Week 4
Node participant note: I am a remote Genspace node listener based in Nigeria without onsite lab access. The Week 4 lab (Protein Design I) was fully computational — ESMFold inference, ESM2 mutational scanning, latent space analysis, ProteinMPNN inverse folding — and I completed all exercises remotely using Google Colab and local tools. The outputs documented below represent my complete engagement with the lab material.
Class Assignment — Week 4
Part A. Conceptual Questions
1) How many molecules of amino acids do you take with a piece of 500 grams of meat?
Assumptions: lean meat is ~20% protein by mass, average amino acid residue ~100 Da (≈100 g/mol).
Step 1: Protein mass in 500 g meat
500 g × 0.20 = 100 g protein
Step 2: Convert to moles of amino acid residues
100 g ÷ (100 g/mol) = 1 mole
Step 3: Convert moles to molecules
1 mole = 6.022 × 10²³ molecules
Answer: approximately 6.0 × 10²³ amino acid molecules (about 600 sextillion) which is actually the Avogadro’s Number in chemistry, or one mole of water
2) Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because eating provides raw materials, not biological identity. Digestion breaks proteins, fats, and nucleic acids into small molecules such as amino acids and fatty acids. By the time nutrients enter the bloodstream, they are no longer “cow” or “fish,” they are shared chemical building blocks used by all life.
What determines what we become is our genome and regulatory systems. Human cells assemble human proteins because human DNA encodes the instructions. Food is like construction material. The same bricks can build different structures depending on the blueprint.
3) Why are there only 20 natural amino acids?
The “20” is an evolutionary, chemical, and informational compromise. The standard amino acids provide enough chemical diversity for folding, catalysis, and signaling while keeping translation machinery stable and error-tolerant. Expanding beyond this set would require major coordinated changes to tRNAs, aminoacyl-tRNA synthetases, and ribosomes, which coul possibly be evolutionarily costly.
Also, the genetic code has 64 codons, which comfortably encodes 20 amino acids plus stop signals. The system stabilized around a set that is chemically sufficient and operationally efficient.
Notably, the set is not absolutely fixed. Biology also uses selenocysteine and pyrrolysine via specialized mechanisms, and synthetic biology can incorporate many noncanonical amino acids in engineered systems.
4) Can you make other non-natural amino acids? Design some new amino acids.
Yes. Chemists and synthetic biologists have created many noncanonical amino acids. Conceptually, you keep the standard amino acid backbone and alter the side chain to introduce new properties. Below are conceptual designs (structural ideas, not synthesis instructions):
Fluoro-leucine variant
Replace a leucine side-chain hydrogen with fluorine to increase stability and hydrophobicity.Photo-switch amino acid
Add a light-responsive group (azobenzene-like) that changes shape under light, enabling reversible control of protein behavior.Metal-binding amino acid
Design a side chain with a strong chelating motif to coordinate metals more tightly than histidine, enabling engineered metalloenzymes.Redox-active amino acid
A side chain designed for reversible electron transfer beyond cysteine/tyrosine chemistry, expanding redox options.Bulky steric-block amino acid
A large aromatic side chain that can restrict folding paths or block active sites to tune structure and function.Synthetic polar-gradient amino acid
A side chain with donor/acceptor geometry not present in the canonical set to enable new hydrogen-bonding patterns.
Practical considerations for synthetic possibility include recognition by synthetases, ribosomal fit, folding effects, toxicity, and translational fidelity.
5) Where did amino acids come from before enzymes and before life started?
Amino acids can arise through prebiotic chemistry. Three common sources are:
Atmospheric chemistry: Early Earth gases plus energy (lightning, UV, heat) can generate amino acids (supported by classic Miller–Urey-type results).
Hydrothermal vents: Mineral surfaces, heat, and gradients can promote organic synthesis and concentration of building blocks.
Extraterrestrial delivery: Meteorites such as Murchison contain amino acids, showing formation can occur beyond Earth and be delivered.
Life later evolved enzymes to produce amino acids more efficiently and selectively.
6) If you make an α-helix using D-amino acids, what handedness would you expect?
A polypeptide made of D-amino acids would form a left-handed α-helix. Natural α-helices are right-handed because proteins use L-amino acids; mirroring chirality mirrors the preferred helix.
7) Can you discover additional helices in proteins?
Within natural peptide chemistry, backbone geometry is constrained by peptide bond planarity, allowed φ/ψ angles, and hydrogen bonding rules. However, we can still expand what we call “helical forms” in practice by:
- identifying less common helical geometries in known proteins
- designing novel helices computationally
- engineering sequences that stabilize alternative helix types under specific conditions
So “new helices” are often new realizations within physical constraints rather than completely new backbone physics.
