Group Final Project
Authored and reviewed by:
- 2026a-john-adeyemo-adedeji
- 2026a-eric-schneider
- 2026a-albert-manrique
- 2026a-tehseen-rubbab
- 2026a-brie-taylor
Introduction
This document captures the full scope of our group work within the Genspace node focused on engineering the MS2 bacteriophage L protein. Group 2 formed around a shared interest in improving the toxicity, stability, and tunability of the L protein through computational design.
Our early brainstorming sessions centered on three broad goals:
- Increased stability
- Higher titers
- Higher toxicity of the lysis protein
After several meetings and independent exploration, the group converged on two main computational directions. The first centered on systematic truncation and mutagenesis of the N-terminal regulatory domain. The second focused on point mutations within conserved regions that could alter electrostatic interactions while preserving structure.
Two major pipelines emerged from that work. John’s pipeline explored N-terminal truncations, DnaJ disruption, sequence redesign, codon optimization, and sequencing validation. Eric’s pipeline focused on charge-based mutations, conservation mapping, structural modeling, ORF overlap analysis, and cross-referencing with experimental lysis data.
Both approaches identified strong but distinct candidates for improving L protein function.
John’s Analysis and Pipeline
Summary
The MS2 lysis protein L is a 75 amino acid single-pass transmembrane protein whose N-terminal region acts as a regulatory brake on lysis. Rather than directly participating in membrane disruption, this region delays insertion and oligomerization of the transmembrane domain.
My pipeline focused on systematically removing portions of that inhibitory region while preserving the membrane-spanning lytic core. The central hypothesis was simple: if the N-terminal domain slows lysis, then partial removal should release that inhibition and produce earlier, stronger lytic activity.
The strongest candidate to emerge from the analysis was L_trunc30, which removes the first 30 amino acids while preserving the entire transmembrane domain.
Working Sequence
Confirmed L protein sequence:
Confirmed DNA sequence:
Core Hypothesis
Three ideas guided the pipeline:
- Partial truncations of the N-terminal region should reduce inhibition and increase lysis efficiency.
- The regulatory function is probably localized to a smaller sub-region rather than spread evenly across the entire N-terminus.
