Week 4 HW: Protein Design I
Week 04 - Protein Design I
Part A
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
6.6 x 1023 molecules of amino acids
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestive system breaks down food components (proteins, fats, and carbohydrates) into their constituent building blocks. For example, proteins are digested into amino acids, which the body then uses to build and maintain its own cells and tissues. We do not directly assimilate cow proteins or cells when eating meat, only the nutrients and molecular components derived from them.
Why are there only 20 natural amino acids?
Technically, more amino acids could exist, but evolution settled on a core set of approximately 20 very early in the history of life. These amino acids became the biological standard because their chemical diversity is sufficient to support the structural and functional complexity required for life.
Where did amino acids come from before enzymes that make them, and before life started?
Before life as we know it started on Earth, amino acids were likely produced through primordial geochemical and extraterrestrial processes. Amino acids and their chemical precursors are commonly detected in meteorites, hydrothermal vent systems, and other geochemically active environments. In the absence of living organisms to metabolize or degrade them, geochemically synthesized amino acids (being relatively stable molecules) could have accumulated over time.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left handed. Biology uses the L-amino acids but these create right handed α-helixes.
Can you discover additional helices in proteins?
Yes, some uncommon helices are only found during molecular binding and not shown in x-ray crystallography
Why are most molecular helices right-handed?
They are more thermodynamically and energetically stable
Why do β-sheets tend to aggregate?
Hydrogen bonding occurs between the ends of the sheets allowing the sheets attach to each other and stack
What is the driving force for β-sheet aggregation?
Hydrophobic effects
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
My protein is the peptide Dermaseptin s4. Dermaseptin S4 is an antimicrobial peptide originally isolated from the skin of the South American leaf frog (Phyllomedusa sauvagii). It is highly regarded in scientific research for its ability to disrupt microbial and fungal cell membranes. I chose this peptide because it will prove to be useful in my project, as I will attempt to use this protein to disrupt the cell membranes of yeast after secretion by a bacteria. This will act as my proof-of-concept model for my project to show the antifungal activity of amphibian skin peptides, which can then later be used on Batrachochytrium dendrobatidis as a potential probiotic to treat the disease.
Identify the amino acid sequence of your protein.
ALWMTLLKKVLKAAAKAALNAVLVGANA
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It is 28 amino acids long, the most frequent amino acid is Alanine (9 times).
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
9 homologs were found.
Does your protein belong to any protein family?
It belongs to the Frog Skin Active Peptide (FSAP) family and is further classified into the Dermaseptin subfamily.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The full protein structure of Dermaseptin S4 has not been solved currently, only partial truncated synthetic derivatives have been solved using NMR. Only the first 14 amino acids of the sequence of 28 have been solved, discovered in 2006. It is an exceptionally high scoring structure in terms of quality derived from NMR structures.
Are there any other molecules in the solved structure apart from protein?
No, all amino acids in the peptide chain.
Does your protein belong to any structure classification family?
Amphipathic alpha-helical antimicrobial peptides family
Open the structure of your protein in any 3D molecule visualization software: PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands) Visualize the protein as “cartoon”, “ribbon” and “ball and stick”. Color the protein by secondary structure. Does it have more helices or sheets?
It only has helices, contains no sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? The hydrophobic and hydrophilic residues are split perfectly into two halves. It forms a class-L amphipathic alpha-helix. Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
It has no binding pockets, as its structure is more of a single uninterrupted alpha helix.
