Hi, I’m Joe - welcome to my page! I am a Canadian-American currently working in the lab extracting DNA for a biotech company in Chicago. I am very interested in the applications of bioengineering and gene editing for biodiversity conservation and ecosystem preservation. In my free time, I enjoy birdwatching, hiking, and playing water polo.
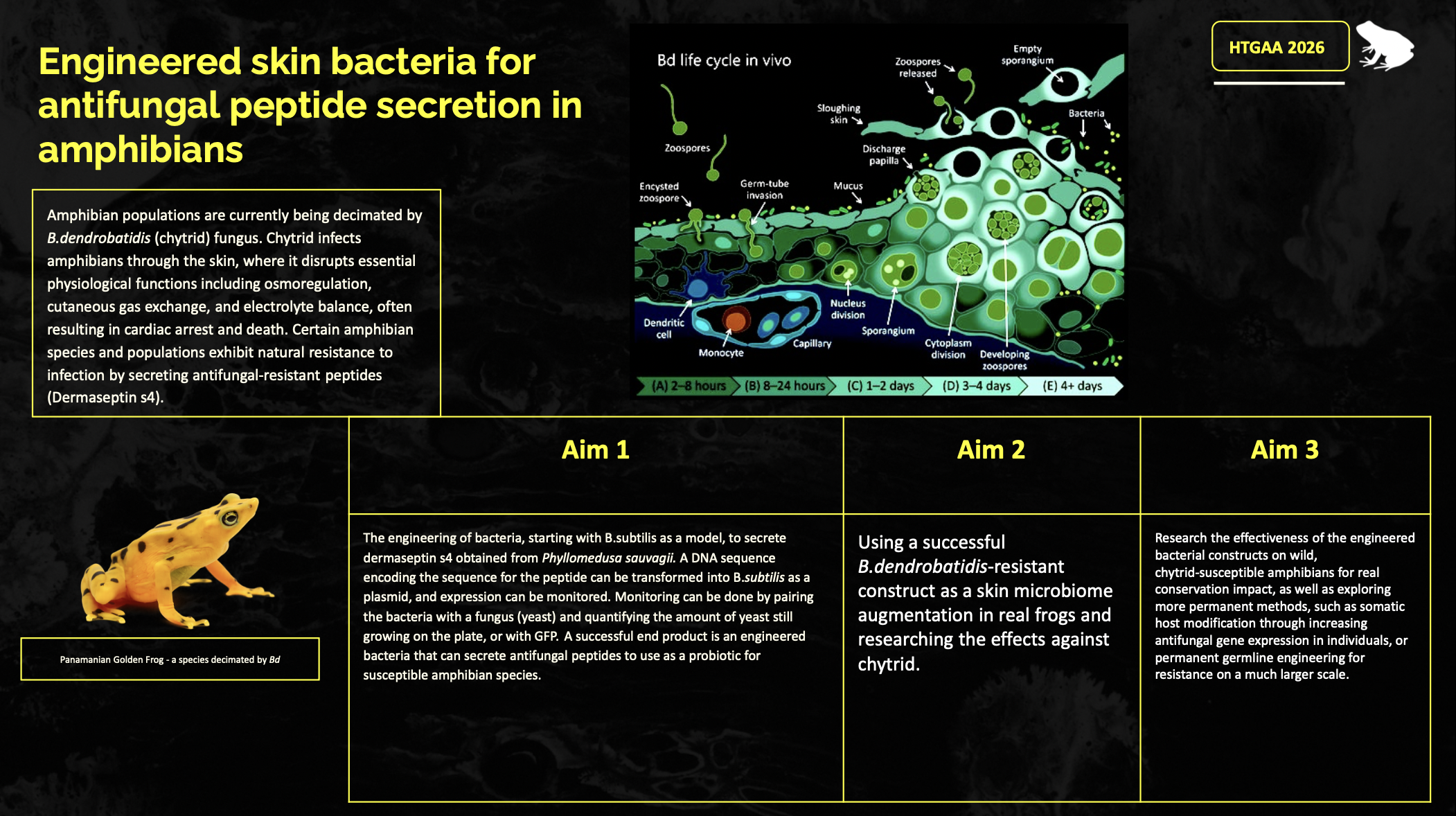
Question 1: A biologically engineered application that I find particularly compelling is the development of gene editing tools for biodiversity conservation. Given the rapid global decline of many species driven by human activity, emerging infectious diseases, and climate change, preserving biodiversity and maintaining ecosystem stability has become an urgent scientific and societal challenge. One promising application of genetic engineering in this context is the germline editing of amphibian genomes to combat the chytrid fungus, Batrachochytrium dendrobatidis, which has caused widespread population declines and driven numerous frog and toad species toward extinction.
Homework Part A: General and Lecturer-Specific Questions
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis removes the constraints we usually face for protein synthesis when working with live cells. For example, working with live cells requires culturing cells throughout the whole cell lifecycle, with all the possibilities for error that this implies (mistakes, unexpected cell behavior due to their non-deterministic nature, etc), as well as the required timelines (speed is limited by the fundamental speed constraints from the cell growth cycle), and costs. Cell-free systems also allow for much greater control, as the main system is boiled down to its most basic functional components, removing a lot of complexity (variables outside of our control), and therefore allowing the possibility of producing much more homogeneous products.
Week 2 Pre-Lecure Homework: Homework Questions from Professor Jacobson: Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? DNA polymerase makes about 1 error per 10⁴ bases during replication. Since the human genome is about 3 billion base pairs, this would result in roughly 300,000 errors per replication if left uncorrected. Biology handles this through proofreading and DNA repair systems, which dramatically reduce the final number of mistakes.
HTGAA Week 03 - Lab Automation Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
https://www.nature.com/articles/s42003-019-0305-x?utm_source=chatgpt.com
This paper used a Tecan liquid handler to enable high-throughput patient-derived tumor organoid drug screening, moving beyond pipetting into a biologically novel precision medicine application. The researchers wanted to determine whether patient-derived tumor organoids (miniature 3D tumors grown from real patient cancer tissue) could be used to rapidly identify effective anti-cancer drugs for individual patients. The Tecan system automated several otherwise tedious and error-prone steps: precise dispensing of organoid cultures into multiwell plates, automated addition of large drug libraries at multiple concentrations, and standardization of timing and reagent handling to reduce variability between samples. This helped make organoid-based drug screening much more scalable as a workflow for research.