8) Why are most molecular helices right-handed?
Because biological polymers are built from chiral monomers that life selected early. L-amino acids favor right-handed α-helices; D-sugars in DNA favor right-handed B-DNA. Once one chirality dominated, evolution locked in downstream structural preferences across biology.
9) Why do β-sheets tend to aggregate? What is the driving force?
β-sheets aggregate because their edges expose backbone hydrogen bond donors and acceptors that can be satisfied by forming intermolecular hydrogen bonds. Aggregation is further stabilized by:
- Backbone hydrogen bonding networks across molecules
- Hydrophobic packing as β-strands often present with alternating polar/hydrophobic patterns
- Planar stacking geometry enabling tight van der Waals packing
These same stabilizing forces underlie amyloid formation when misregulated.
Part B. Protein Analysis and Visualization
1) Why TolC: Structural Proxy for MccM
MccM (the current ÌṢỌ effector candidate) lacks a solved crystal structure in the PDB, making it unsuitable as the direct target for structure-guided computational exercises requiring an experimental backbone. TolC was selected as the structural anchor because it is the confirmed outer membrane export channel for MccH47 and related microcins, is crystallographically well-resolved at 2.10 Å (PDB: 1EK9), and represents a biologically justified choice for studying the efflux arm of the same microcin system I am engineering.
2) Amino acid sequence and basic properties
Sequence (73 AA):
MRKLSENEIKQISGGDGNDGQAELIAIGSLAGTFISPGFGSIAGAYIGDKVHSWATTATVSPSMSPSGIGLSS
- Length: 73 amino acids
- Molecular weight (calculated): ~8.03 kDa
- Most frequent amino acids: Serine(S) and Glycine(G) both occuring 12 times
- Homologs (UniProt BLAST): ~100 protein sequence homologs
- Protein family: Microcin (Class II) antimicrobial peptide family
Amino acid frequencies
| Amino acid | Count | Percent |
|---|---|---|
| S | 12 | 16.44% |
| G | 12 | 16.44% |
