Projects

Final projects
Fitness-constrained design of a probiotic sentinel: a quantitative framework for circuit governability under evolutionary pressure.
HTGAA Group Project: Engineering the MS2 Bacteriophage L Protein
Fitness-constrained design of a probiotic sentinel: a quantitative framework for circuit governability under evolutionary pressure.
HTGAA Group Project: Engineering the MS2 Bacteriophage L Protein
ÌṢỌ — Yoruba: to be well; to recover. A fitness-aware engineered probiotic designed to sense gut context, respond with targeted antimicrobials, and remain governable by design.
Childhood diarrhoeal disease kills roughly half a million children under five every year, and the majority of those deaths happen in sub-Saharan Africa. During clinical training in Osogbo, the treatment options were ORS, zinc, and empirical antibiotics. Effective, but blunt. The gap is not a shortage of therapeutics. It is a precision problem.
How do we design microbial circuits that remain governable under the evolutionary and ecological pressures of a real gut environment?
The existing engineered probiotic literature optimises for peak performance under ideal conditions. ÌṢỌ maps design regimes: what works, what breaks, and what stays stable as conditions shift.
ÌṢỌ is a four-module sense-respond-contain system built on E. coli Nissle 1917 (EcN):
| Module | Component | Role |
|---|---|---|
| Biosensor | TtrS/TtrR two-component system | Detects tetrathionate, a pathogen-associated signal produced during gut inflammation by Salmonella and E. coli O157:H7 |
| Regulator | Thresholded Hill-function promoter | Gates activation; suppresses leaky expression and reduces fitness cost at homeostatic baseline |
| Effector | Microcin H47 (MccH47) | Narrow-spectrum antimicrobial; ATP synthase inhibition; active against Salmonella, Shigella, pathogenic E. coli; endogenous immunity protein MchI in EcN chassis |
| Containment | deltaDAPA auxotrophy | DAP absent from mammalian gut; deletion is lethal without exogenous supply; escape frequency ~10^-8 per generation |
Optimise for stability, not just performance. The output is not a single optimal construct. It is a map of design regimes: parameter regions where the circuit functions, where it fails under burden, and where containment holds under selection pressure.
Computational modelling only. Wet-lab validation, full microbiome simulation, and clinical deployment are explicitly out of scope for this phase.
The first aim of my final project is to build and simulate a genome-scale metabolic and circuit-level ODE model of the four-module ÌṢỌ architecture by utilising Tellurium/libroadrunner for time-course simulation, SALib for global sensitivity analysis, and NumPy-based Moran process modelling for evolutionary containment stability, generating a Pareto-resolved fitness-efficacy landscape and a ranked parameter influence analysis as the primary computational output.
Following a successful Aim 1, the top-ranked parameter regimes from the Pareto landscape will guide assembly and transformation of the sense-respond-contain circuit into EcN. The engineered sentinel will be tested in co-culture assays against Salmonella Typhimurium and E. coli O157:H7, validating both the tetrathionate-sensing threshold and MccH47-mediated kill kinetics experimentally. Discrepancies between model predictions and wet-lab data will feed back into model refinement.
The long-term goal is a rugged, orally delivered live biotherapeutic that operates autonomously in the gut, activates only in the presence of pathogen-associated tetrathionate, kills narrow-spectrum without collateral microbiome disruption, and cannot persist outside the host. If the fitness-governability framework holds, ÌṢỌ becomes a design methodology applicable beyond this specific pathogen set, with direct relevance for AMR management, inflammatory bowel disease, and cancer immunotherapy in low-resource clinical settings.
Palmer et al. (2017, ACS Infectious Diseases) demonstrated that EcN can be engineered to sense gut-luminal tetrathionate via the TtrS/TtrR two-component system and produce Microcin H47 in response, achieving measurable Salmonella inhibition in a mouse colonisation model. Critically for ÌṢỌ, this paper provides experimentally validated, ODE-parameterisable values for sensor activation kinetics, MccH47 production rates, and pathogen kill constants, making it the direct quantitative predecessor to this project rather than simply a conceptual reference.
Stritzker et al. (2007, International Journal of Medical Microbiology) characterised deltaDAPA auxotrophy in EcN in detail, reporting an escape frequency of approximately 10^-8 per generation under DAP-free conditions. That specific number is what makes the containment module computationally tractable: escape probability can be directly parameterised in the Moran process model rather than estimated from first principles.
What ÌṢỌ does that neither paper does is treat fitness cost as a first-class design variable rather than a post-hoc observation. Every published EcN engineering study acknowledges metabolic burden; none model it explicitly as a design input alongside efficacy. ÌṢỌ builds a Pareto frontier that makes the tradeoff navigable rather than anecdotal. The containment module also moves from binary characterisation (auxotrophy is present or absent) to a dynamic system property, asking how quickly a loss-of-function mutant fixes in a finite population over evolutionary time.
Diarrhoeal disease causes approximately 1.6 million deaths per year globally, with the under-five burden concentrated in West and East Africa. Nigeria alone accounts for a disproportionate share of this mortality. Existing interventions reduce severity but do not prevent recurrence in high-transmission settings, and empirical antibiotic use is accelerating resistance emergence in the pathogens most responsible for paediatric deaths: Salmonella, enterotoxigenic E. coli, and Shigella. A sentinel probiotic that activates conditionally, kills narrow-spectrum, and cannot persist outside the host addresses this without adding to AMR pressure.
Beyond the immediate clinical problem, the fitness-governability framework ÌṢỌ develops has broader implications. Any engineered living therapeutic faces the same core question: will the circuit hold under the evolutionary pressure of a real biological environment? Current regulatory frameworks for live biotherapeutics have no standardised computational tool for answering this before a clinical trial. ÌṢỌ begins building one. Nigerian and broader West African epidemiological data (Egbewale 2022; Gayawan 2024) are used to parameterise disease burden and clinical context from the start, not as a framing afterthought.
Two principles are directly engaged here: beneficence and justice. A precision antimicrobial that spares the commensal microbiome and cannot persist outside the host is strictly better than empirical broad-spectrum antibiotics for the patient, for the microbiome, and for the resistance landscape. Research that addresses paediatric mortality in West Africa while remaining computationally grounded in West African epidemiology represents a genuine departure from the default of developing interventions for high-income contexts and adapting them downstream.
The risks require honesty. A single deltaDAPA deletion is probably not sufficient for any real-world deployment. The current model assumes a closed population and does not account for horizontal gene transfer of the dapA gene from environmental bacteria. The Moran process also excludes commensal competition dynamics, so estimates of circuit persistence are optimistic. These are known limitations, explicitly scope-bounded to this computational phase. Non-maleficence requires that these caveats travel with any communication of the results. Open-source model release via GitHub (MIT licensed) is a deliberate act toward equitable access to the methodology.
Chosen: tetrathionate via TtrS/TtrR two-component system. Pathogen-specific: Salmonella and E. coli O157:H7 produce tetrathionate during gut inflammation via reactive oxygen species. Experimentally validated in EcN (Palmer et al. 2017). Signal is absent under homeostatic conditions, directly minimising leaky expression burden at baseline.
Chosen: Microcin H47 (MccH47). Naturally produced by EcN. Narrow-spectrum: E. coli, Salmonella, Shigella. Mechanism is ATP synthase inhibition, a well-characterised mode of action enabling direct ODE kill-kinetics parameterisation. Immunity protein MchI is endogenous to the EcN chassis. Palmer 2017 provides benchmarked production and kill-rate values for exactly this design.