- There is likely an optimal truncation point where toxicity increases without destabilizing the membrane-spanning domain.
Pipeline Overview
| Stage | Tool | Purpose |
|---|---|---|
| 1 | ESM2 | Mutational scanning across all 75 residues |
| 2 | ESMFold | Structural prediction of truncation variants |
| 3 | AlphaFold-Multimer | Modeling interaction with DnaJ |
| 4 | GROMACS | Molecular dynamics and RMSF analysis |
| 5 | ProteinMPNN | Junction redesign and charge reduction |
| 6 | Codon optimization | Prepare E. coli expression constructs |
| 7 | Synthetic construct design | Assemble expression cassette |
| 8 | Bowtie2 + BCFtools | Variant calling and sequencing validation |
| 9 | IGV | Manual inspection of called variants |
Major Findings
ESM2 Mutagenesis Scan
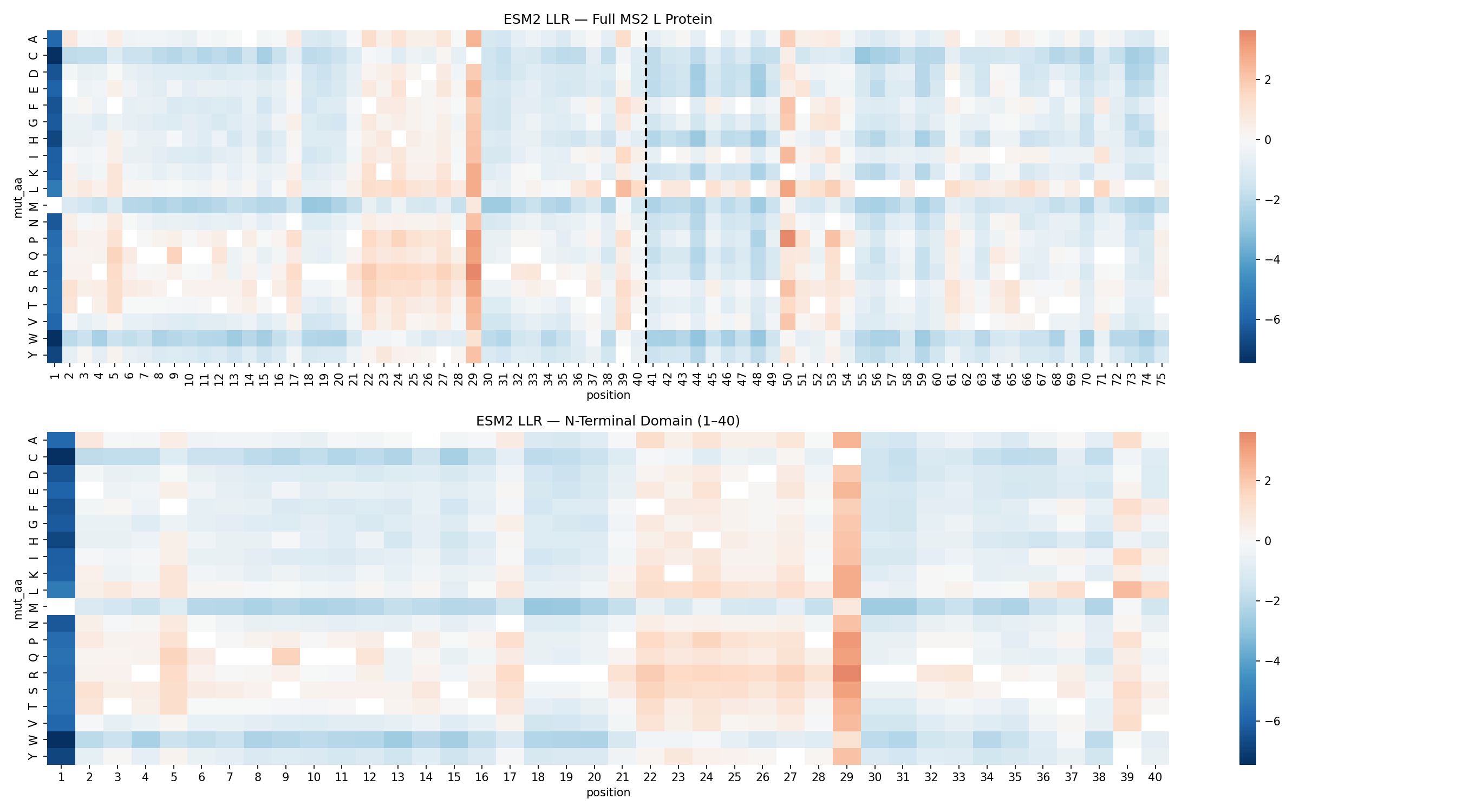
The ESM2 scan identified position C29 as the dominant mutational hotspot in the N-terminal domain.
| Mutation | LLR | Notes |
|---|---|---|
| C29R | 3.64 | Top-ranked substitution |
| C29P | 3.17 | Strong helix-disrupting mutation |
| C29Q | 3.06 | Conservative but highly favored |
| F22R | 1.86 | Introduces basic charge |
| S9Q | 1.69 | Recovered independently in prior work |
C29 accounted for 12 of the top 20 substitutions. That concentration strongly suggested that the wild-type residue at this site is not ideal for maximizing toxicity outside the native viral context.
Structural Findings
ESMFold predictions for all truncation variants suggested that the N-terminal domain is highly disordered in solution. Interdomain contact analysis returned essentially zero contacts across all variants, which fits with the known biology of the L protein.
The more useful signal came from molecular dynamics.
For L_trunc30:
- Remaining N-terminal stub RMSF: ~1.87 nm
- Transmembrane domain RMSF: ~0.27 nm
That sharp drop in flexibility confirmed that the transmembrane region remains stable even after removing 30 amino acids from the N-terminus.
Charge Analysis
The wild-type N-terminal region is strongly basic due to motifs like RRRPFK and RRQQR.
L_trunc30 reverses the overall charge profile:
| Variant | Net charge | Interpretation |
|---|---|---|
| Wild-type L | Approximately +8 | Strong DnaJ interaction expected |
| L_trunc30 | -2 | Reduced DnaJ binding and earlier lysis expected |
This was important mechanistically because DnaJ binding depends heavily on electrostatic interactions with the positively charged N-terminal region.