Week 04 - Protein Design I Part A How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
6.6 x 1023 molecules of amino acids
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Part A: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Sequence with mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
WRYYAAALRHKG WRYYAVAARHKK WRSYVVVLELGG HHYPAVAVALKG FLYRWLPSRRGG
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Part B
Subsections of Homework
Week 1 HW: Principles and Practices
Question 1:
A biologically engineered application that I find particularly compelling is the development of gene editing tools for biodiversity conservation. Given the rapid global decline of many species driven by human activity, emerging infectious diseases, and climate change, preserving biodiversity and maintaining ecosystem stability has become an urgent scientific and societal challenge. One promising application of genetic engineering in this context is the germline editing of amphibian genomes to combat the chytrid fungus, Batrachochytrium dendrobatidis, which has caused widespread population declines and driven numerous frog and toad species toward extinction.
Chytrid infects amphibians through the skin, where it disrupts essential physiological functions including osmoregulation, cutaneous gas exchange, and electrolyte balance, often resulting in cardiac arrest and death. Notably, certain amphibian species and populations exhibit natural resistance or tolerance to infection. I would begin by studying these resistant taxa to identify antifungal genes and immune pathways that are differentially expressed and to determine the molecular mechanisms by which resistance is conferred.
Using this information, I would develop a targeted gene editing strategy, most likely using CRISPR based tools, to introduce resistance associated alleles into the germline of chytrid susceptible species. Edited individuals would then be raised and monitored in controlled captive environments to assess disease resistance, overall fitness, reproductive success, and potential off target effects across multiple generations. Only after rigorous validation would these individuals be considered for carefully regulated reintroduction into natural habitats. If successful, this approach could enable the spread of chytrid resistance through vulnerable populations and contribute to the recovery of amphibian species and the ecosystems they support.
Question 2:
To ensure that germline gene editing for amphibian conservation contributes to an ethical future, a central governance goal must be the prevention of harm while enabling responsible intervention in biodiversity crises. Because releasing genetically edited organisms into the wild introduces permanent and potentially far-reaching changes, policy frameworks must prioritize ecological safety, long-term accountability, and proportional use of the technology. Ethical governance with this technology should aim to ensure that interventions are scientifically justified and carefully controlled, without halting innovation outright.
One key policy objective is ecological and biological safety. This includes requiring extensive pre-release testing of edited amphibians across multiple generations in captivity to evaluate fitness, behavior, reproduction, and unintended physiological effects beyond disease resistance. In addition, governance frameworks should mandate ecological risk assessments that examine how chytrid-resistant individuals might alter ecosystem dynamics or pathogen evolution, including the possibility of selecting for more virulent fungal strains. Policies should also require clear containment and mitigation strategies prior to release, such as geographically limited trials or genetic mechanisms that reduce the risk of uncontrolled spread.
Another major governance goal is strong oversight and accountability. Decisions about releasing gene-edited organisms should be evaluated by independent, interdisciplinary review bodies that include not only molecular biologists, but also ecologists, ethicists, conservation practitioners, and representatives of local or Indigenous communities. Clear legal responsibility must be established for long-term monitoring and potential ecological harm, recognizing that unintended consequences may arise years or decades after release. Because amphibians and pathogens frequently cross political borders, international coordination will also be necessary to prevent unilateral actions that could impact shared ecosystems.
Ethical governance must also address proportionality and justice. Gene editing should be framed as a last-resort conservation tool, deployed only when less invasive strategies (such as habitat protection, captive breeding, or antifungal treatments) have proven insufficient. Each proposed intervention should be evaluated on a species-by-species basis, taking into account ecological role, conservation status, and cultural significance. Finally, long-term monitoring and adaptive governance should be mandatory, treating gene-edited conservation organisms as an ongoing responsibility rather than a one-time solution. By embedding safety, transparency, and equity into policy from the outset, conservation gene editing can be developed as an ethical and legitimate response to the accelerating loss of global biodiversity.
Question 3:
One potential governance action is the creation of a mandatory staged release regulatory framework for gene edited conservation organisms, overseen by federal environmental regulators in coordination with academic researchers. Currently, conservation gene editing proposals fall into regulatory gray areas and are often evaluated under frameworks designed for agriculture or laboratory research rather than irreversible ecological interventions. This proposal would introduce a formal, stepwise approval process similar to phased clinical trials, requiring progression from laboratory validation to contained semi natural environments, then to limited field trials, and only then to broader release. To work effectively, this system would require regulators such as the U.S. Fish and Wildlife Service or equivalent international bodies to develop species specific guidelines, while researchers and institutions opt in by designing studies that meet these staged criteria. This approach assumes that ecological risks can be meaningfully assessed at each stage and that regulators have sufficient expertise and funding to evaluate complex genetic and ecological data. A major risk of failure is regulatory bottlenecking, as overly slow or conservative approvals could delay interventions until species are already extinct. Conversely, a successful rollout could unintentionally normalize gene editing as a default conservation tool, reducing investment in habitat protection or other non genetic solutions.
A second governance action involves the use of funding based incentives and requirements, driven by public research funders and philanthropic conservation organizations. At present, much conservation biotechnology research is funded without standardized requirements for long term ecological monitoring or community engagement. Under this approach, funders would require applicants working on chytrid resistant amphibians to commit to extended post release monitoring, open data sharing, and collaboration with local stakeholders as conditions of grant funding. This is analogous to how clinical research increasingly ties funding to transparency and post market surveillance. For this strategy to work, funding agencies must be willing to allocate resources not only for innovation but also for long term monitoring, and researchers must accept longer timelines and broader accountability. This approach assumes that financial leverage is sufficient to shape researcher behavior and that public or nonprofit funders remain dominant actors in this space. Risks include the possibility that private actors bypass these norms entirely or that monitoring requirements become superficial compliance exercises rather than meaningful safeguards. If highly successful, this action could also skew research toward species or regions that are easier to monitor, leaving the most vulnerable ecosystems under served.
A third governance action is the development of technical safeguards embedded directly into gene editing designs, pursued primarily by academic laboratories and biotechnology companies but guided by regulatory expectations. Rather than relying solely on policy restrictions, researchers would be encouraged or required to design edits that limit spread or persistence, such as conditional gene expression, geographically constrained release strategies, or inheritance patterns that reduce uncontrolled dissemination. Making this approach effective would require advances in genetic engineering tools, agreement on acceptable design standards, and regulatory recognition of these safeguards as meaningful risk reduction measures. This strategy assumes that ecological complexity can be partially managed through genetic design and that engineered constraints will function as intended in natural environments. Failure could occur if these safeguards break down under natural selection, mutation, or environmental variability. Even apparent success carries risks, as confidence in technical controls could lead regulators or researchers to underestimate broader ecological uncertainties and create a false sense of security around releases that remain fundamentally irreversible.