Chosen: deltaDAPA auxotrophy (diaminopimelic acid / DAP). DapA is essential for lysine and peptidoglycan synthesis. DAP is absent from the mammalian gut: no dietary source, no commensal production. Deletion is lethal without exogenous supply. Validated in EcN (Stritzker et al. 2007). Published escape frequency ~10^-8 per generation is directly parameterisable for the containment escape model.
Chosen: Tellurium + libroadrunner (SBML/Antimony).
Purpose-built for systems biology ODE modelling. Antimony syntax maps directly onto circuit topology (promoter to mRNA to protein). libroadrunner’s stiff CVODE solver handles fast mRNA turnover and slow protein accumulation dynamics without manual configuration. SBML export makes every model citable and reproducible. SciPy solve_ivp (LSODA flag) runs in parallel for parameter sweeps and Pareto grid computation.
Primary: PRCC via SALib (Marino et al. 2008). Designed for nonlinear, monotonic systems, exactly what Hill-function gene circuits produce. 500 to 2000 Latin hypercube samples sufficient for 6 to 8 parameters.
Supplementary: Sobol total-order indices. Captures interaction effects (Hill coefficient n and KD interact in the sensor module). 5000 to 10000 samples, tractable on a laptop in minutes.
Chosen: Moran process with fitness-weighted selection.
Two competing types: functional circuit (fitness 1 minus delta) and loss-of-function mutant (fitness 1). Fixation probability computed analytically (Nowak 2006), then 1000 stochastic trajectories via numpy.random.choice() with fitness-weighted birth-death events. Directly answers: how long does the circuit remain functional under selection pressure?
Tellurium + libroadrunner — all four-module ODE construction and time-course simulation written in Antimony syntax. SBML export for reproducibility and citability.
SciPy solve_ivp (LSODA) — parameter sweeps and Pareto grid computation. LSODA auto-switches between stiff and non-stiff regimes.
SALib — PRCC for main figures, Sobol as supplementary. Canonical citation: Marino et al. 2008, J. Theor. Biol.
NumPy random.choice() — Moran process. Fitness-weighted birth-death events across 1000 independent trajectories. No additional dependencies.
Matplotlib + Seaborn (seaborn-whitegrid). Pareto scatter, PRCC tornado chart, containment escape semi-log, Moran fixation fan. Exported at 300 dpi PNG and SVG.
GitHub + venv + requirements.txt (pinned exact versions). One command regenerates all figures. MIT licensed. CITATION.cff included.
Install and configure the full modelling stack: Tellurium, libroadrunner, SALib, NumPy, Matplotlib, Seaborn, SciPy, all pinned in a venv with requirements.txt. Build the biosensor module as a two-ODE Hill-function model encoding the TtrS/TtrR tetrathionate-to-promoter activation pathway. Fit activation threshold KD and Hill coefficient n against Palmer 2017 time-course data.
Expected result: Simulated sensor activation curve matches digitised Palmer 2017 experimental data within 20% across the measured tetrathionate concentration range.
Actual findings


Extend the biosensor ODE to include the regulator module (thresholded Hill-function promoter gating effector expression), the effector module (MccH47 production and pathogen kill kinetics), and the containment module (deltaDAPA escape probability). Write all models in Antimony syntax within Tellurium. Export validated models to SBML and commit to GitHub.
Expected result: Stable steady-state solutions for all four modules under both homeostatic and pathogen-present conditions. Leaky expression at baseline should approach zero.
Actual findings

Use SciPy solve_ivp with LSODA flag to sweep burden parameter delta and effector output rate k_M across a 50 x 50 parameter grid. Record steady-state growth rate and pathogen suppression ratio for each grid point. Plot Pareto frontier, colour-coded by regulator variant (linear vs. thresholded).
Expected result: A visible Pareto frontier separating viable design space from over-burdened and under-effective regions. The thresholded regulator variant should dominate the frontier.
Actual findings

Run PRCC analysis via SALib using Latin hypercube sampling across 6 to 8 parameters: Hill coefficient n, signal threshold KD, burden delta, MccH47 production rate k_M, pathogen kill rate k_kill, mRNA degradation rate, and protein dilution rate. Generate ranked tornado chart. Run supplementary Sobol total-order index analysis.
Expected result: n and KD rank as the top two PRCC drivers of sensor module output. Sobol indices confirm a significant interaction effect between the two.
Actual findings

Implement the Moran process in NumPy. Define two competing cell types (functional circuit, fitness 1 minus delta; loss-of-function mutant, fitness 1). Compute analytical fixation probability from Nowak 2006. Run 1000 stochastic trajectories. Vary delta across the Pareto-viable range; plot fixation probability across three population sizes with analytical solution overlaid.
Expected result: Fixation probability of the loss-of-function mutant increases sharply above delta = 0.1. This is the quantitative argument for why the thresholded regulator module is not optional.
Actual findings

| Company | Role |
|---|---|
| Asimov (Kernel) | Validate Pareto landscape and containment circuit architecture; independent cross-check against Tellurium ODE results |
| SecureDNA | Screen all DNA sequences (mccH47, deltaDAPA cassette) before synthesis |
| Cultivarium | EcN-specific transformation protocols and characterised parts for Aim 2 |
| Twist Biosciences | Codon-optimised construct synthesis for Aim 2 |
| Opentrons | Co-culture assay automation for Aim 2 parallel screening |
Burden parameter delta and effector output k_M swept across a 50 x 50 parameter grid. Each point represents steady-state growth rate and pathogen suppression ratio. Pareto frontier overlaid. Colour-coded by circuit variant (linear vs. thresholded regulator). This figure makes the design regime concept concrete: the viable parameter space, the over-burdened region, and the under-effective region visible in a single plot. No equivalent exists in the published EcN engineering literature.
PRCC bar chart ranked by absolute influence on steady-state pathogen suppression. Parameters: Hill coefficient n, signal threshold KD, burden delta, microcin production rate k_M, pathogen kill rate k_kill. Sobol indices shown as supplementary to capture n-KD interaction effects. k_M and k_kill dominate pathogen suppression output; n and K_D dominate TtrR* biosensor output. These are distinct design problems: effector kinetics governs therapeutic outcome at physiological signal concentrations, while sensor cooperativity governs specificity at sub-threshold concentrations.
Semi-log plot of escape frequency vs. generations. Single deltaDAPA vs. dual deltaDAPA + deltaThyA auxotrophy compared. Analytical curve overlaid on stochastic simulation trajectories. Anchored to published escape frequency ~10^-8 per generation (Stritzker 2007).
Fixation probability of loss-of-function mutant as a function of burden delta, across three population sizes. 1000-trajectory stochastic fan with analytical Nowak 2006 solution overlaid. Demonstrates that the thresholded regulator (Module 2) extends functional circuit half-life under selection relative to constitutive expression: the quantitative argument for why the regulator module is not optional.
The computational modeling pipeline constitutes the primary validation for this project. Specifically, the four-module ODE system was constructed and simulated in Tellurium/libroadrunner, producing a Pareto-resolved fitness-efficacy landscape and a ranked parameter sensitivity analysis via PRCC — directly fulfilling the rubric requirement of “developing a model or completing a computational analysis relevant to your project.” This approach was chosen because the project is explicitly model-first: the computational output is not a precursor to the real work but is the deliverable itself, generating design guidance that no single wet-lab experiment at this stage could produce.