Part C
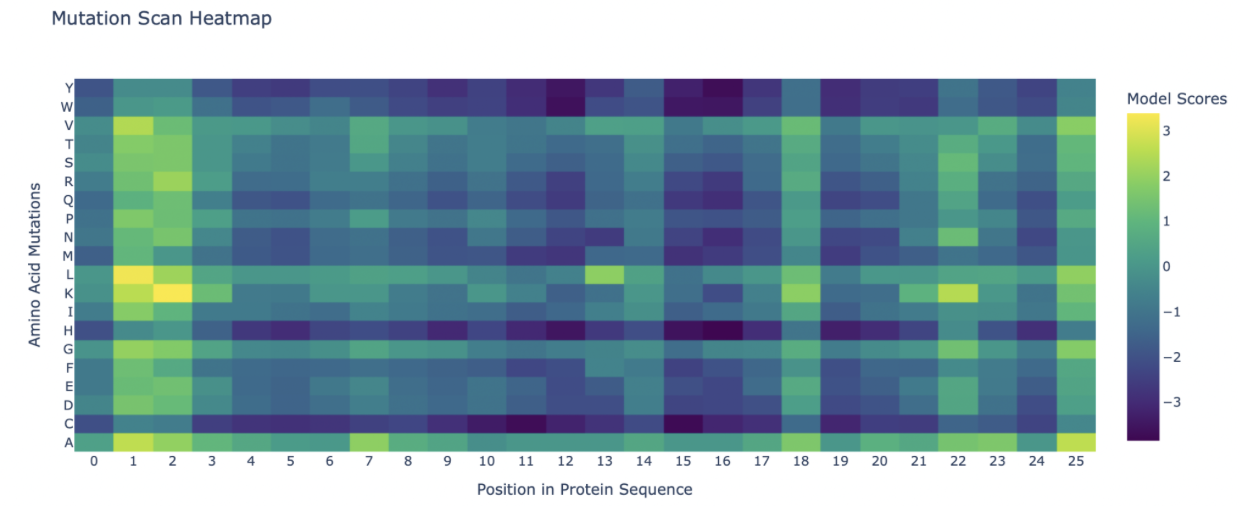
Deep Mutational Scans Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Attached is the mutation scan, I noticed that position 1 and 2 have extremely positive LLR values, indicating mutations that are beneficial to the function of Dermaseptin s4.
Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins?
Couldn’t figure out a way to visualize the latent space analysis correctly. The downloaded file from the web seems to be a broken link, or I have a faulty setting on my end. Place your protein in the resulting map and explain its position and similarity to its neighbors.
Part D
Goal 1: Stabilize MS2 L so it remains correctly folded and membrane-competent under a wider range of conditions (temperature, expression levels), which should support more robust lysis and higher effective titers.
Goal 2: Increase intrinsic toxicity by promoting oligomerization and pore formation of L in the bacterial envelope.
We will focus on improving both the stability and toxicity of the MS2 L lysis protein using a purely computational design-and-screen pipeline before any wet-lab work.
Our first goal is increased stability. We want MS2 L to remain correctly folded and membrane-competent across a broad range of expression and environmental conditions so that lysis is more reliable and supports higher effective titers.
Our second goal is increased toxicity. We aim to increase the intrinsic lytic potency of L by promoting oligomerization and pore formation in the bacterial envelope.
To do this, we will use protein language models (such as ESM or ProtT5) as in silico mutagenesis tools, scoring single and selected double mutants of L and ranking substitutions predicted to improve stability or fitness.
We will combine this with structure prediction and modeling. AlphaFold will be used on full-length and truncated L variants to generate consistent 3D models of the soluble and transmembrane regions, while AlphaFold-Multimer or docking tools will help model L oligomers in a membrane-like context.
Membrane-aware stability analysis will further guide our designs. TMHMM-like predictors will define transmembrane boundaries and disorder regions, while Rosetta- or FoldX-style ΔΔG calculations will help refine promising mutation sites, especially around the LS motif and transmembrane helix.
We will also incorporate coevolution and functional hotspot information by overlaying published mutagenesis data for L. Residues already known to be essential for function will be locked and excluded from substitution.
These approaches are important because mutational studies show that even conservative substitutions near the LS motif can abolish function. We need a guided way to search sequence space while respecting both evolutionary constraints and protein stability.
Protein language models provide a fast way to scan mutation space, while ΔΔG calculations and structure predictions help eliminate variants that appear incompatible with proper helix packing or overall folding, leaving a smaller set of promising stabilizing mutations.
For increased toxicity, MS2 L activity depends on efficient membrane insertion and higher-order oligomer formation through its C-terminal transmembrane segment. Modeling and optimizing helix–helix interactions should therefore influence pore stability and lytic potency.
By favoring small or helix-packing residues at predicted contact sites, while avoiding unfavorable charges in the membrane, we may be able to design variants that form more stable oligomers without disrupting the LS motif or essential hydrophobic patterning.
In single-gene lysis systems, lysis timing and efficiency emerge from a balance of expression level, folding, and toxicity. Improving stability may therefore reduce misfolding and allow higher levels of functional L protein, ultimately improving titers under comparable induction conditions.
Our overall objective is to computationally design MS2 L variants that combine improved membrane stability with increased intrinsic toxicity.