| I | 8 | 10.96% |
| A | 7 | 9.59% |
| L | 4 | 5.48% |
| T | 4 | 5.48% |
| K | 3 | 4.11% |
| E | 3 | 4.11% |
| D | 3 | 4.11% |
| P | 3 | 4.11% |
| M | 2 | 2.74% |
| N | 2 | 2.74% |
| Q | 2 | 2.74% |
| F | 2 | 2.74% |
| V | 2 | 2.74% |
| R | 1 | 1.37% |
| Y | 1 | 1.37% |
| H | 1 | 1.37% |
| W | 1 | 1.37% |
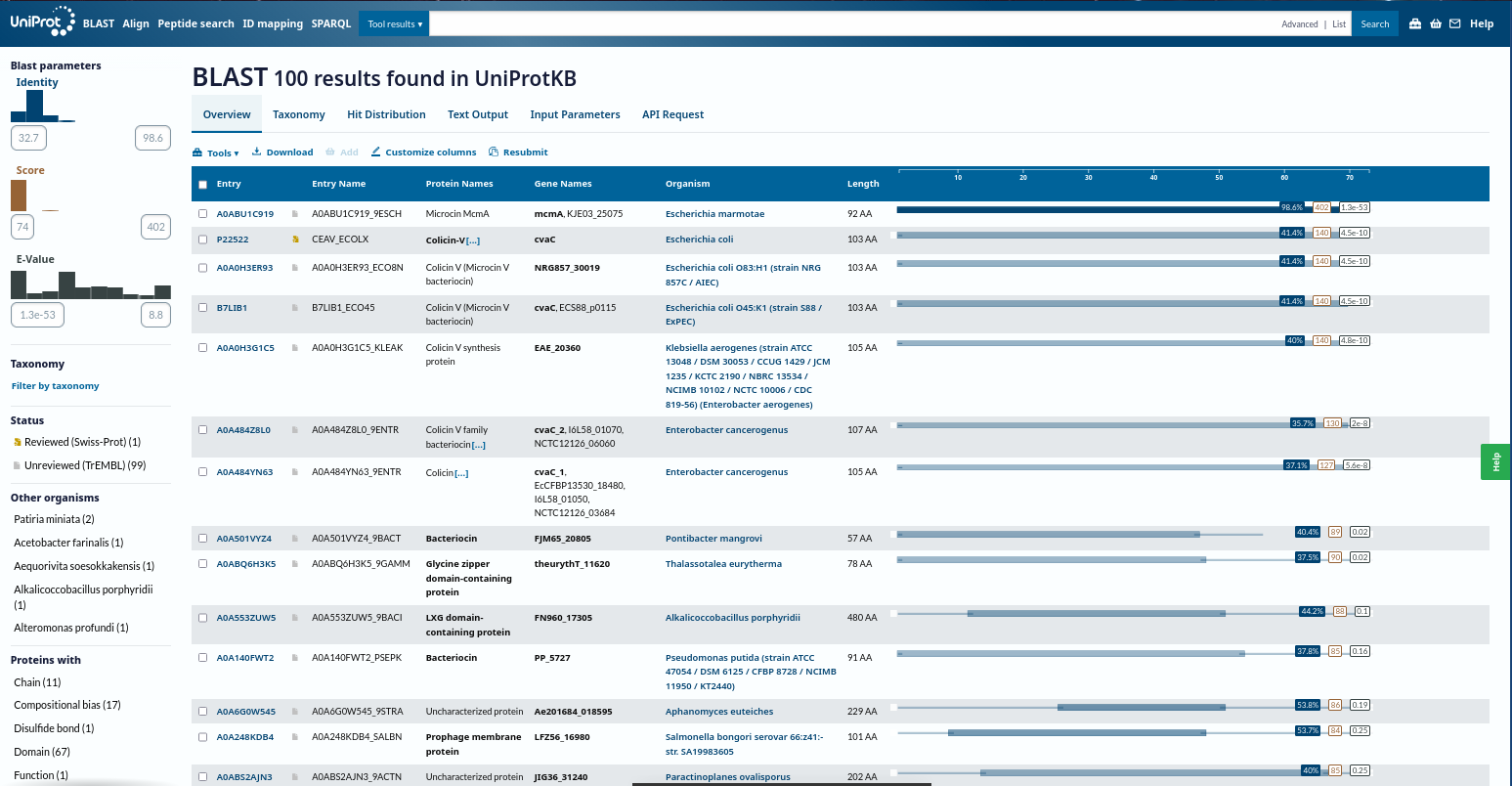
3) Structure Page of My Choice Microcin Protein (RCSB)
Microcin systems, especially my initial Microcin A systems could not be resolved as standalone structures in a way that supports the expected full visualization. To meet the requirements for a high-quality structure with clear visualization features, I used TolC as the structural anchor because it is directly relevant to microcin export and is well characterized in the literature.
- Protein: TolC (E. coli outer membrane export channel)
- PDB: 1EK9
- Resolution: 2.10 Å
- Classification: Outer membrane channel, efflux pump component
Other molecules present experimentally apart from protein include:
- Solvent molecules: 1,508 solvent atoms
- Detergents/Surfactants: Dodecyl glucopyranoside, hexyl glucopyranoside, heptyl glucopyranoside, and octyl glucopyranoside
- Salts/Buffers: Sodium chloride, magnesium chloride, and Tris buffer
- Additives: PEG 400, PEG 2000 MME, and 1,2,3-heptanetriol
RCSB links:
https://www.rcsb.org/structure/1EK9
https://doi.org/10.2210/pdb1EK9/pdb
4) 3D Molecular Visualization
Trimer architecture, surface envelope with internal helical core
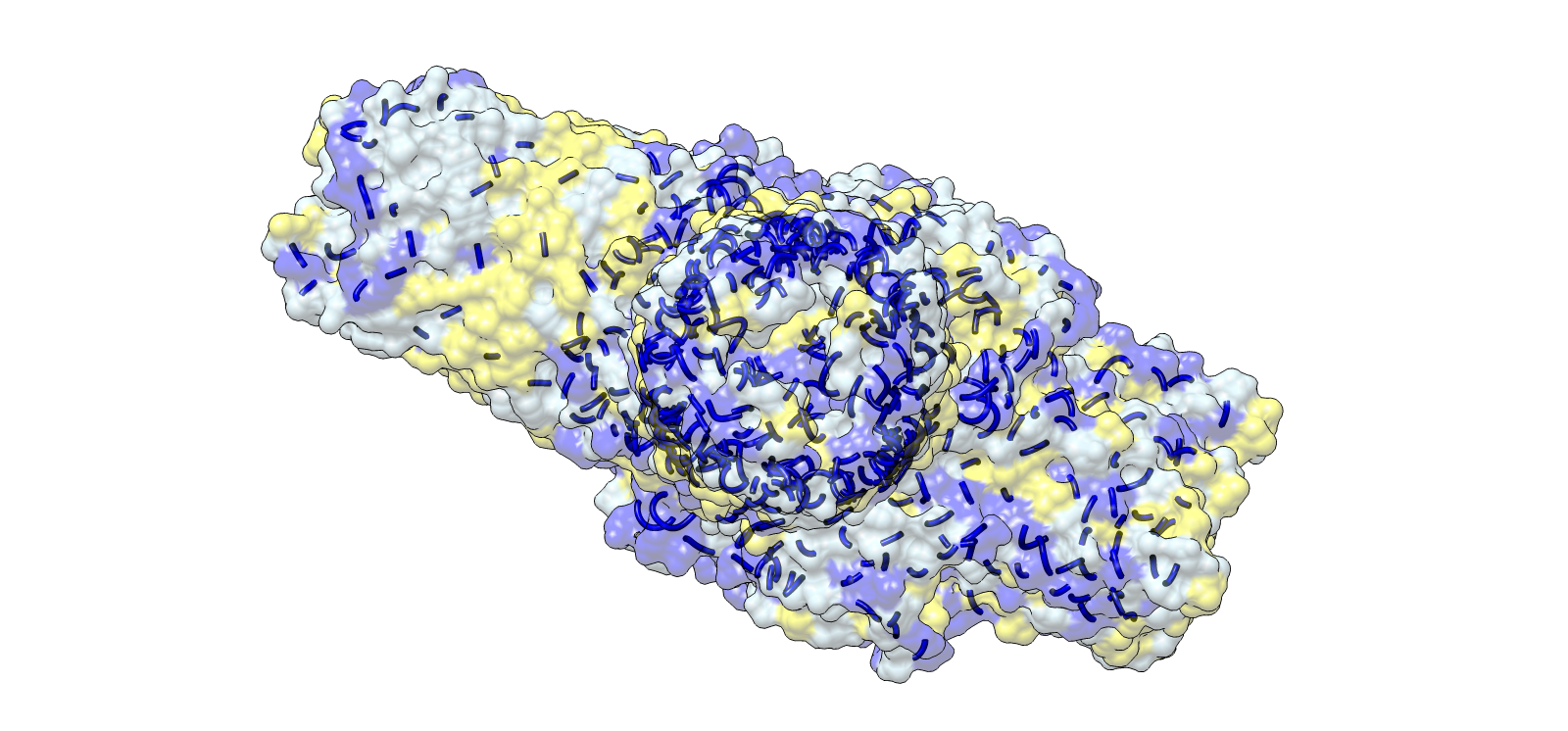
Axial top view highlighting symmetry and central channel
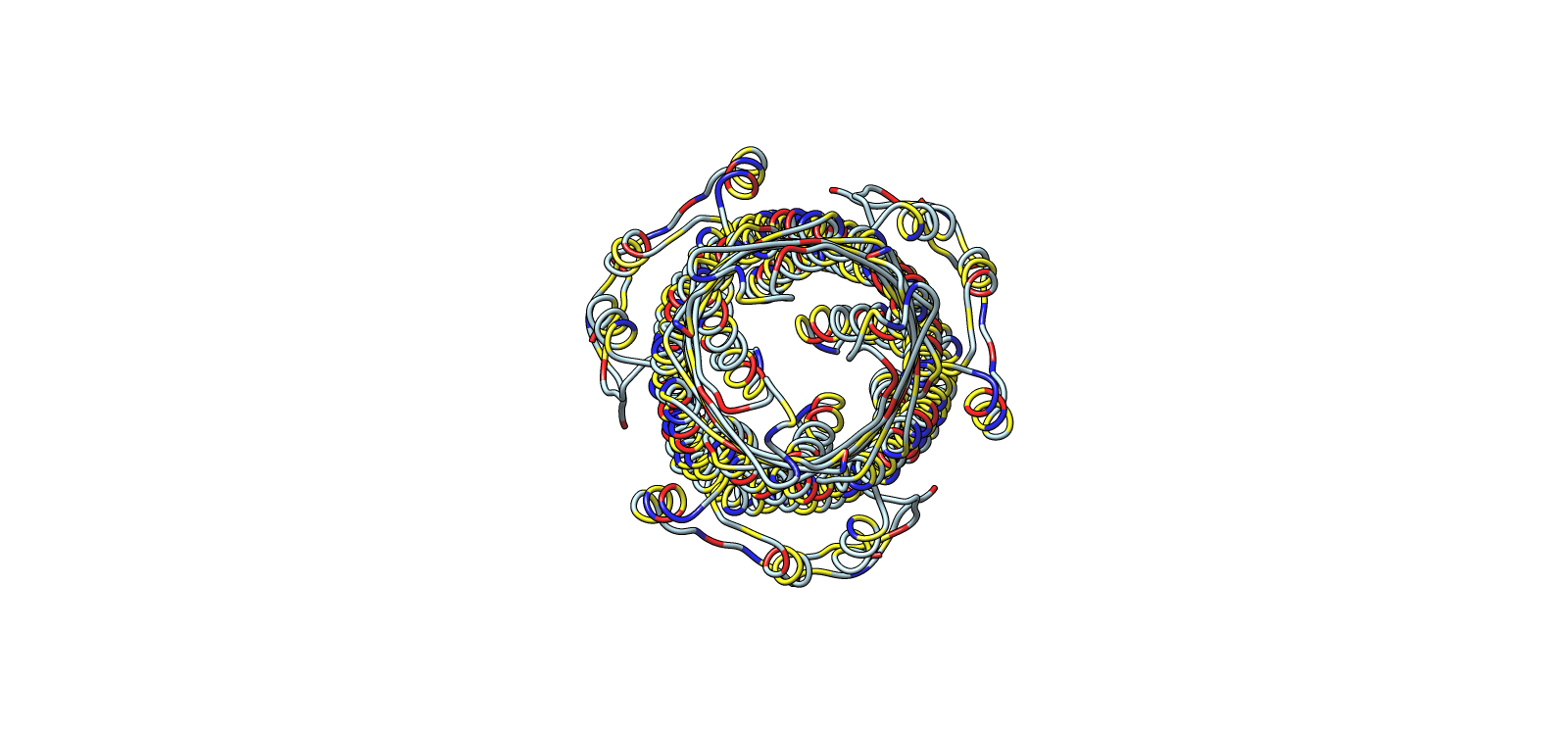
Surface electrochemical landscape showing charge distribution
Lateral chemical view emphasizing membrane-facing hydrophobics
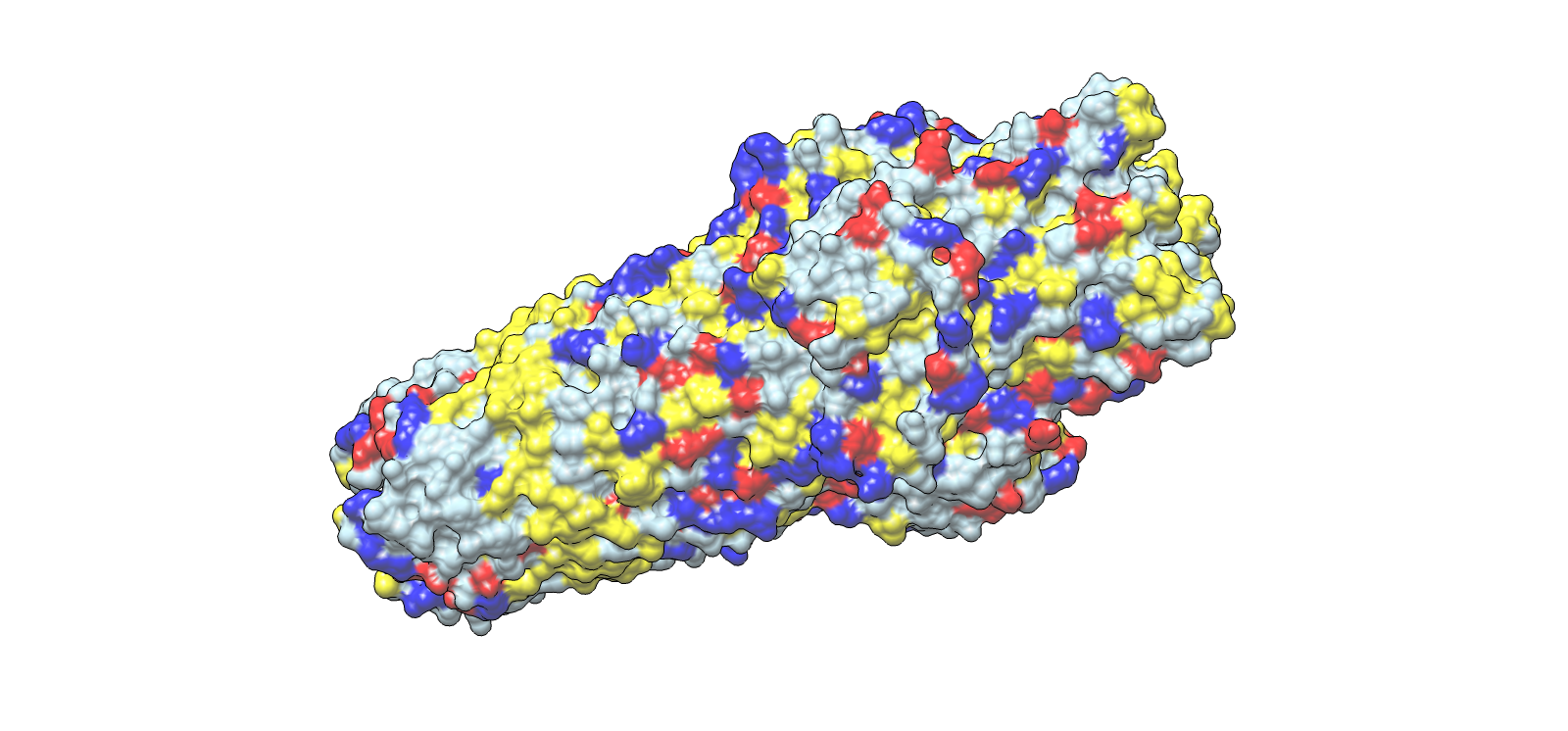
Ribbon colored by residue chemistry to show lumen and interfaces
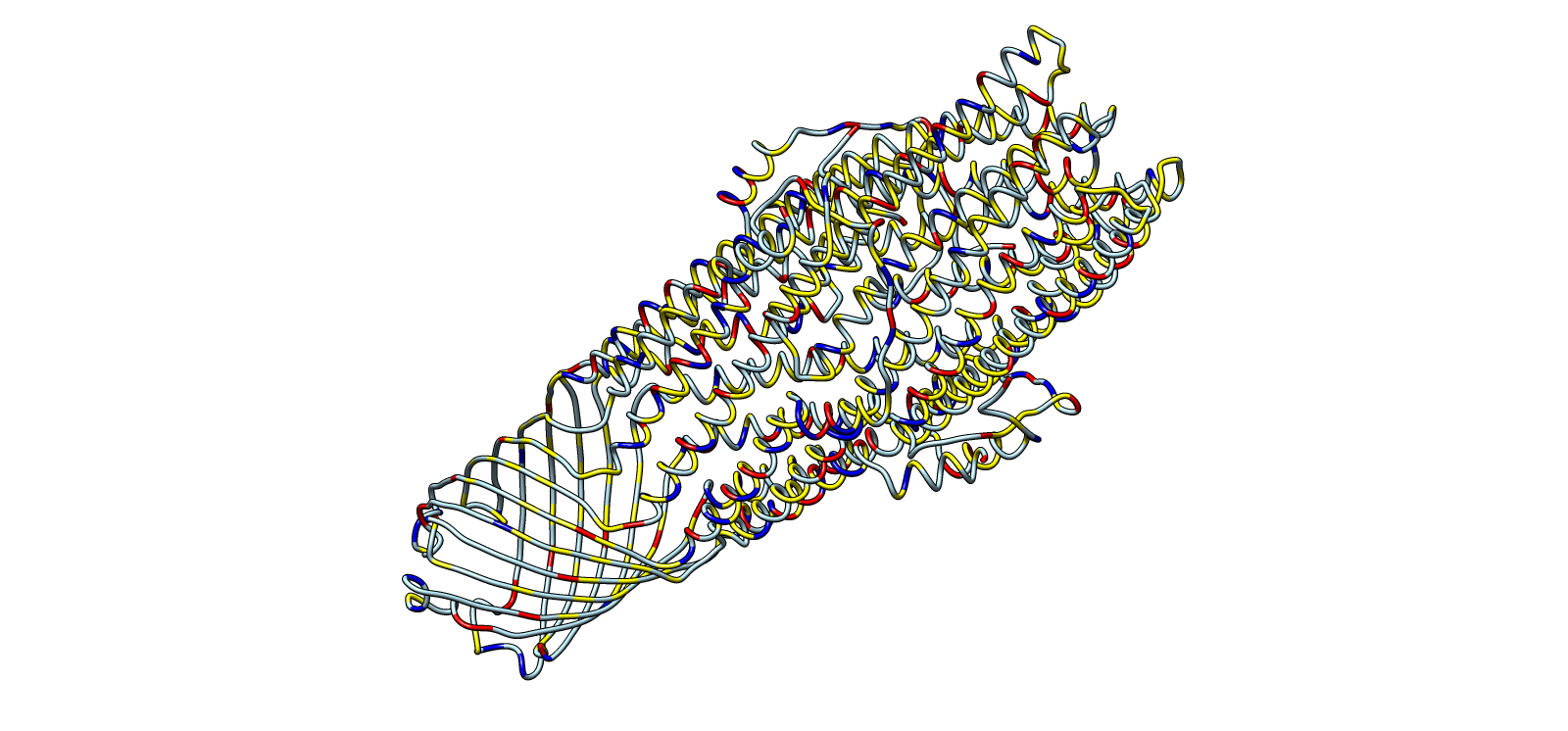
Ribbon-only structural architecture for fold clarity

Color Representation of Selected Images