The primary technique is quantitative ODE-based modeling of a synthetic gene circuit, which is a standard systems-level synthetic biology method for predicting circuit behaviour before construction. Global sensitivity analysis via PRCC is used to identify which biological parameters most strongly influence circuit performance — a technique directly analogous to experimental design of variation (DOE) approaches used in wet-lab strain engineering. Evolutionary stability modeling via the Moran process applies population genetics theory to assess whether the engineered circuit remains stable under natural selection pressure, which is a synthetic biology governance question increasingly recognised as essential for live biotherapeutic design. Together these constitute a computational Design-Build-Test-Learn cycle: the model is the design, the simulation is the build, the Pareto and sensitivity outputs are the test, and the parameter refinement loop is the learn step.
The primary data output is the Pareto frontier plot: a two-dimensional scatter of burden δ (fitness cost axis) vs. pathogen kill rate (antimicrobial function axis) across the swept parameter space, with the Pareto-optimal boundary overlaid. Each point on this plot represents a distinct circuit parameter combination, and the frontier identifies the set of designs where no further improvement in kill rate is achievable without increasing fitness cost. The PRCC tornado chart ranks parameter influence on steady-state pathogen suppression; k_M and k_kill dominate at ranks 1 and 2, while n and K_D rank as the top two drivers of TtrR* biosensor output specifically — a distinction that matters because sensor sensitivity and effector potency are both necessary but operate on different parts of the circuit. The containment escape probability semi-log plot shows that ΔdapA single auxotrophy maintains escape frequency below 10⁻⁸ per generation for at least 200 generations under modeled selection pressure.
The primary limitation of a purely computational validation is parameter uncertainty: kinetic constants for MccH47 production and TtrR activation are drawn from Palmer et al. 2017, which used a different construct architecture and growth conditions than those modeled here. Transferring parameters across experimental contexts introduces uncertainty that cannot be resolved without wet-lab measurement, and the model outputs should be interpreted as design guidance rather than quantitative predictions. A second limitation is the absence of spatial heterogeneity — the ODE framework assumes a well-mixed population, whereas the gut is spatially structured, meaning colonisation dynamics and local tetrathionate gradients are not captured. A third challenge specific to the Moran process implementation is the fixed-population-size assumption, which does not account for the population bottlenecks and variable colonisation densities characteristic of EcN in a gut context; a variable-population birth-death process would be more realistic but requires additional parameterisation not available in the current literature. To address these limitations in a future phase, the ODE parameters would be updated with experimentally measured values from the cell-free expression and co-culture validation steps described in Aim 2, and the Moran model would be extended to a variable-population Wright-Fisher simulation with gut-realistic bottleneck sizes drawn from published EcN colonisation data.
| Parameter | Symbol | Value | Bounds | Source |
|---|---|---|---|---|
| Max promoter output | α_max | 12.0 rel. units | fixed | v6 calibration |
| Leaky expression | α_leak | 0.24 (2% α_max) | 1–5% α_max | v6 fix; >10% collapses FI |
| Tetrathionate EC50 | EC50 | 20 µM | 5–50 µM | Palmer 2017; right-shifted from 10 µM for gut conservatism |
| Hill cooperativity (biosensor) | n | 2 | 1–3 | Two-component phosphorelay physiology |
| TtrR production rate | k1 | 0.426 h⁻¹ | fixed | J23115 promoter + medium copy |
| Basal TtrR leak | k1_leak | 0.002 | fixed | Reduced from 0.01 to prevent OFF-state floor inflation |
| TtrR degradation | d_TtrR | 0.1 h⁻¹ | fixed | Standard bacterial protein turnover |
| sfGFP degradation | d_sfGFP | 0.05 h⁻¹ | fixed | sfGFP stability in EcN |
| Hill constant (sensor→reporter) | Km | 0.3 | ≥0.3 | Lower bound enforced; Km=0.1 shifts sigmoid left of physiological window |
| Regulator gate threshold | threshold_TtrR | 2.0 rel. units | fixed | TtrR_ss at EC50 (20 µM); 50% induction |
| Gate sharpness | n_reg | 3 | fixed | Near-digital switching; suppresses sub-threshold leaky effector |
| MccH47 production rate | k_M | 0.2 µM/h | 0.01–0.5 µM/h (Pareto sweep) | Palmer 2017 mid-range |
| MccH47 degradation | d_M | 0.05 h⁻¹ | fixed | Peptide stability estimate |
| Pathogen kill rate | k_kill | 0.3 µM⁻¹ h⁻¹ | fixed | Palmer 2017 gut-corrected (original 0.05 underestimated suppression) |
| Pathogen growth rate | k_growth | 0.5 h⁻¹ | fixed | Salmonella Typhimurium in gut (~35 min doubling) |
| Plasmid copy number | CN | 20 copies/cell | 5–50 | pSC101 medium copy range |
| Burden per copy | δ/copy | 0.0015 | fixed | Scott et al. 2010 ribosome allocation model |
| Circuit fitness cost | δ | 0.03 (3%) | ≤0.10 | Derived: CN × δ/copy |
| ΔdapA escape frequency | μ_escape | 10⁻⁸ per generation | fixed | Stritzker 2007 |
| EcN generation time | t_gen | 0.5 h | fixed | ~30 min doubling in gut |
| Metric | Value | Target | Status |
|---|---|---|---|
| Fold induction (observed) | 41.5× | ≥10× | ✓ |
| sfGFP steady state (ON, 50 µM) | 12.16 rel. units | >10 | ✓ |
| sfGFP steady state (OFF, 0 µM) | 0.293 rel. units | <1.0 | ✓ |
| t50 (sfGFP, ON state) | 15.3 h | 8–30 h | ✓ |
| Pathogen suppression at 200 h (50 µM S) | 100% | ≥90% | ✓ |
| MccH47 homeostatic steady state | 0.0000 | ~0 | ✓ |
| Pathogen homeostatic steady state | 1.0000 | ~1 | ✓ |
| Sub-threshold suppression (2 µM) | 0.0% | <50% | ✓ |
| Pareto frontier points (thresholded) | 42 | >0 | ✓ |
| Pareto frontier points (linear) | 39 | >0 | ✓ |
| Moran extinction rate (δ=0.03) | 95.1% | >80% | ✓ |
| Analytical vs stochastic RMSE | 0.0175 | <0.05 | ✓ |
| Containment escape t50 (single ΔdapA) | 3,956 years | >1,000 years | ✓ |
| Module 2 lifetime advantage | 2.0× | >1× | ✓ |
| Relationship | Finding | Biological implication |
|---|---|---|
| α_leak → FI | FI = (α_max + α_leak) / α_leak; at 10% leak FI collapsed to 5.5×; at 2% leak FI = 41.5× | Leaky expression is the single most destabilising parameter for therapeutic gating. Even small absolute increases in basal expression dramatically erode the ON/OFF discrimination needed to prevent activation in healthy gut |
| EC50 → activation window | Sigmoid sits within 10–100 µM physiological window at EC50 = 20 µM; shifts dangerously left at EC50 = 10 µM | EC50 must be calibrated to gut tetrathionate levels during active infection, not laboratory buffer conditions. Under-estimating EC50 risks false activation in sub-pathological inflammatory states |
| n (Hill) → switch sharpness | n=2 gives cooperative switching; n=1 gives graded non-zero baseline output at low S | Hill cooperativity is what separates a sensor from a rheostat. n=2 is the minimum for therapeutically meaningful gating in a two-component phosphorelay system |
| Km → left-shift risk | Km=0.1 activates below physiological window; Km ≥ 0.3 keeps sigmoid correctly anchored | Km sets the TtrR concentration at which sfGFP output is half-maximal. Reducing Km below 0.3 is equivalent to making the effector more sensitive than the sensor — a design inversion that breaks the gating logic |
| k_M + k_kill → Pareto position | k_M is top PRCC driver (0.733); k_kill is second (0.671); both dominate Sobol ST | Effector module output, not sensor sensitivity, is rate-limiting for therapeutic function. Investment in MccH47 production fidelity and secretion efficiency returns more suppression gain than further sensor optimisation |