Codon Optimization and Construct Design
All major truncation variants were codon-optimized for E. coli K-12.
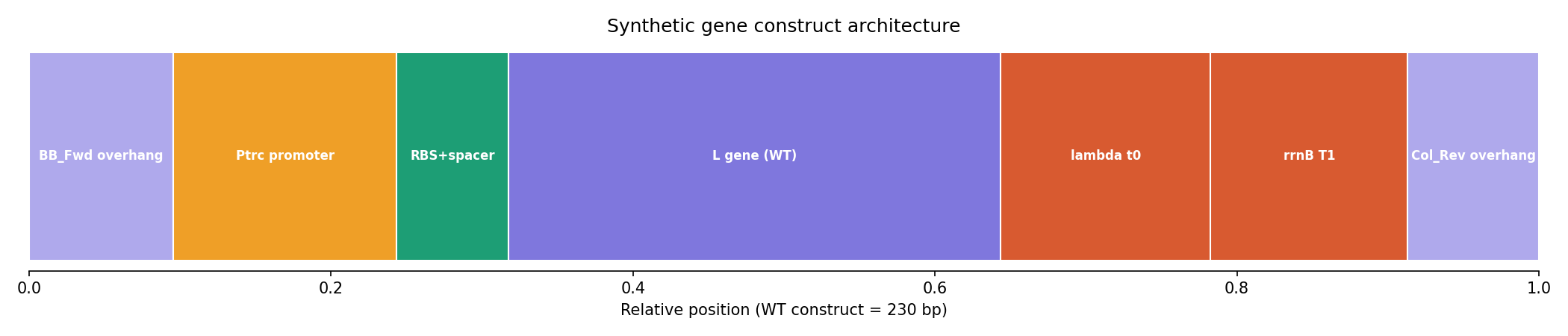
The lead construct, L_trunc30, preserved the essential LS motif and was assembled into a complete 230 bp expression cassette with:
- Ptrc promoter
- Optimized RBS
- Lambda t0 terminator
- rrnB T1 terminator
- Gibson overhangs compatible with the mUAV backbone
Lead Candidate
| Candidate | Key Feature | Reason |
|---|---|---|
| L_trunc30 | Removes aa 1-30 | Strongest balance of toxicity, structural stability, and DnaJ disruption |
Secondary Candidates
| Candidate | Reason for Inclusion |
|---|---|
| C29R | Highest ESM2 score overall |
| F22R | Adds positive charge in N-terminal region |
| S9Q | Recovered independently in previous scans |
| L_trunc40 | Most aggressive truncation, likely strongest toxicity |
GDrive Folder Depo: https://drive.google.com/drive/folders/17TE8ES8jUfnYL5irekBBFF2hsXrgr9lT?usp=sharing
Eric’s Analysis and Pipeline
Eric approached the same problem from a different angle. Instead of removing large sections of the N-terminus, he focused on identifying individual amino acid substitutions that could improve toxicity while preserving the overall structure of the protein.
His strongest candidate was P13L, a single amino acid change in the N-terminal region.
Pipeline Overview
| Stage | Tool | Purpose |
|---|---|---|
| 1 | UniProt + BLAST | Sequence retrieval and homolog identification |
| 2 | Clustal Omega | Conservation mapping |
| 3 | AlphaFold-Multimer | Oligomer modeling |
| 4 | ESM2 | Mutation scoring |
| 5 | ESMFold | Structural confidence and pTM analysis |
| 6 | ChimeraX | Electrostatic visualization |
| 7 | Benchling | ORF overlap analysis |
Major Findings
Conservation Analysis
Eric identified a relatively unconstrained region between amino acids 16 and 28 that could tolerate mutation without damaging essential structure.
| Position | Wild-type residue | Interpretation |
|---|---|---|
| 18 | R | Fully conserved, avoid |
| 21 | P | Fully conserved, avoid |
| 23 | K | Fully conserved, avoid |
| 26 | D | Variable, strong candidate |
| 13 | P | Weakly conserved, potentially safe |
Structural Modeling
P13L produced the strongest ESMFold result among all variants tested.
| Variant | pTM | Change vs WT |
|---|---|---|
| Wild-type | 0.273 | Reference |
| D26R | 0.267 | Slight decrease |
| P13L | 0.420 | Strong increase |
The jump from 0.273 to 0.420 made P13L the most structurally favorable point mutation in Eric’s pipeline.