Together, these governance actions, including regulatory staging, funding based incentives, and built in technical safeguards, illustrate how ethical oversight of chytrid focused conservation gene editing can be distributed across multiple actors and mechanisms. Rather than relying on a single authority or rule, this approach acknowledges uncertainty, spreads responsibility, and attempts to balance urgency with caution to combat accelerating amphibian decline.
Question 4:
Does This Option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
1
• By helping respond
2
1
3
Foster Lab Safety
• By preventing incident
2
1
2
• By helping respond
2
1
3
Protect the environment
• By preventing incidents
1
2
1
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
2
• Feasibility?
2
1
3
• Not impede research
2
1
1
• Promote constructive applications
2
1
1
Question 5:
Based on the scoring across biosecurity, environmental protection, feasibility, and impact on research, I would prioritize a combined governance approach centered on funding based incentives and requirements, complemented by a staged release regulatory framework, with embedded technical safeguards treated as a supporting rather than primary control. This recommendation is aimed primarily at national environmental regulators and public research funders, such as the U.S. Fish and Wildlife Service, the National Science Foundation, and equivalent agencies internationally. Together, these actors are well positioned to shape researcher behavior, ensure ecological responsibility, and maintain scientific momentum in conservation gene editing.
Funding based incentives emerge as the strongest foundational governance option because they score highly on feasibility, responsiveness, and their ability to promote constructive applications without excessively impeding research. Tying grant funding to long term ecological monitoring, transparency, and stakeholder engagement allows governance to operate early in the research lifecycle while remaining flexible across species and ecosystems. This approach also scales well internationally and avoids the delays associated with creating entirely new regulatory regimes. However, this strategy assumes that public and philanthropic funders remain influential in conservation biotechnology and that compliance requirements are meaningfully enforced rather than treated as procedural formalities.
The staged release regulatory framework should function as a second layer of governance, particularly at the point of environmental release, where irreversible ecological risks are highest. While this option imposes higher costs and may slow deployment, its strong performance in preventing environmental harm justifies its use at later stages rather than as a blanket constraint on early research. A key trade off here is timing. Overly conservative approvals could delay intervention beyond the point where species recovery is feasible, yet insufficient oversight risks ecological damage that cannot be reversed. This framework also depends on the assumption that regulators have adequate expertise, funding, and legal authority to evaluate gene edited organisms in ecological rather than agricultural contexts.
Embedded technical safeguards should be encouraged but not relied upon as the primary governance mechanism. While they score highly in preventing incidents and promoting innovation, they perform poorly in response capacity and carry substantial uncertainty under real world ecological conditions. Natural selection, mutation, or environmental variability may erode designed constraints over time. Treating technical safeguards as a supplement or last resort rather than a substitute for oversight avoids the risk of false confidence and maintains institutional responsibility for long term outcomes.
Overall, this layered governance strategy reflects an assumption that no single tool can adequately manage the uncertainty inherent in releasing gene edited organisms into complex ecosystems. By prioritizing funding based governance to shape norms and practices, reserving regulatory staging for high risk transitions, and supporting technical safeguards as risk reducing features, policymakers can balance urgency with caution. This approach accepts trade offs in speed and cost in exchange for legitimacy, adaptability, and long term ecological responsibility in the use of gene editing for amphibian conservation.
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis removes the constraints we usually face for protein synthesis when working with live cells. For example, working with live cells requires culturing cells throughout the whole cell lifecycle, with all the possibilities for error that this implies (mistakes, unexpected cell behavior due to their non-deterministic nature, etc), as well as the required timelines (speed is limited by the fundamental speed constraints from the cell growth cycle), and costs. Cell-free systems also allow for much greater control, as the main system is boiled down to its most basic functional components, removing a lot of complexity (variables outside of our control), and therefore allowing the possibility of producing much more homogeneous products.
Two special cases that CFS allow are the use of molecules that would usually be toxic for cells, and working with a much wider variety of molecules that would otherwise be destroyed by the cell, as well as modified versions of the most basic components of this machinery (that could not work in a live cell), such as amino acids that don’t exist in nature.
Describe the main components of a cell-free expression system and explain the role of each component.
The basic components are:
The lysate, which includes the “machinery”: basically ground up cells (prokaryotic or eukaryotic), providing the cellular components we need for the reactions, like ribosomes, enzymes, tRNAs and cofactors, which will translate mRNA into protein
The genetic template (DNA or RNA) that encodes the product we want
The “building blocks” (amino acids and nucleotides)
The energy system (needed to provide and replenish the ATP and GTP needed for the reactions) (ie Mg^2+ and K+)
The “environmental tuning” factors (salts, buffers, temperature, etc)
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your
cell-free experiment.
Because protein synthesis is an energy-intensive process. In living cells, mitochondria handle the production of ATP, but in CFS we need to set up systems that can continually obtain the phosphates needed to convert ADP into ATP. Not being able to meet these energy needs will cause the reactions to stall or provide low yield. A method to ensure a continuous ATP supply is to include an ATP regeneration system, such as the phosphocreatine–creatine kinase system. Creatine kinase will transfer a phosphate group from phosphocreatine to ADP, regenerating ATP as it is consumed.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Only eukaryotic cells can perform PTM post-translational modifications (ie glycosylation), so any desired product requiring this process will require the use of eukaryotic cell lysate. This will also be required if the final desired product requires the usage of mammalian regulators. However, when these requirements are absent, prokaryotic systems are faster and cheaper.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The main challenge when working with membrane proteins is that they tend to aggregate or misfold in aqueous environments (due to their hydrophobic regions trying to “escape” it)
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
The required energy needs are not being met.
The proteins might be misfolding. In this case, we would check and tune the environmental variables, like the temperature or the salts, and possibly add chaperones or use different membrane-mimicking systems.
The final or intermediary products might be suffering degradation. In this case, I would check for the presence of nucleases, proteases, etc.
Homework Question from Kate Adamala:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would be built to capture environmental CO2. The minimal inputs would be an energy source for carbon fixation (ATP), CO2, inorganic nutrients (phosphates, salts, enzymatic cofactors), and a carbon concentration mechanism. I would also need a metabolic mechanism for carbon fixation, such as a rubisco-based Calvin cycle.The output would be a stable storage mechanism in the form of carbonates, to safely lock away carbon in the system.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Probably not, as carbon fixation and compartmentalization would require additional metabolic processes beyond basic protein translation.