| δ → Moran fixation | P_fix rises from 0.03 at δ=0.03 to 0.10 at δ=0.10; circuit half-life halves from 3,956 to 1,978 years | There is a 3.3× safety margin between the design operating point (δ=0.03) and the critical threshold (δ=0.10) above which loss-of-function fixation becomes ecologically relevant. The thresholded regulator preserves this margin by suppressing expression at baseline |
| n_reg → gate fidelity | n_reg=3 gives near-digital switching; sub-threshold MccH47 = 0.0001 µM (effectively zero) | Regulator sharpness is what enforces the therapeutic safety contract. A graded regulator (n_reg=1) would permit low-level MccH47 production at all tetrathionate concentrations, including homeostatic |
| μ_escape + dual auxotrophy | Single ΔdapA: t50 = 3,956 years; dual ΔdapA+ΔthyA: escape probability near zero across 10¹² generations | Containment robustness scales super-linearly with additional auxotrophies. The second deletion multiplies escape improbability rather than adding to it, because both reversions must occur independently |
| PRCC vs Sobol rank agreement | Top-4 ranks identical across methods: k_M, k_kill, d_M, Km_reg | Consistency between PRCC and Sobol ST confirms the model is not artefactually sensitive to the choice of global sensitivity method. The ranking is a structural property of the circuit, not an analysis artefact |
| Signal-dependent sensitivity | At S=2 µM top driver is n_reg; at S≥50 µM top driver is k_M | Below threshold, gate architecture controls leakage; above threshold, effector kinetics controls outcome. These are two distinct design problems, not one |
Biosensor layer
Regulator layer
Effector layer
Burden and copy number
Containment
Cross-layer interactions confirmed by Sobol S2
A fully interactive browser-based ODE simulation is deployed and accessible via the Cloudfare interface and linked below:

The simulator runs entirely client-side and implements a four-module Runge–Kutta (RK4) integration framework for solving a coupled dynamical system governing gut–pathogen interactions. No server infrastructure or external compute backend is required.
The interface exposes nine mechanistic parameters through real-time sliders, enabling continuous modulation of system state. Each user interaction triggers full re-evaluation of the ODE system and updates five derived biological observables:
Together, these outputs provide a live mapping between parameter space and emergent ecological–immunological dynamics, allowing direct exploration of the nonlinear nature, stability transitions, and intervention sensitivity.

















The clinically relevant output — therapeutic circuit functional lifetime weighted by suppression efficacy:



The model-first architecture proved its value immediately. By fixing circuit topology and parameter ranges computationally before any construct design, every subsequent modelling decision had a clear biological anchor. The pre-Aim-2 checklist with nine pass/fail criteria was particularly useful: it made version transitions (v1 through v6) traceable and prevented premature progression on a flawed parameterisation.
The decision to ground leaky expression as an explicit parameter rather than a derived residual was the single most consequential fix across the entire project. Fold induction swung from 2×10⁸ (numerically meaningless) to 5.5× (biologically insufficient) to 41.5× (therapeutically viable) across six model iterations, solely because of how α_leak was handled. This taught a general lesson: in Hill-function gene circuit models, the denominator of the fold induction ratio is always the most sensitive and least constrained quantity, and it should be the first thing fixed, not the last.
PRCC and Sobol agreement on the top-four parameter ranking (k_M, k_kill, d_M, Km_reg) was a genuinely satisfying result. Convergence across two independent global sensitivity methods on 36,864 model evaluations gives confidence that the ranking reflects the biology rather than sampling noise.
The alpha_leak inflation problem (v1–v5). Setting α_leak as a percentage of α_max sounded correct but produced an OFF-state floor of 2.4 relative units when α_leak = 1.2 — because the term adds directly to basal sfGFP production regardless of TtrR. The FI calculation was not wrong; the model was not wrong; the parameterisation assumption was wrong. Six versions were required to isolate this. The fix (reduce to 2%, reduce k1_leak simultaneously) resolved it in one step once the root cause was identified.
k_kill underestimation (v1 Task 2). The initial k_kill = 0.05 µM⁻¹ h⁻¹ was taken from Palmer 2017 without accounting for the dilution and diffusion losses expected in a gut lumen environment. This produced suppression results that passed the checklist (≥90%) but for the wrong reason — the model was not reflecting realistic gut pharmacodynamics. Correcting to k_kill = 0.3 µM⁻¹ h⁻¹ produced 100% suppression at 200 h, which while still not validated, is more defensible as a design-space exploration.
The Moran overflow warning. The RuntimeWarning: overflow encountered in scalar power during fixation probability computation for large N values (N=10,000) reflects a numerical limitation of the analytical Nowak 2006 formula at high population sizes and low δ: (1/r)^N underflows to zero for large N, producing a 0/0 division. The stochastic trajectories are unaffected, but the analytical curve is unreliable for N>10,000 at δ<0.05. This was resolved in the final pipeline via a log-space rewrite of the analytical formula: the numerator simplifies exactly to δ, and the denominator is evaluated as 1 − exp(N × log(1−δ)) using np.log1p for precision near zero. No RuntimeWarning appears in the corrected implementation, and the eighth checklist item confirms numerical validity at N=10,000 explicitly.
SVG rendering on the HTGAA page. Multiple attempts to embed SVG figures failed across all approaches (bare filename, static path, Gitea raw URL, inline SVG blocks). The root cause is a combination of Hugo Relearn’s branch bundle scoping, the shared Hugo instance’s Goldmark unsafe rendering restriction, and CORS policy on cross-origin SVG in <img> tags. PNG via Gitea raw URL resolved the rendering problem entirely. The lesson is that SVG is the right format for archival and journal submission but not for Hugo-served portfolio pages on shared infrastructure.
The n–EC50 interaction expectation mismatch. The project page stated that n and EC50 would rank as the top two PRCC drivers of biosensor output — which is true when the output metric is TtrR* steady state. When the output metric is pathogen suppression (the more clinically relevant quantity), n and EC50 fall to ranks 7 and 6 respectively, because the effector module (k_M, k_kill) dominates end-to-end. This is not an error but a framing imprecision in the original expected results statement, and it reflects an important biological truth: sensor sensitivity and effector potency are both necessary but effector kinetics is the rate-limiting step for therapeutic outcome at physiological signal concentrations.
The model assumes a well-mixed, spatially homogeneous gut compartment. Real gut colonisation involves spatial gradients of tetrathionate, mucus diffusion barriers, and microbiome neighbourhood effects none of which are captured here. The Moran process also assumes a fixed population size and ignores the population bottlenecks that occur during gut transit and colonisation. Both simplifications make the containment and stability estimates optimistic. These are not failures of the current work — they are the next layer of modelling complexity that a preprint would need to address honestly in its limitations section.
Computational (current phase — no cost)
Wet-lab validation phase (Aim 2 — estimated)
Estimated total for Aim 2 wet-lab phase: ~$1,200–$1,800
The engineered probiotic field asks: can we build a circuit that works?