| Image | Title | Representation | Color | Meaning |
|---|---|---|---|---|
| 1 | Surface envelope with helical core overlay | Transparent surface + ribbon | Light grey | Outer surface |
| Yellow | Hydrophobic surface regions | |||
| Blue | Helical channel core | |||
| 2 | Central channel, axial top view | Ribbon | Yellow | Chain A |
| Blue | Chain B | |||
| Light grey | Chain C | |||
| 3 | Surface electrochemical landscape | Surface | Red | Acidic residues |
| Blue | Basic residues | |||
| Yellow | Hydrophobic residues | |||
| Light grey | Neutral/other | |||
| 4 | Outer membrane barrel, lateral chemical view | Surface | Red/Blue/Yellow/Grey | Same chemistry scheme |
| 5 | Ribbon colored by residue type | Ribbon | Red/Blue/Yellow/Grey | Residue chemistry |
| 6 | Secondary structure architecture | Ribbon | Light cyan | Backbone only |
Microcin A processing pathway (my initial microcin protein choice)
| Step | Protein | Function | Role in pathway | Stage |
|---|---|---|---|---|
| 1 | MccA | Precursor peptide | Scaffold for toxin | Precursor |
| 2 | MccB | Adenyltransferase | Adds AMP to C-terminus | Modification |
| 3 | MccD | Aminopropyltransferase | Adds aminopropyl group | Modification |
| 4 | MccC | Efflux pump | Exports mature microcin | Export / Resistance |
| 5 | MccE | Acetyltransferase | Detoxifies microcin in producer | Immunity |
| 6 | MccF | Serine peptidase | Cleaves toxic moiety | Immunity |
Microcin M processing pathway (my current choice after further exploring the literature)
| Step | Gene / protein | Function | Role in pathway |
|---|---|---|---|
| 1 | mcmA | MccM precursor peptide | Ribosomal scaffold |
| 2 | mcmI | Immunity protein | Producer self-protection |
| 3 | mcmL | Glycosyltransferase-like | Supports siderophore moiety preparation |
| 4 | mcmK | Esterase-like | Supports siderophore processing |
| 5 | mchC / mchD | Linker proteins | Attachment steps (biochemistry not fully resolved) |
| 6 | mchF | ABC transporter | Exports mature microcin |
