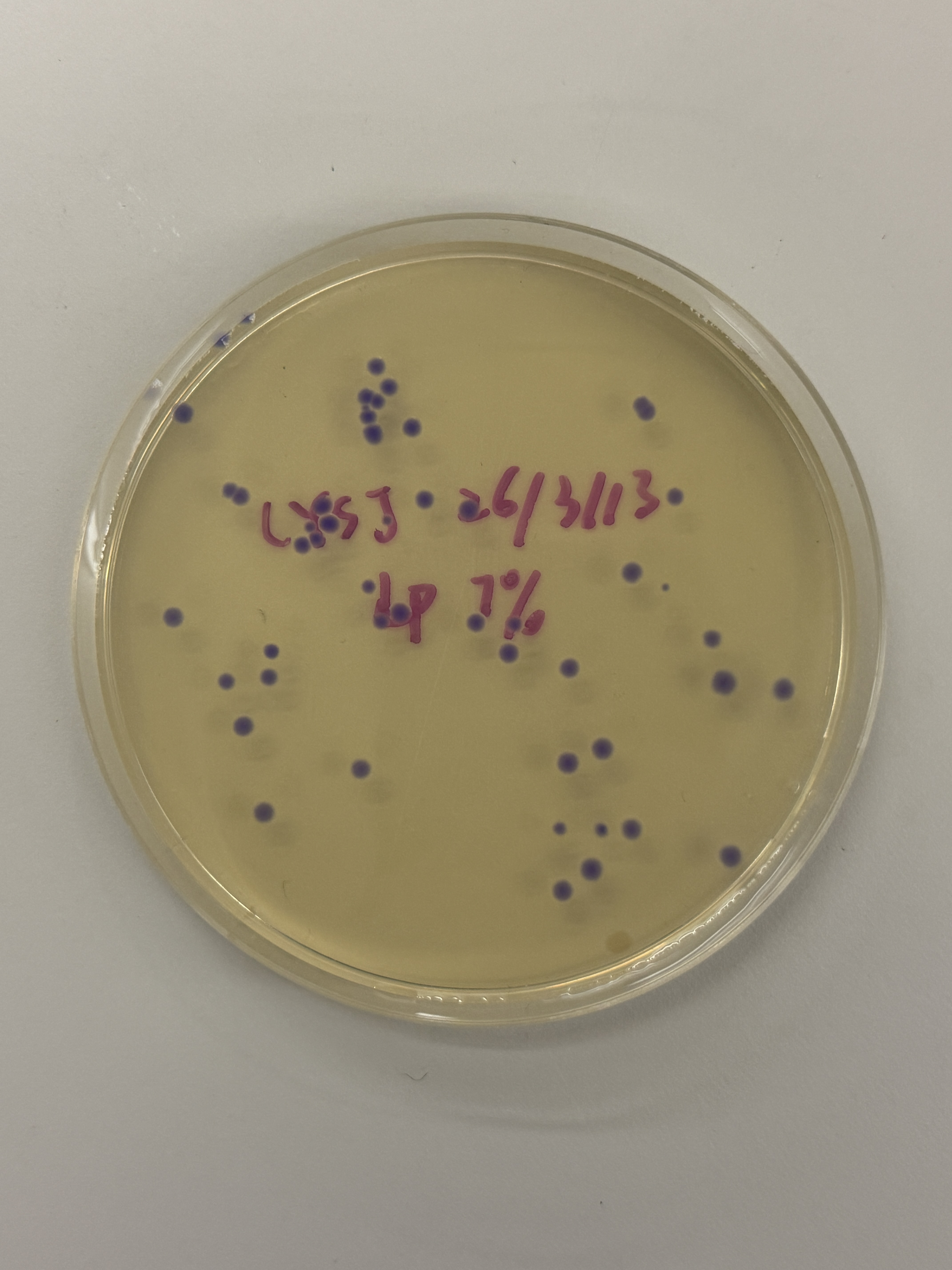
~40–50 colonies

~8–10 colonies

⭕ transparent colony
Observation
All colonies across every plate — regardless of intended color variant (blue, pink, light pink) — express a uniform blue-purple color consistent with wildtype amilCP. The intended color shifts to pink or blue did not appear. One notable exception is the red-circled colony in the final plate (Fig. 5h), which is transparent/colorless.
Analysis
Imbalanced Insert:Backbone Molar Ratio
Gibson Assembly outcome is highly sensitive to the molar ratio of insert to backbone, not just the volumes used. The protocol specifies 0.5 µL backbone and 1.0 µL insert — but those volumes assume both fragments are at exactly the stated concentrations after purification.
When backbone is in excess, the probability of the two backbone ends annealing to each other increases sharply — rather than each end finding the insert:
Because the ratio imbalance originates in the Gibson reaction before transformation, it would affect all three volume groups (2µL, 4µL, 7µL) equally — explaining the consistency of the wildtype purple outcome across all plates.
transparent Colony
Partially succeeded, as this is consistent with a scenario where the backbone reassembled without the color insert — the Gibson exonuclease chewed back both ends of the backbone, they annealed to each other rather than to the insert, and ligase sealed the nick. The result is a backbone-only plasmid that carries CmR but lacks the amilCP CDS entirely, hence no color.
Alternatively, the insert was incorporated but with a frameshift or premature stop codon introduced during the Gibson join, knocking out chromoprotein expression without replacing it with a new color.
Either way, this colony is evidence that the Gibson Assembly chemistry was active and processing DNA correctly. The colorless result is not a failure — it is a partial success where the backbone was modified but the color swap did not complete as intended.
the wildtype blue-purple across all other colonies most likely reflects surviving template from incomplete DpnI digestion, while the single transparent colony shows that at least one genuine assembly event occurred.