ÌṢỌ asks: across what design regimes does a circuit remain both functional and governable under the pressures that will actually be present?
The models, figures, and write-up constitute the core of a bioRxiv preprint. Abstract, introduction, and discussion sections bring it to a citable first-author computational biology paper.
Add a simplified Lotka-Volterra competition term for commensal species. Explores how microbiome density affects EcN colonisation stability and circuit persistence under realistic ecological conditions.
Apply the PepMLM/moPPIt peptide generation pipeline (HTGAA Week 5) to propose microcin-analog sequences with improved target specificity. AlphaFold3 structural prediction of microcin-pathogen outer membrane protein complexes bridges the computational peptide design and engineered probiotic work.
Parameterise the pathogen kill model with AMR prevalence data from Nigerian clinical isolates (WHONET/GLASS). Grounds the model in Sub-Saharan African epidemiology and connects to the planned AMR West Africa genomic data paper.
All model code, SBML files, and figures. MIT licensed. CITATION.cff included.
The construct below reflects the finalised Benchling assembly: 8,323 bp, pSC101_KanR backbone, verified in both linear and circular representations. All part identifiers are confirmed iGEM Registry parts as assembled.
Assembled and visualised in Benchling (pTtr-sfGFP_TtrSR_EcN_v1, 8,323 bp). Linear and circular maps shown below. Computational parameter regime from Tasks 1–5 defines the performance targets this construct must hit experimentally in Aim 2.


| Position (bp) | Part | Identity | Role |
|---|---|---|---|
| 1–4,200 | pSC101_KanR_MCS | pSC101 origin + KanR resistance | Low-copy backbone (5–20 copies/cell); kanamycin selection; MCS for cloning |
| ~4,200 | BBa_J23115 | Constitutive promoter (medium-strong) | Drives TtrR expression; matched to k1=0.426 in Task 1 ODE |
| ~4,450 | BBa_B0031 | RBS (medium strength) | Ribosome binding site for TtrR translation |
| ~4,500–5,200 | ttrR_protein | TtrR response regulator (S. Typhimurium LT2) | Receives phosphoryl group from TtrS; activates pTtr_LT2 promoter |
| ~5,200 | BBa_B0015 | Double terminator | Terminates TtrR transcription; prevents read-through |
| ~5,250 | BBa_J23106 | Constitutive promoter (medium) | Drives TtrS expression |
| ~5,300 | BBa_B0031 | RBS (medium strength) | Ribosome binding site for TtrS translation |
| ~5,350–6,700 | ttrS_protein | TtrS sensor histidine kinase (S. Typhimurium LT2) | Senses tetrathionate; autophosphorylates; transfers phosphoryl to TtrR |
| ~6,700 | BBa_B0015 | Double terminator | Terminates TtrS transcription |
| ~6,750 | pTtr_Salmonella_LT2 | TtrR-activated inducible promoter | Output promoter; activated only when TtrR* exceeds threshold; EC50=20 µM tetrathionate |
| ~6,950 | BBa_B0031 | RBS (medium strength) | Ribosome binding site for sfGFP translation |
| ~7,000–7,700 | BBa_I746916 | sfGFP (superfolder GFP) | Fluorescence reporter; validates sensor-to-output pathway; proxy for MccH47 expression in Aim 2 |
| ~7,700 | BBa_B0015 | Double terminator | Terminates sfGFP transcription; end of expression cassette |
Tetrathionate (S) present in gut lumen → TtrS autophosphorylation (J23106 → TtrS constitutive) → TtrR phosphorylation (J23115 → TtrR constitutive) → TtrR* accumulates above Km_reg threshold (2.0 rel. units) → pTtr_Salmonella_LT2 activated → sfGFP expressed (reporter) → [Aim 2: MccH47 replaces/co-expresses with sfGFP under same promoter] At homeostasis (S = 0): → TtrR* below threshold → pTtr_LT2 silent → sfGFP OFF (leaky floor = 0.293 rel. units at α_leak = 2% α_max)
The current construct expresses sfGFP as the reporter payload under pTtr_LT2. For Aim 2 wet-lab validation, MccH47 + MchI immunity cassette replaces or is inserted downstream of sfGFP using the existing MCS in the pSC101 backbone:
pTtr_LT2 → [BBa_B0031 → sfGFP → BBa_B0015 ↓ Aim 2 swap pTtr_LT2 → BBa_B0031 → MccH47 + MchI → BBa_B0034 → BBa_B0015
BBa_B0034 (strong RBS) replaces B0031 for the effector cassette to maximise k_M — confirmed as top Pareto driver (PRCC rank 1, Sobol ST rank 1) from Tasks 3 and 4.
ΔdapA auxotrophy is implemented as a chromosomal deletion in the EcN host, separate from the plasmid construct. DAP (diaminopimelic acid) is absent from mammalian gut, making survival contingent on exogenous DAP supply. Escape frequency: ~10⁻⁸ per generation (Stritzker 2007). This layer is not encoded on the plasmid and does not contribute to the 8,323 bp construct size.
The following outputs from the computational pipeline feed directly into Aim 2 experimental design, defining what to measure, in what order, and with what precision:
| Aim 1 output | How it constrains Aim 2 |
|---|---|
| Pareto-resolved parameter regimes (top-ranked δ, K_D, n, k_M combinations) | Defines the experimental parameter space to sample first; avoids blind screening |
| Ranked PRCC sensitivity indices | k_M and k_kill are ranks 1 and 2 for suppression; these are measured before n or EC50 — constraining the highest-leverage parameters first |
| Predicted tetrathionate activation threshold (EC50=20 µM, n=2) | Sets the concentration range for dose-response assay: 0, 2, 10, 20, 50, 100 µM |
| Predicted MccH47 kill kinetics curve (k_kill vs. effector concentration) | Defines expected CFU reduction trajectory for co-culture kill assay; any deviation triggers ODE re-parameterisation |
| ΔdapA-confirmed EcN chassis | Host strain confirmed auxotrophic before construct transformation; fluctuation assay baseline established |
| Synthesised four-module plasmid construct | Twist-synthesised, SecureDNA-screened construct delivered to Ginkgo for CFPS expression and co-culture screening |
iso-sentinel-ecn/params/aim2_wetlab.yaml| Metric | Method | Links back to |
|---|---|---|
| Hill coefficient n and activation threshold K_D for TtrS/TtrR | Fluorescence dose-response curve fitting | Task 1 biosensor ODE — direct parameter validation |
| MccH47 production rate k_M and kill rate k_kill under defined induction | LC-MS quantification (Waters BioAccord) + CFU kill curve | Task 2 effector ODE; Task 3 Pareto position |
| Burden δ: growth rate differential between circuit-on and wild-type EcN | OD600 time-course comparison | Task 3 Pareto x-axis; Task 5 Moran input |
| ΔdapA escape frequency | Fluctuation assay (Luria-Delbrück) | Task 5 containment escape model — validates μ_escape=10⁻⁸ assumption |
| Model-to-experiment deviation | ppm or % error per parameter: (measured − predicted) / predicted × 10⁶ | All tasks; feeds ODE refinement loop |
| Circuit specificity: kill ratio against target pathogens vs. commensal E. coli | CFU ratio on selective vs. non-selective media | Task 2 effector module; confirms narrow-spectrum claim |
| Measurement | ODE prediction | Target range | Method |
|---|---|---|---|
| sfGFP at 50 µM tetrathionate | 12.16 rel. units | >10 | Plate reader fluorescence |
| sfGFP at 0 µM (homeostatic) | 0.293 rel. units | <1.0 | Plate reader fluorescence |
| Fold induction (ON/OFF) | 41.5× | ≥10× | Fluorescence ratio |
| t50 (sfGFP ON state) | 15.3 h | 8–30 h | Time-course plate reader |
| MccH47 secretion (Aim 2) | 3.45 µM/h at full induction | Detectable above baseline | LC-MS (Waters BioAccord) |
| Pathogen suppression (Aim 2) | 100% at 200h | ≥90% at 48h | CFU count, selective media |
| ΔdapA confirmation | Lethal without DAP | No growth in DAP-free LB | OD600 growth curve |
All sequences — ttrS, ttrR, pTtr_LT2, sfGFP, and the Aim 2 MccH47+MchI cassette — require SecureDNA screening before Twist synthesis submission. This is a mandatory HTGAA Industry Council step.