Could this function be realized by genetically modified natural cell?
Yes, and pretty easily as there are already many organisms that perform these functions.
Describe the desired outcome of your synthetic cell operation.
I would aim to make this operation as stripped down as possible, trying to create the minimal viable carbon capturing mechanism from a cell-free system. The desired outcome should be a minimal measurable function of carbon processing and storage, the cell being “alive” is not needed. I would aim for the synthetic cell to continuously convert dissolved CO2 into measurable reduced carbon for several hours while maintaining internal biochemical activity.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
I’d want a stable membrane permeable to CO2, such as a lipid membrane inspired by bacteria, composed of mostly POPC and POPE.
What would you encapsulate inside? Enzymes, small molecules.
I would need plasmids encoding DNA and tRNAs + ribosomes for translation, PEP and pyruvate for ATP regeneration, salts and cofactors such as Mg, phosphate, potassium etc.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
A bacterial system will suffice.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Membrane pores for passive diffusion will work well for small molecules, membrane channels to retain larger proteins (Outer Membrane Protein F)
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
HPLC assays can provide product quantification to show if CO2 has been converted into reduced carbon in the synthetic system.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
Portable pathogen sensor patches embedded in public transport vehicles to monitor the load of airborne pathogens
How will the idea work, in more detail? Write 3-4 sentences or more.
By using a freeze-dried detection system coupled with an RNA switch or CRISPR-based sensor, this device could be carried by rideshare or transport service providers to periodically monitor pathogen levels inside a vehicle using a simple swab test. The system could potentially be integrated into seatbelts, door panels, or other interior surfaces as a small patch or sticker.
What societal challenge or market need will this address?
This technology could help identify when a vehicle requires more thorough disinfection, improving both driver and passenger safety. For example, immunocompromised riders could potentially request vehicles with lower measured pathogen loads. In addition, the system could provide useful public health data for applications such as outbreak monitoring, epidemic detection, and prevention.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The device itself could take the form of a disposable patch containing a cartridge with the required water and reaction buffers, activated through a simple button or switch that mixes the components (such as at the end of a driver’s shift). A single-use format replaced daily would help minimize contamination risks, while the freeze-dried design could maintain stability for approximately 3–6 months at room temperature.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Future human missions to Mars will require sustainable systems for food production, waste recycling, and oxygen generation. However, Mars presents extreme conditions, including high radiation, low temperatures, and limited nutrients, making microbial survival difficult. My proposal investigates how bacterial stress-resistance pathways could be engineered and tested using the BioBits cell-free protein expression system to identify genes that improve survival under Mars-like conditions. Understanding how to design hardier microbes is significant for supporting long-term space habitation, scientifically interesting for studying adaptation to extreme environments, and relevant to developing biotechnology for extraterrestrial living.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Genes and proteins involved in bacterial salt tolerance, including osmotic stress-response pathways, to understand survival under high-salinity Mars-like conditions.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Mars soil is believed to contain high concentrations of salts, which can damage cells by causing dehydration and disrupting biological processes. By studying bacterial salt-tolerance genes and proteins, my project aims to identify mechanisms that help microbes survive in high-salinity environments. Using the BioBits cell-free system, we can test stress-response pathways under simulated Mars-like salt conditions to better understand how bacteria might be engineered to survive and support future human missions through waste recycling, food production, or oxygen generation.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
The goal of this project is to determine whether bacterial genes associated with salt resistance can help maintain biological activity in environments with high salinity, similar to conditions expected on Mars. I predict that introducing these stress-response genes into the cell-free system will result in more effective protein production under salty conditions compared to systems without them. This idea is based on the observation that some Earth bacteria naturally survive in highly saline habitats by activating protective mechanisms against osmotic stress. Identifying genes that remain functional in harsh environments could contribute to developing microbes better suited for supporting long-term human space exploration.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
This experiment will use the BioBits cell-free protein expression system to test bacterial salt-tolerance genes under increasing salt concentrations that mimic Mars-like conditions. Samples will include reactions containing salt-resistance genes and control reactions without these genes. Multiple salinity levels will be tested to compare protein expression across environments. Protein production will be measured using fluorescent reporters and visualized with a P51 Molecular Fluorescence Viewer. Data collected will include fluorescence intensity under different salt conditions to determine whether salt-tolerance genes improve biological activity in high-salinity environments.
Week 2 HW: DNA Read, Write, Edit
Week 2 Pre-Lecure Homework:
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase makes about 1 error per 10⁴ bases during replication. Since the human genome is about 3 billion base pairs, this would result in roughly 300,000 errors per replication if left uncorrected. Biology handles this through proofreading and DNA repair systems, which dramatically reduce the final number of mistakes.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Because 61 codons code for 20 amino acids, there are many possible DNA sequences that can encode the same protein. In reality, not all work well due to factors like tRNA availability, mRNA structure, and protein folding during translation, which affect how efficiently and accurately a protein is made.
Sources: ChatGPT Prompt: “Detail the most common different factors in which variant amino acid codes may fail and explain each”
ChatGPT Prompt: “How does biology deal with the error rate of DNA polymerase on a molecular level?”
Homework Questions from Dr. LeProust:
Most common oligo synthesis method:
The most widely used method is solid-phase synthesis using phosphoramidite chemistry.
Why are oligos longer than ~200 nt hard to make?
Errors build up during synthesis, making long sequences increasingly inaccurate.
Why can’t a 2000 bp gene be made directly?
The error rate becomes too high, so genes are instead built from shorter oligos that are assembled together.
Homework Question from George Church:
The ten essential amino acids in animals are phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, arginine, leucine, and lysine.
Because animals must get all of these from their diet, the “lysine contingency” proposed in Jurassic Park is not a strong safety mechanism as limiting lysine alone would not effectively control an organism.
Week 2 Homework
1. Benchling & In-silico Gel Art
I made the letter M! (I grew up in Michigan, Go Blue!)
2. Gel Art - Restriction Digests and Gel Electrophoresis
I ran a gel in lab! I pipetted DNA into 10 wells, only 1 band shows up clearly besides the ladder, but there are a couple of fainter bands as well.
3. DNA Design Challenge
3.1: I chose the protein Magainin-2, an antimicrobial peptide isolated from the skin of the African Clawed Frog, Xenopus Laevis. I thought this would be an interesting protein to study because of its antibiotic properties and possible function as a resistance to chytrid fungus in amphibians.