Experimental Cross-Reference
Unlike my pipeline, Eric cross-referenced computational candidates with available lysis data.
| Mutation | Replicate A | Replicate B | Result |
|---|---|---|---|
| P13L | 1 | 1 | Confirmed lytic |
| D26G | 1 | 0 | Mixed |
| K23E | 1 | 0 | Mixed |
| E25G | 1 | 0 | Mixed |
P13L was the only candidate to remain consistently positive across both replicates.
ORF Overlap Analysis
One of the more interesting parts of Eric’s work was the DNA-level overlap analysis.
P13L falls within the overlap region between the coat protein and the L protein, which initially made it look risky. After codon-level analysis, though, the mutation turned out to be safe.
| Gene | WT codon | Mutant codon | Result |
|---|---|---|---|
| L protein | CCG | CTG | Pro → Leu |
| Coat protein | TCC | TCT | Ser → Ser |
That synonymous change in the coat protein meant the mutation could proceed without disrupting the overlapping reading frame.
Lead Candidate
| Candidate | Key Feature | Reason |
|---|---|---|
| P13L | Single amino acid substitution | Best structural score and strongest experimental support |
Secondary Candidates
| Candidate | Status |
|---|---|
| D26R | Untested but promising |
| D26G | Mixed experimental results |
| N17R | Open candidate |
| H24R | Open candidate |
Albert’s Notes
Albert focused primarily on structural stability.
His workflow emphasized:
- Sequence retrieval from UniProt
- BLAST and Clustal Omega for conservation mapping
- ESM2 mutational scanning
- ESMFold structure prediction
- AlphaFold-Multimer confirmation of DnaJ interactions
- Wet lab validation of top-ranked variants
His key concern was preserving structure while introducing beneficial mutations.
He also pointed out an important limitation that kept showing up across the project: membrane proteins are underrepresented in both structural databases and protein language model training sets. That means even high-scoring mutations should still be interpreted cautiously.
Tehseen’s Notes
Tehseen’s approach aligned closely with my truncation-based strategy but focused more on identifying the smallest regulatory segment required for precise control over lysis timing.
The central idea was not simply to remove the N-terminal region, but to identify exactly which residues are responsible for slowing lysis.
That led to three closely related hypotheses:
- Partial truncations can increase lysis gradually rather than all at once.
- Regulatory effects are probably localized to a smaller sub-region.
- There is likely an optimal balance point between stronger toxicity and preserved protein stability.
Comparative Summary
| Aspect | John’s Pipeline | Eric’s Pipeline |
|---|---|---|
| Main strategy | Progressive N-terminal truncation | Point mutation design |
| Lead candidate | L_trunc30 | P13L |
| Core hypothesis | Remove inhibitory domain | Increase local electrostatic effects |
| ESM2 scope | Full 1,425-substitution scan | Single-site targeted analysis |
| Structural analysis | ESMFold + GROMACS RMSF | ESMFold + ChimeraX |
| DnaJ interaction | Central to model | Considered indirectly |
| Experimental validation | Not yet completed | P13L confirmed experimentally |
| Construct design | Fully assembled | Still planned |
| Sequencing workflow | Fully designed with Bowtie2, BCFtools, IGV | Listed as future step |
Final Interpretation
The project ended up producing two very different but complementary engineering directions.
L_trunc30 represents the stronger systems-level redesign. It removes the inhibitory N-terminal region, reduces DnaJ engagement, preserves the transmembrane core, and provides a fully buildable expression construct ready for synthesis and sequencing validation.
P13L represents the cleaner minimal-change strategy. It preserves the full-length protein, improves structural confidence, survives ORF overlap analysis, and already has positive experimental support.
If the goal is maximum disruption of the native regulatory system, L_trunc30 is the stronger candidate.
If the goal is a simpler mutation with lower engineering risk and existing wet lab support, P13L is the better starting point.
The most practical next step would be to synthesize and compare both side by side.