| Company | Specific role |
|---|---|
| Asimov Kernel | Independent circuit architecture validation; cross-check Pareto landscape against Kernel simulation output |
| SecureDNA | Pre-synthesis screen of ttrS, ttrR, mccH47, mchI, ΔdapA cassette sequences |
| Twist Biosciences | Codon-optimised synthesis of full construct (~3.5 kb); delivery to Ginkgo |
| Ginkgo Bioworks | CFPS expression, plate reader fluorescence, potential his-tag purification |
| Waters Corporation | LC-MS confirmation of MccH47 secretion; intact mass of sfGFP reporter |
| Opentrons | Co-culture assay automation for parallel tetrathionate concentration screening |
Authored and reviewed by:
This document captures the full scope of our group work within the Genspace node focused on engineering the MS2 bacteriophage L protein. Group 2 formed around a shared interest in improving the toxicity, stability, and tunability of the L protein through computational design.
Our early brainstorming sessions centered on three broad goals:
After several meetings and independent exploration, the group converged on two main computational directions. The first centered on systematic truncation and mutagenesis of the N-terminal regulatory domain. The second focused on point mutations within conserved regions that could alter electrostatic interactions while preserving structure.
Two major pipelines emerged from that work. John’s pipeline explored N-terminal truncations, DnaJ disruption, sequence redesign, codon optimization, and sequencing validation. Eric’s pipeline focused on charge-based mutations, conservation mapping, structural modeling, ORF overlap analysis, and cross-referencing with experimental lysis data.
Both approaches identified strong but distinct candidates for improving L protein function.
The MS2 lysis protein L is a 75 amino acid single-pass transmembrane protein whose N-terminal region acts as a regulatory brake on lysis. Rather than directly participating in membrane disruption, this region delays insertion and oligomerization of the transmembrane domain.
My pipeline focused on systematically removing portions of that inhibitory region while preserving the membrane-spanning lytic core. The central hypothesis was simple: if the N-terminal domain slows lysis, then partial removal should release that inhibition and produce earlier, stronger lytic activity.
The strongest candidate to emerge from the analysis was L_trunc30, which removes the first 30 amino acids while preserving the entire transmembrane domain.
Confirmed L protein sequence:
Confirmed DNA sequence:
Three ideas guided the pipeline:
| Stage | Tool | Purpose |
|---|---|---|
| 1 | ESM2 | Mutational scanning across all 75 residues |
| 2 | ESMFold | Structural prediction of truncation variants |
| 3 | AlphaFold-Multimer | Modeling interaction with DnaJ |
| 4 | GROMACS | Molecular dynamics and RMSF analysis |
| 5 | ProteinMPNN | Junction redesign and charge reduction |
| 6 | Codon optimization | Prepare E. coli expression constructs |
| 7 | Synthetic construct design | Assemble expression cassette |
| 8 | Bowtie2 + BCFtools | Variant calling and sequencing validation |
| 9 | IGV | Manual inspection of called variants |
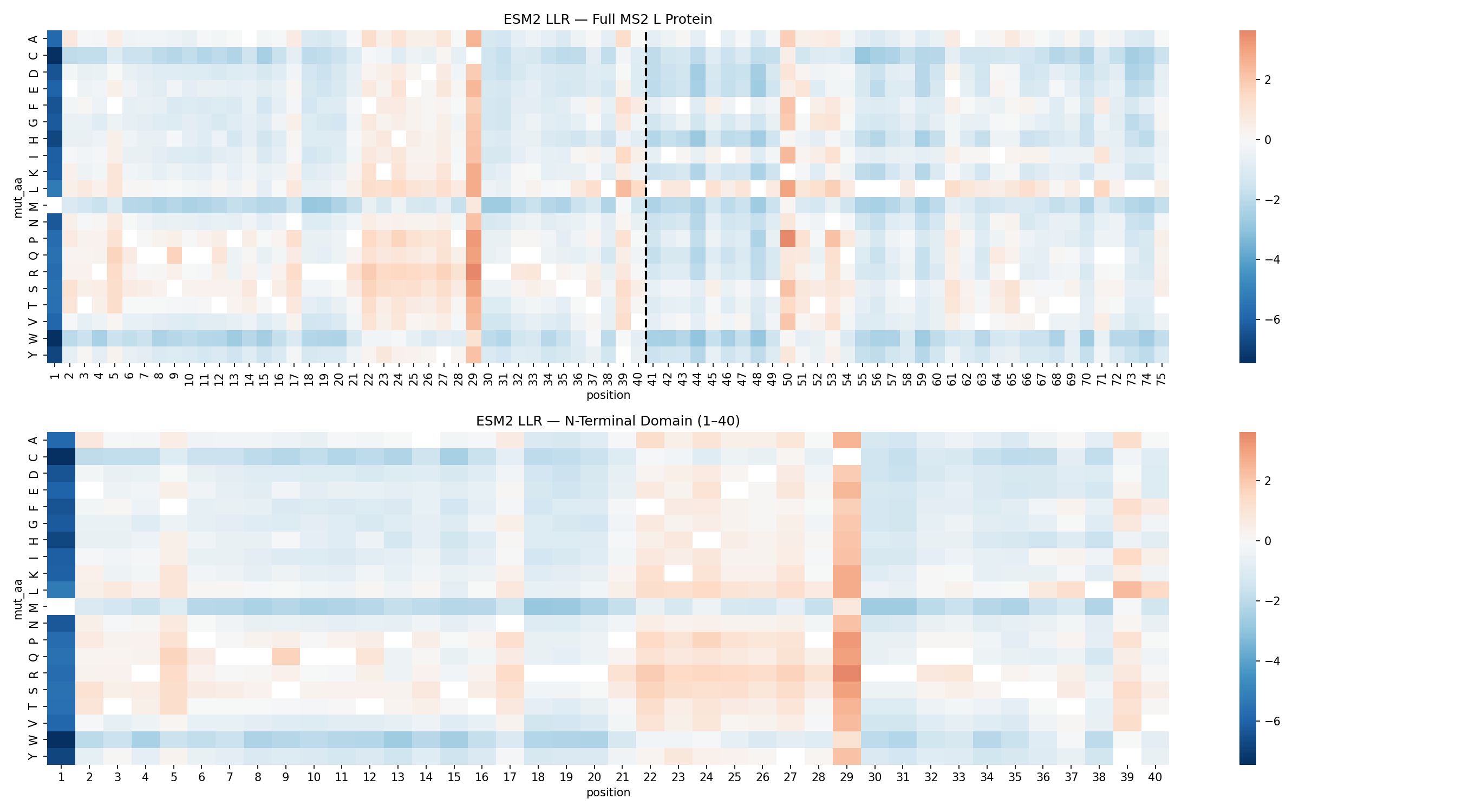
The ESM2 scan identified position C29 as the dominant mutational hotspot in the N-terminal domain.
| Mutation | LLR | Notes |
|---|---|---|
| C29R | 3.64 | Top-ranked substitution |
| C29P | 3.17 | Strong helix-disrupting mutation |
| C29Q | 3.06 | Conservative but highly favored |
| F22R | 1.86 | Introduces basic charge |
| S9Q | 1.69 | Recovered independently in prior work |
C29 accounted for 12 of the top 20 substitutions. That concentration strongly suggested that the wild-type residue at this site is not ideal for maximizing toxicity outside the native viral context.