Amino Acid Sequence: GIGKFLHSAKKFGKAFVGEIMNS
3.2:
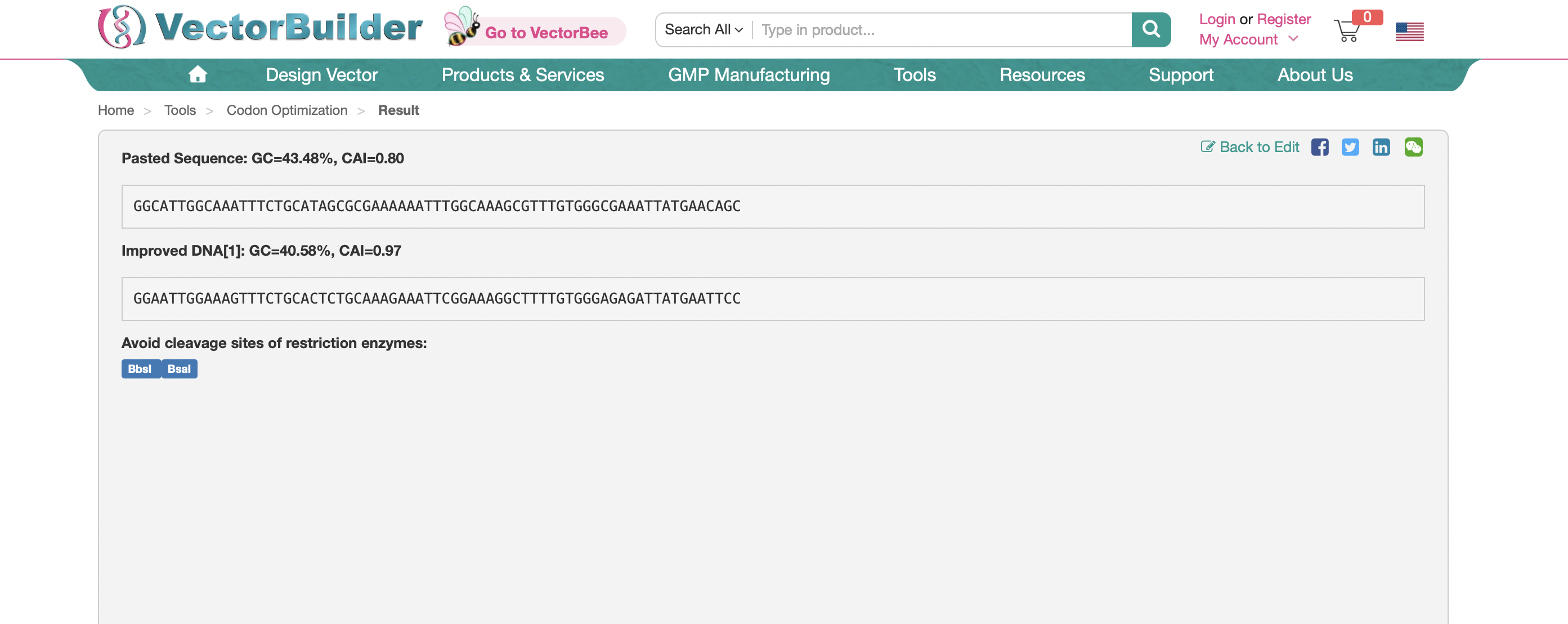
reverse translation of Untitled to a 69 base sequence of most likely codons.
ggcattggcaaatttctgcatagcgcgaaaaaatttggcaaagcgtttgtgggcgaaatt
atgaacagc
3.3: Codon Optomization
Here is the optomized sequence using Xenopus Laevis as my chosen model organism.
Here is the sequence using E.coli as the model organism for codon optomization: GGCATTGGCAAATTTCTGCATAGCGCGAAAAAATTTGGCAAAGCGTTTGTGGGCGAAATTATGAACAGC
E.Coli would be preferable to create this protein in for ease-of-use, widespread availability, and fast growth.
3.4 You Have A Sequence! Now What?
To synthesize a protein from this sequence, a common method used is cell-dependent, where I create a plasmid vector that includes a promoter, ribosome-binding sequence, my sequence, a terminator, and possibly a selection marker that helps verify that the plasmid is being expressed (such as an antibiotic resistance sequence). The plasmid can then be transformed into a host organism such as a bacteria or yeast, via electroporation or heat shock. The sequence is now able to express my protein!
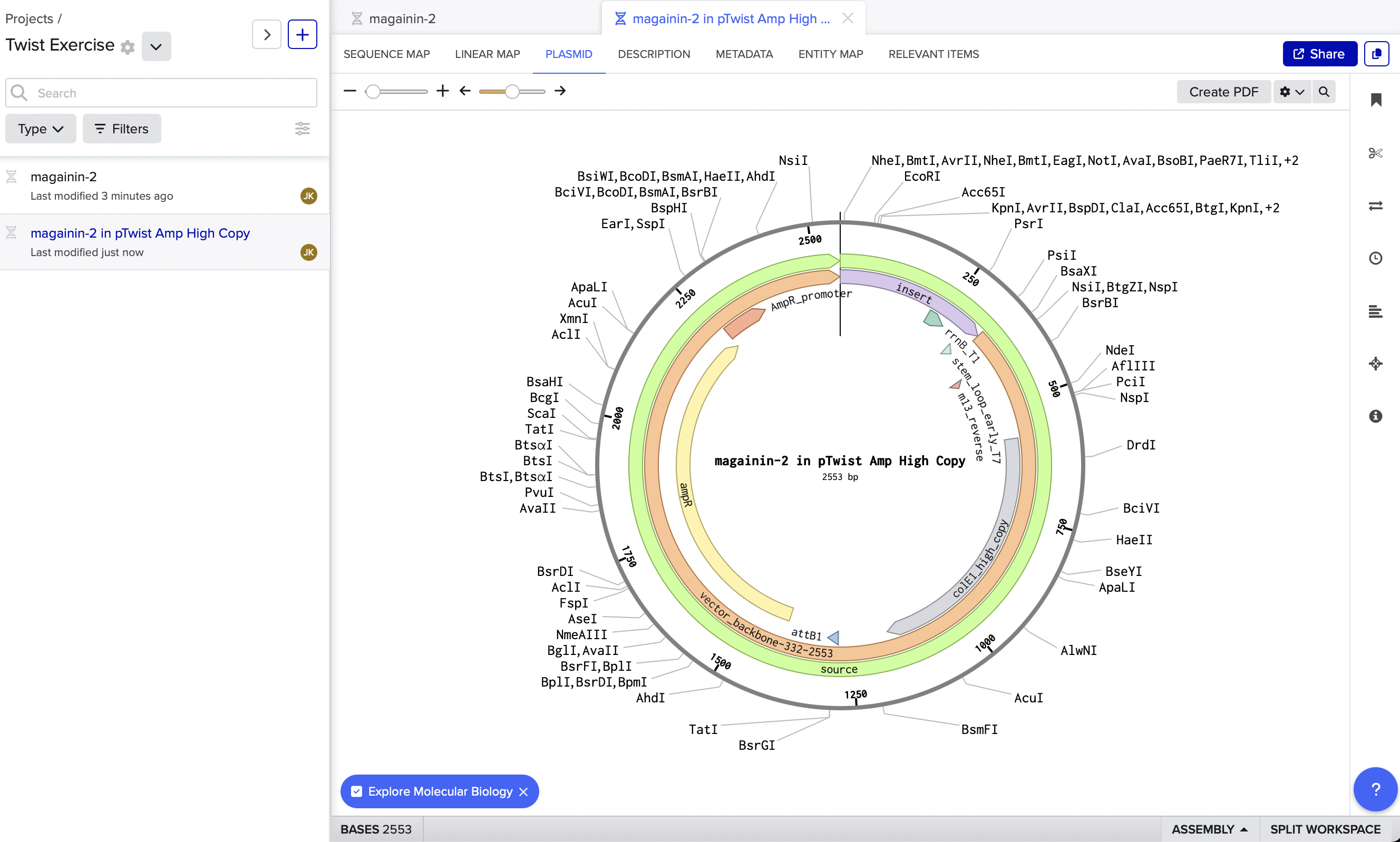
4. Prepare a Twist DNA Synthesis Order
Here is my Twist - generated plasmid in Benchling. My coding sequence was short, so I had to add in a neutral regulatory element upstream of my coding region so I had enough BP for Twist to generate the rest of the plasmid.
5. DNA Read/Write/Edit
5.1: If I were to sequence and read any DNA, I would love to read a full genome of an extinct species that still has available tissue for ancient DNA sequencing. It would be interesting to compare the genome of these extinct species to their closest relatives and see what genes are conserved and how they differ genetically. This would give some insight into the evolutionary history behind the speciation of that particular lineage and could give clues on how to safely and ethically bring back the species from extinction if it would help a current ecosystem ecologically.
5.2:
Week 3 HW: Lab Automation
HTGAA Week 03 - Lab Automation
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
This paper used a Tecan liquid handler to enable high-throughput patient-derived tumor organoid drug screening, moving beyond pipetting into a biologically novel precision medicine application. The researchers wanted to determine whether patient-derived tumor organoids (miniature 3D tumors grown from real patient cancer tissue) could be used to rapidly identify effective anti-cancer drugs for individual patients. The Tecan system automated several otherwise tedious and error-prone steps: precise dispensing of organoid cultures into multiwell plates, automated addition of large drug libraries at multiple concentrations, and standardization of timing and reagent handling to reduce variability between samples. This helped make organoid-based drug screening much more scalable as a workflow for research.
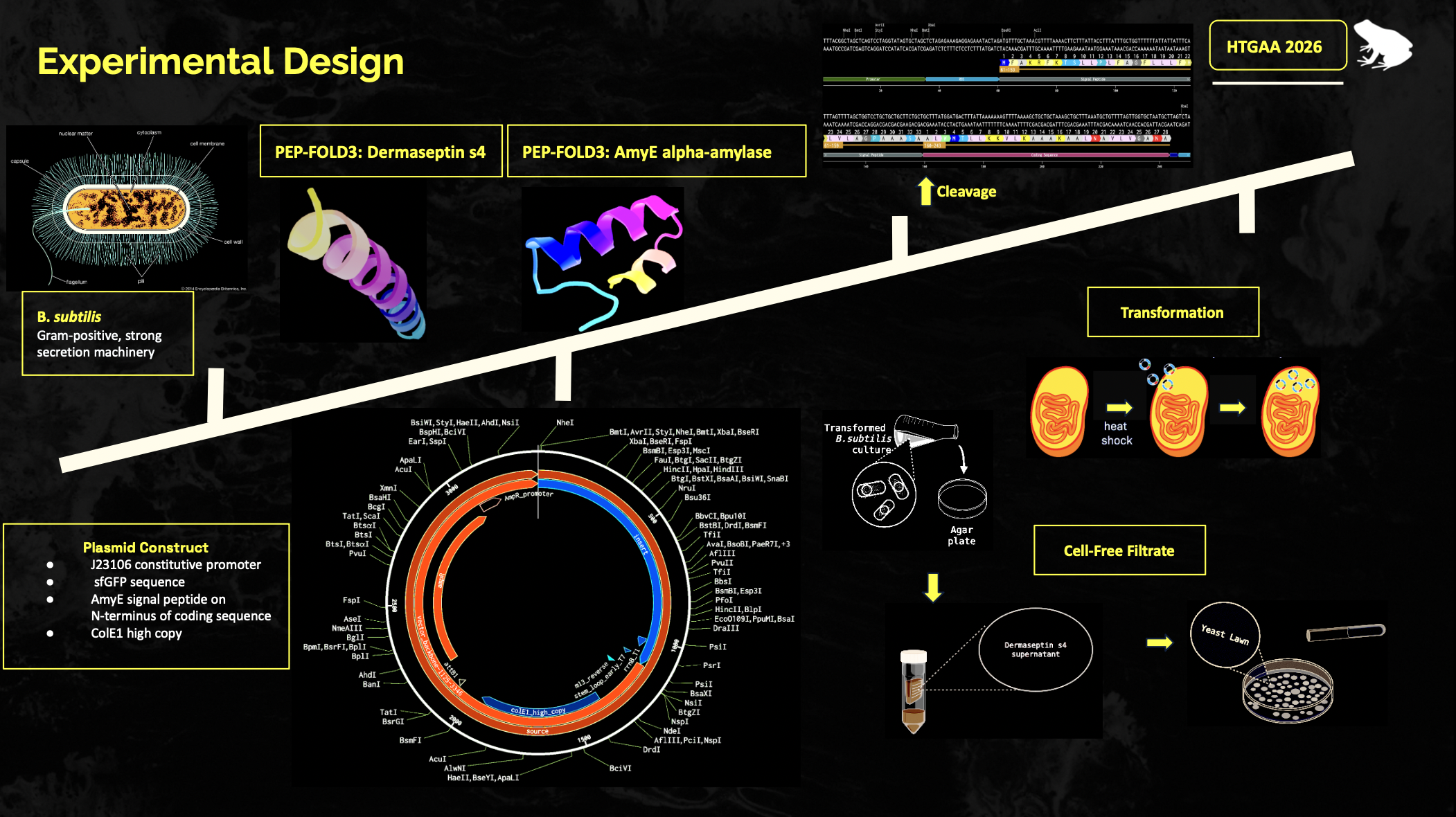
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
One possible application would involve automated liquid handling platforms, such as Tecan or Opentrons systems, to standardize microbial growth assays and peptide screening. For example, engineered bacterial strains expressing candidate antifungal peptides (dermaseptins or related amphibian antimicrobial peptides) could be cultured in multiwell plates and automatically co-incubated with fungal models or Batrachochytrium dendrobatidis analog systems. Optical density measurements and inhibition assays would improve experimental consistency while enabling larger-scale screening of secretion strategies with varying peptides.