ESMFold predictions for all truncation variants suggested that the N-terminal domain is highly disordered in solution. Interdomain contact analysis returned essentially zero contacts across all variants, which fits with the known biology of the L protein.
The more useful signal came from molecular dynamics.
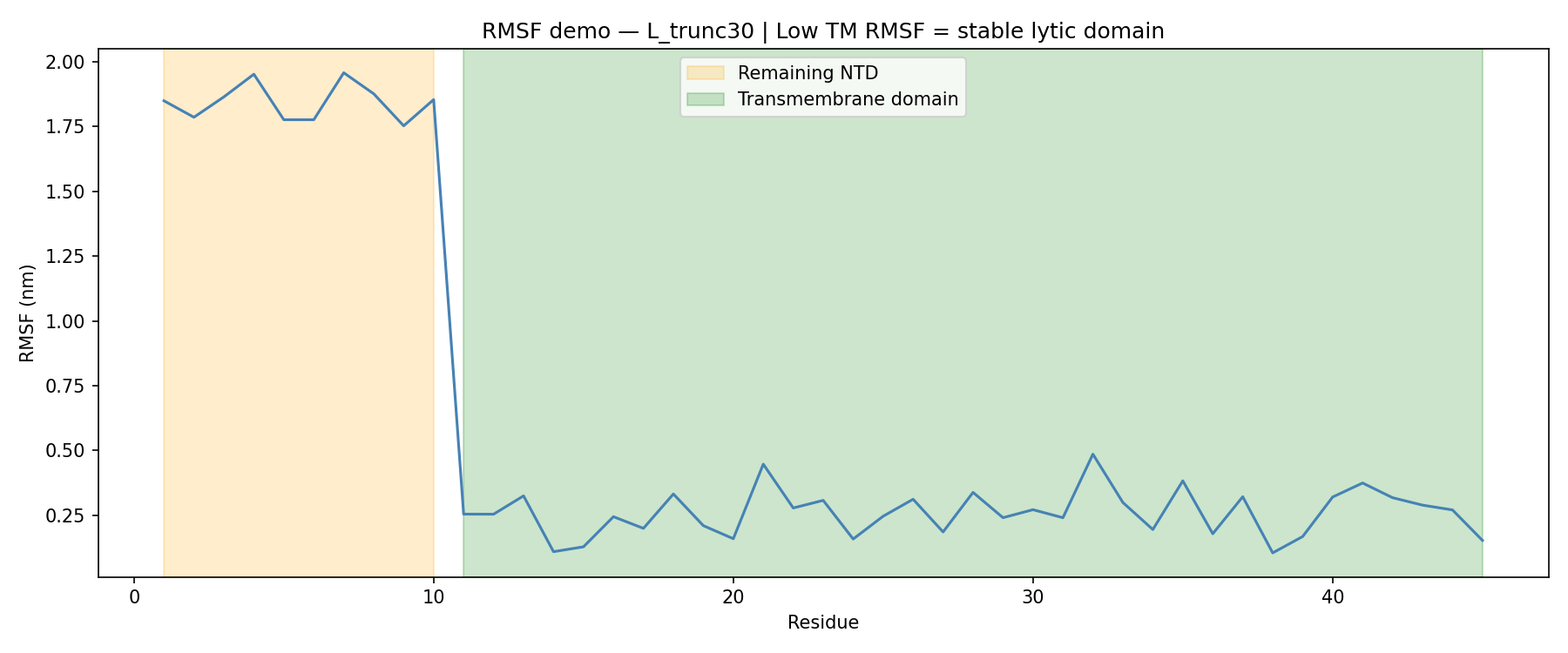
For L_trunc30:
That sharp drop in flexibility confirmed that the transmembrane region remains stable even after removing 30 amino acids from the N-terminus.
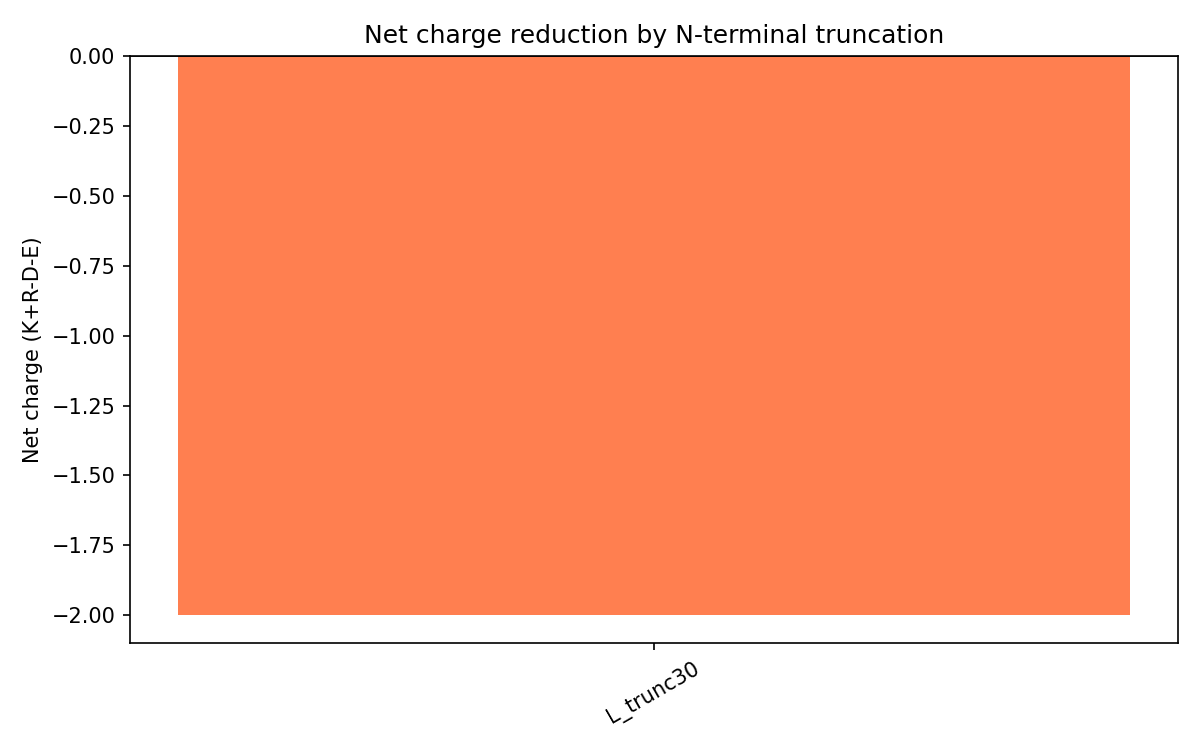
The wild-type N-terminal region is strongly basic due to motifs like RRRPFK and RRQQR.