Computational automation could also support the project through Python-based analysis pipelines. Example pseudocode may be developed to automate experimental data processing, including colony growth quantification, fluorescence analysis of GFP expression, inhibition zone measurement from plate images, and statistical comparisons between engineered strains. Computer vision tools could also potentially be used to quantify fungal inhibition from photographs of agar plates, reducing subjectivity in phenotype scoring.
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
6.6 x 1023 molecules of amino acids
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestive system breaks down food components (proteins, fats, and carbohydrates) into their constituent building blocks. For example, proteins are digested into amino acids, which the body then uses to build and maintain its own cells and tissues. We do not directly assimilate cow proteins or cells when eating meat, only the nutrients and molecular components derived from them.
Why are there only 20 natural amino acids?
Technically, more amino acids could exist, but evolution settled on a core set of approximately 20 very early in the history of life. These amino acids became the biological standard because their chemical diversity is sufficient to support the structural and functional complexity required for life.
Where did amino acids come from before enzymes that make them, and before life started?
Before life as we know it started on Earth, amino acids were likely produced through primordial geochemical and extraterrestrial processes. Amino acids and their chemical precursors are commonly detected in meteorites, hydrothermal vent systems, and other geochemically active environments. In the absence of living organisms to metabolize or degrade them, geochemically synthesized amino acids (being relatively stable molecules) could have accumulated over time.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left handed. Biology uses the L-amino acids but these create right handed α-helixes.
Can you discover additional helices in proteins?
Yes, some uncommon helices are only found during molecular binding and not shown in x-ray crystallography
Why are most molecular helices right-handed?
They are more thermodynamically and energetically stable
Why do β-sheets tend to aggregate?
Hydrogen bonding occurs between the ends of the sheets allowing the sheets attach to each other and stack
What is the driving force for β-sheet aggregation?
Hydrophobic effects
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
My protein is the peptide Dermaseptin s4. Dermaseptin S4 is an antimicrobial peptide originally isolated from the skin of the South American leaf frog (Phyllomedusa sauvagii). It is highly regarded in scientific research for its ability to disrupt microbial and fungal cell membranes. I chose this peptide because it will prove to be useful in my project, as I will attempt to use this protein to disrupt the cell membranes of yeast after secretion by a bacteria. This will act as my proof-of-concept model for my project to show the antifungal activity of amphibian skin peptides, which can then later be used on Batrachochytrium dendrobatidis as a potential probiotic to treat the disease.
Identify the amino acid sequence of your protein.
ALWMTLLKKVLKAAAKAALNAVLVGANA
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It is 28 amino acids long, the most frequent amino acid is Alanine (9 times).
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
9 homologs were found.
Does your protein belong to any protein family?
It belongs to the Frog Skin Active Peptide (FSAP) family and is further classified into the Dermaseptin subfamily.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The full protein structure of Dermaseptin S4 has not been solved currently, only partial truncated synthetic derivatives have been solved using NMR. Only the first 14 amino acids of the sequence of 28 have been solved, discovered in 2006. It is an exceptionally high scoring structure in terms of quality derived from NMR structures.
Are there any other molecules in the solved structure apart from protein?
No, all amino acids in the peptide chain.
Does your protein belong to any structure classification family?
Amphipathic alpha-helical antimicrobial peptides family
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
It only has helices, contains no sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic and hydrophilic residues are split perfectly into two halves. It forms a class-L amphipathic alpha-helix.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
It has no binding pockets, as its structure is more of a single uninterrupted alpha helix.
Part C
Deep Mutational Scans
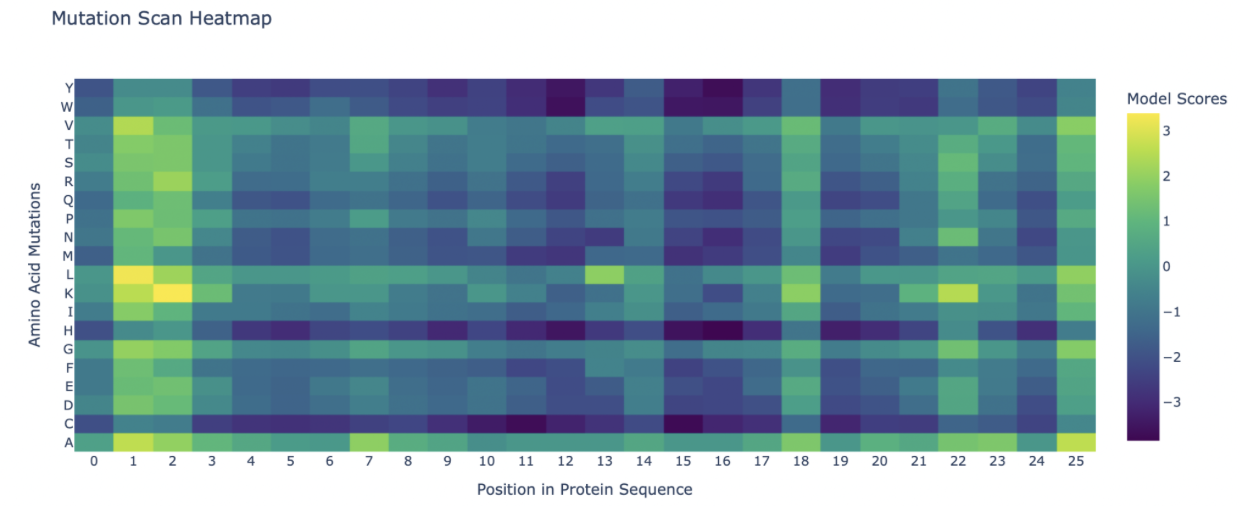
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Attached is the mutation scan, I noticed that position 1 and 2 have extremely positive LLR values, indicating mutations that are beneficial to the function of Dermaseptin s4.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Couldn’t figure out a way to visualize the latent space analysis correctly. The downloaded file from the web seems to be a broken link, or I have a faulty setting on my end.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Part D
Goal 1: Stabilize MS2 L so it remains correctly folded and membrane-competent under a wider range of conditions (temperature, expression levels), which should support more robust lysis and higher effective titers.
Goal 2: Increase intrinsic toxicity by promoting oligomerization and pore formation of L in the bacterial envelope.
We will focus on improving both the stability and toxicity of the MS2 L lysis protein using a purely computational design-and-screen pipeline before any wet-lab work.
Our first goal is increased stability. We want MS2 L to remain correctly folded and membrane-competent across a broad range of expression and environmental conditions so that lysis is more reliable and supports higher effective titers.
Our second goal is increased toxicity. We aim to increase the intrinsic lytic potency of L by promoting oligomerization and pore formation in the bacterial envelope.
To do this, we will use protein language models (such as ESM or ProtT5) as in silico mutagenesis tools, scoring single and selected double mutants of L and ranking substitutions predicted to improve stability or fitness.
We will combine this with structure prediction and modeling. AlphaFold will be used on full-length and truncated L variants to generate consistent 3D models of the soluble and transmembrane regions, while AlphaFold-Multimer or docking tools will help model L oligomers in a membrane-like context.
Membrane-aware stability analysis will further guide our designs. TMHMM-like predictors will define transmembrane boundaries and disorder regions, while Rosetta- or FoldX-style ΔΔG calculations will help refine promising mutation sites, especially around the LS motif and transmembrane helix.
We will also incorporate coevolution and functional hotspot information by overlaying published mutagenesis data for L. Residues already known to be essential for function will be locked and excluded from substitution.
These approaches are important because mutational studies show that even conservative substitutions near the LS motif can abolish function. We need a guided way to search sequence space while respecting both evolutionary constraints and protein stability.
Protein language models provide a fast way to scan mutation space, while ΔΔG calculations and structure predictions help eliminate variants that appear incompatible with proper helix packing or overall folding, leaving a smaller set of promising stabilizing mutations.
For increased toxicity, MS2 L activity depends on efficient membrane insertion and higher-order oligomer formation through its C-terminal transmembrane segment. Modeling and optimizing helix–helix interactions should therefore influence pore stability and lytic potency.
By favoring small or helix-packing residues at predicted contact sites, while avoiding unfavorable charges in the membrane, we may be able to design variants that form more stable oligomers without disrupting the LS motif or essential hydrophobic patterning.
In single-gene lysis systems, lysis timing and efficiency emerge from a balance of expression level, folding, and toxicity. Improving stability may therefore reduce misfolding and allow higher levels of functional L protein, ultimately improving titers under comparable induction conditions.
Our overall objective is to computationally design MS2 L variants that combine improved membrane stability with increased intrinsic toxicity.
Week 5 HW: Protein Design II
Part A: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Sequence with mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Part B
Peptide #1: WRYYAAALRHKG AlphaFold: ipTM = 0.38 pTM = 0.8 Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ipTM = 0.38 is low confidence of binding. It would be surface-bound and far from the beta-barrel region. Number #2 WRYYAVAARHKK Alphafold: ipTM = 0.31 pTM = 0.75
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? Surface-bound, relatively close to the mutation site. It does not engage the beta barrel site. iPTM = 0.31 which shows low confidence.
Number #3 WRSYVVVLELGG Alphafold: ipTM = 0.75 pTM = 0.86
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? This one is bound on the surface. ipTM = 0.75 which is somewhat confident (we treat a score between 70 and 90 as “confident”). It is somewhat close to the beta-barrel area. It isn’t too far from the mutation area, but also not super close.
Number 4: HHYPAVAVALKG Alphafold: ipTM = 0.29 pTM = 0.86
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? ipTM = 0.29 which is very low confidence of binding. No, it’s not close to the mutation site. It’s on the opposite side of where the beta-barrel sits. It is surface-bound and closest to the positions 86-87.
Number 5: FLYRWLPSRRGG Alphafold: ipTM = 0.35 pTM = 0.8
Part C
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ipTM = 0.35, hence confidence of binding is low. It’s far from the N-terminus that has the A4V. It is surface-bound. It is close to the beta-barrel region (the arrows).
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Yes, WRSYVVVLELGG has a higher iPTM (0.75 vs 0.35) plus a lower perplexity score (18 vs 20), even though it has the highest perplexity score out of the other generated ones. In general, the iPTM values for all of them except this one are low (~0.30), meaning that AlphaFold is not very confident about their vinding affinity.
In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Yes, WRSYVVVLELGG performs better overall, with a higher iPTM score (0.75 vs 0.35) and a lower perplexity score (18 vs 20), even though it still has the highest perplexity among the generated candidates. In general, though, the iPTM values for most of the peptides are fairly low (~0.30), suggesting that AlphaFold is not especially confident in their binding interactions.
WRSYVVVLELGG also scores the highest iPTM in AlphaFold and the strongest predicted binding affinity in PeptiVerse. The trends are pretty consistent between both platforms, with stronger binders generally showing higher scores across methods. Solubility is close to 1.0 for all peptides, so none seem to have major solubility concerns.
Most of the peptides also show low hemolytic probabilities (<0.1), with one exception: the strongest predicted binder. Interestingly, this peptide is also one of only two with a positive GRAVY score, meaning it is more hydrophobic. Since hydrophobic peptides are more likely to disrupt membranes, this raises some concerns from a toxicity standpoint. In other words, the peptide with the strongest predicted binding also appears to carry the highest risk for toxicity and aggregation.
Peptide #1 (WRYYAAALRHKG) has the lowest predicted hemolysis score, giving it a strong safety profile, but its binding affinity is relatively modest, making it a weaker candidate overall. Peptide #2 shows weak binding affinity and a high positive charge, so it is likely less favorable. Highly positively charged peptides can increase nonspecific interactions with negatively charged membranes and biomolecules, which may increase toxicity, hemolysis, and off-target effects.
Choose one peptide you would advance and justify your decision briefly.
I would choose peptide #1 because it has the second highest binding affinity (despite still being moderately low) but has a good safety profile.