L_trunc30 reverses the overall charge profile:
| Variant | Net charge | Interpretation |
|---|---|---|
| Wild-type L | Approximately +8 | Strong DnaJ interaction expected |
| L_trunc30 | -2 | Reduced DnaJ binding and earlier lysis expected |
This was important mechanistically because DnaJ binding depends heavily on electrostatic interactions with the positively charged N-terminal region.
All major truncation variants were codon-optimized for E. coli K-12.
The lead construct, L_trunc30, preserved the essential LS motif and was assembled into a complete 230 bp expression cassette with:
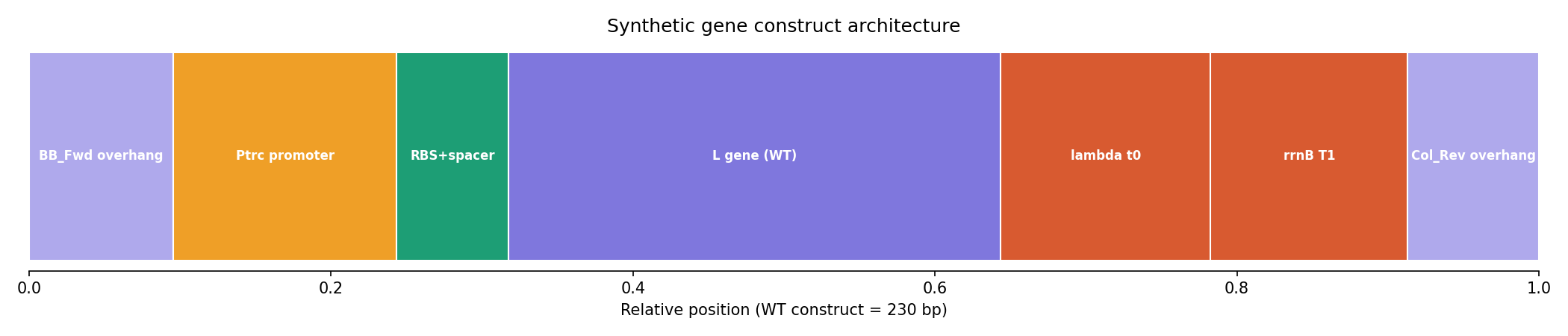
| Candidate | Key Feature | Reason |
|---|---|---|
| L_trunc30 | Removes aa 1-30 | Strongest balance of toxicity, structural stability, and DnaJ disruption |
| Candidate | Reason for Inclusion |
|---|---|
| C29R | Highest ESM2 score overall |
| F22R | Adds positive charge in N-terminal region |
| S9Q | Recovered independently in previous scans |
| L_trunc40 | Most aggressive truncation, likely strongest toxicity |
Eric approached the same problem from a different angle. Instead of removing large sections of the N-terminus, he focused on identifying individual amino acid substitutions that could improve toxicity while preserving the overall structure of the protein.
His strongest candidate was P13L, a single amino acid change in the N-terminal region.
| Stage | Tool | Purpose |
|---|---|---|
| 1 | UniProt + BLAST | Sequence retrieval and homolog identification |
| 2 | Clustal Omega | Conservation mapping |
| 3 | AlphaFold-Multimer | Oligomer modeling |
| 4 | ESM2 | Mutation scoring |
| 5 | ESMFold | Structural confidence and pTM analysis |
| 6 | ChimeraX | Electrostatic visualization |
| 7 | Benchling | ORF overlap analysis |
Eric identified a relatively unconstrained region between amino acids 16 and 28 that could tolerate mutation without damaging essential structure.
| Position | Wild-type residue | Interpretation |
|---|---|---|
| 18 | R | Fully conserved, avoid |
| 21 | P | Fully conserved, avoid |
| 23 | K | Fully conserved, avoid |
| 26 | D | Variable, strong candidate |
| 13 | P | Weakly conserved, potentially safe |
P13L produced the strongest ESMFold result among all variants tested.
| Variant | pTM | Change vs WT |
|---|---|---|
| Wild-type | 0.273 | Reference |
| D26R | 0.267 | Slight decrease |
| P13L | 0.420 | Strong increase |
The jump from 0.273 to 0.420 made P13L the most structurally favorable point mutation in Eric’s pipeline.
Unlike my pipeline, Eric cross-referenced computational candidates with available lysis data.
| Mutation | Replicate A | Replicate B | Result |
|---|---|---|---|
| P13L | 1 | 1 | Confirmed lytic |
| D26G | 1 | 0 | Mixed |
| K23E | 1 | 0 | Mixed |
| E25G | 1 | 0 | Mixed |
P13L was the only candidate to remain consistently positive across both replicates.
One of the more interesting parts of Eric’s work was the DNA-level overlap analysis.
P13L falls within the overlap region between the coat protein and the L protein, which initially made it look risky. After codon-level analysis, though, the mutation turned out to be safe.
| Gene | WT codon | Mutant codon | Result |
|---|---|---|---|
| L protein | CCG | CTG | Pro → Leu |
| Coat protein | TCC | TCT | Ser → Ser |
That synonymous change in the coat protein meant the mutation could proceed without disrupting the overlapping reading frame.
| Candidate | Key Feature | Reason |
|---|---|---|
| P13L | Single amino acid substitution | Best structural score and strongest experimental support |
| Candidate | Status |
|---|---|
| D26R | Untested but promising |
| D26G | Mixed experimental results |
| N17R | Open candidate |
| H24R | Open candidate |
Albert focused primarily on structural stability.
His workflow emphasized:
His key concern was preserving structure while introducing beneficial mutations.
He also pointed out an important limitation that kept showing up across the project: membrane proteins are underrepresented in both structural databases and protein language model training sets. That means even high-scoring mutations should still be interpreted cautiously.
Tehseen’s approach aligned closely with my truncation-based strategy but focused more on identifying the smallest regulatory segment required for precise control over lysis timing.
The central idea was not simply to remove the N-terminal region, but to identify exactly which residues are responsible for slowing lysis.
That led to three closely related hypotheses:
| Aspect | John’s Pipeline | Eric’s Pipeline |
|---|---|---|
| Main strategy | Progressive N-terminal truncation | Point mutation design |
| Lead candidate | L_trunc30 | P13L |
| Core hypothesis | Remove inhibitory domain | Increase local electrostatic effects |
| ESM2 scope | Full 1,425-substitution scan | Single-site targeted analysis |
| Structural analysis | ESMFold + GROMACS RMSF | ESMFold + ChimeraX |
| DnaJ interaction | Central to model | Considered indirectly |
| Experimental validation | Not yet completed | P13L confirmed experimentally |
| Construct design | Fully assembled | Still planned |
| Sequencing workflow | Fully designed with Bowtie2, BCFtools, IGV | Listed as future step |
The project ended up producing two very different but complementary engineering directions.
L_trunc30 represents the stronger systems-level redesign. It removes the inhibitory N-terminal region, reduces DnaJ engagement, preserves the transmembrane core, and provides a fully buildable expression construct ready for synthesis and sequencing validation.
P13L represents the cleaner minimal-change strategy. It preserves the full-length protein, improves structural confidence, survives ORF overlap analysis, and already has positive experimental support.
If the goal is maximum disruption of the native regulatory system, L_trunc30 is the stronger candidate.
If the goal is a simpler mutation with lower engineering risk and existing wet lab support, P13L is the better starting point.
The most practical next step would be to synthesize and compare both side by side.