| 7 | mchE | Membrane fusion protein | Works with export machinery |
| 8 | tolC | Outer membrane channel | Final export conduit |
Part C. Using ML-Based Protein Design Tools
1A) Deep Mutational Scan (ESM2)
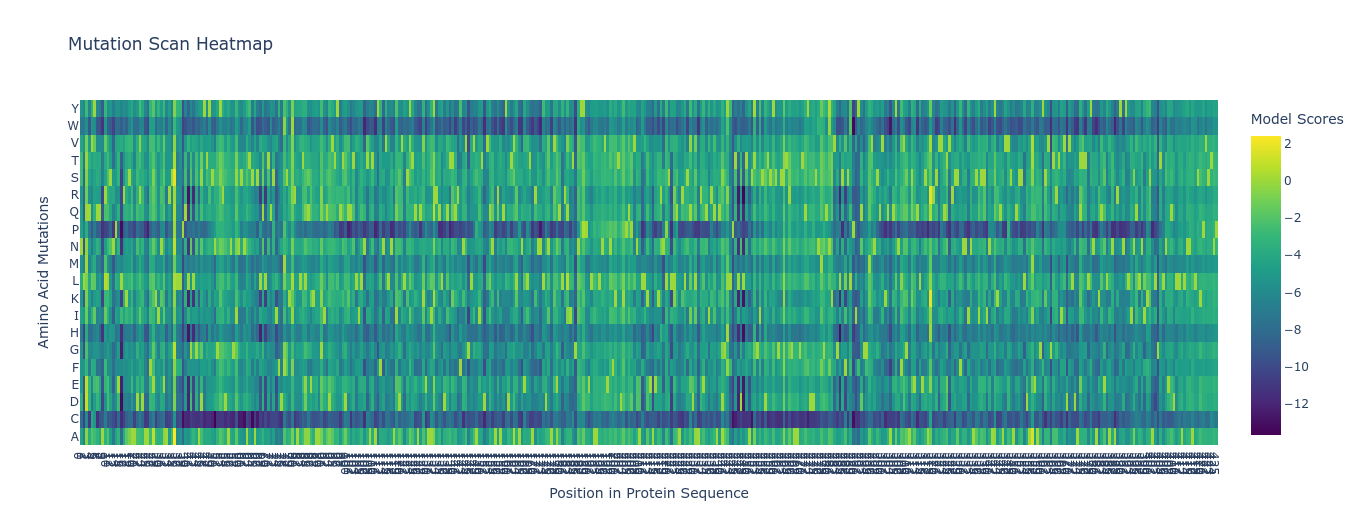
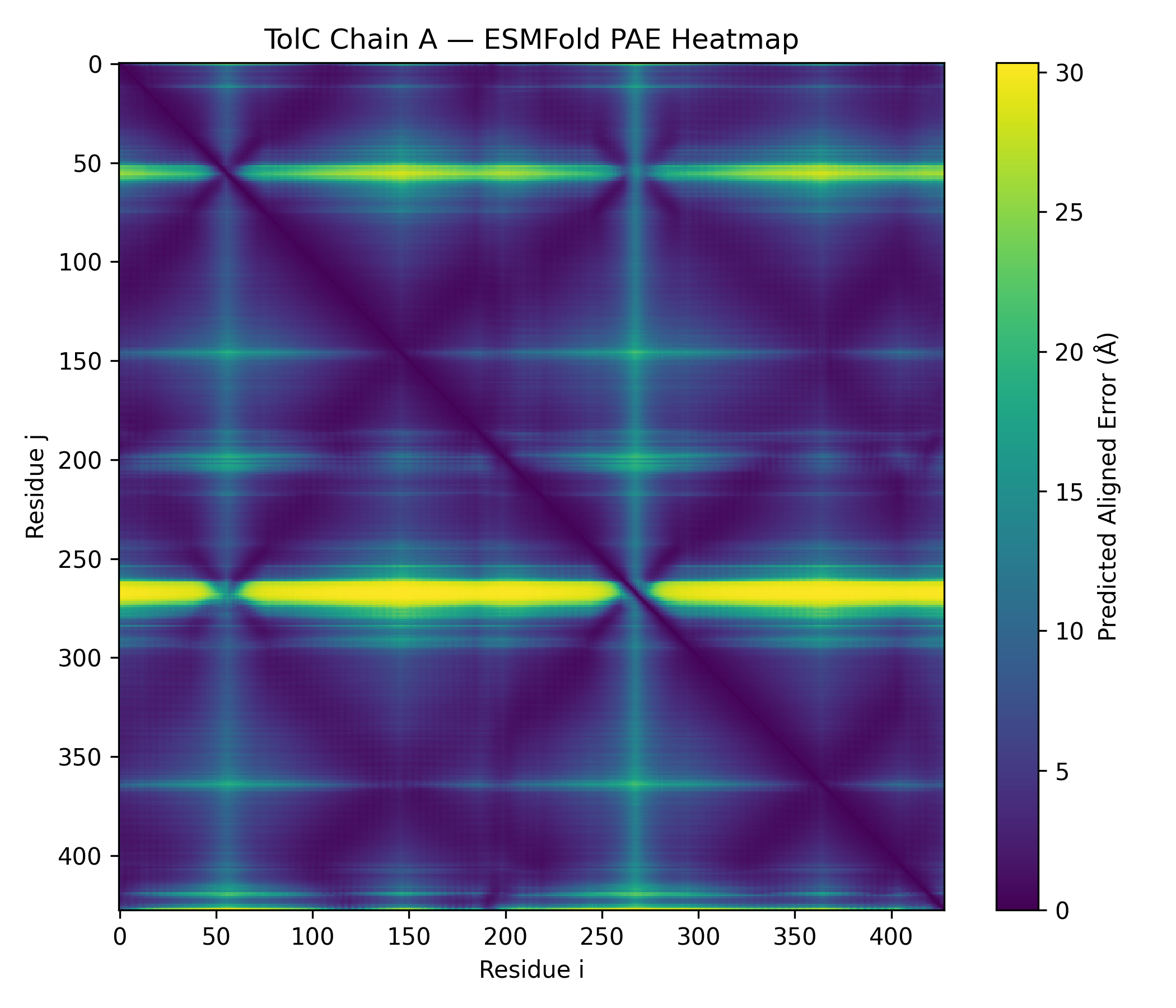
Using ESM2, I generated an unsupervised deep mutational scan across the TolC sequence. The heatmap showed multiple constrained regions, visible as vertical bands, suggesting positions that are broadly intolerant to mutation.

A clear example was residue 178. The wild-type residue is tryptophan (W). The mutation W178D produced a relative log-likelihood score of −2.38, indicating a strong model penalty. Structural inspection supports this: W178 is buried within the TolC trimeric structure. Replacing a bulky hydrophobic aromatic residue with a negatively charged aspartate is expected to disrupt local hydrophobic packing and weaken the inter-chain interface.
Supporting snapshots:
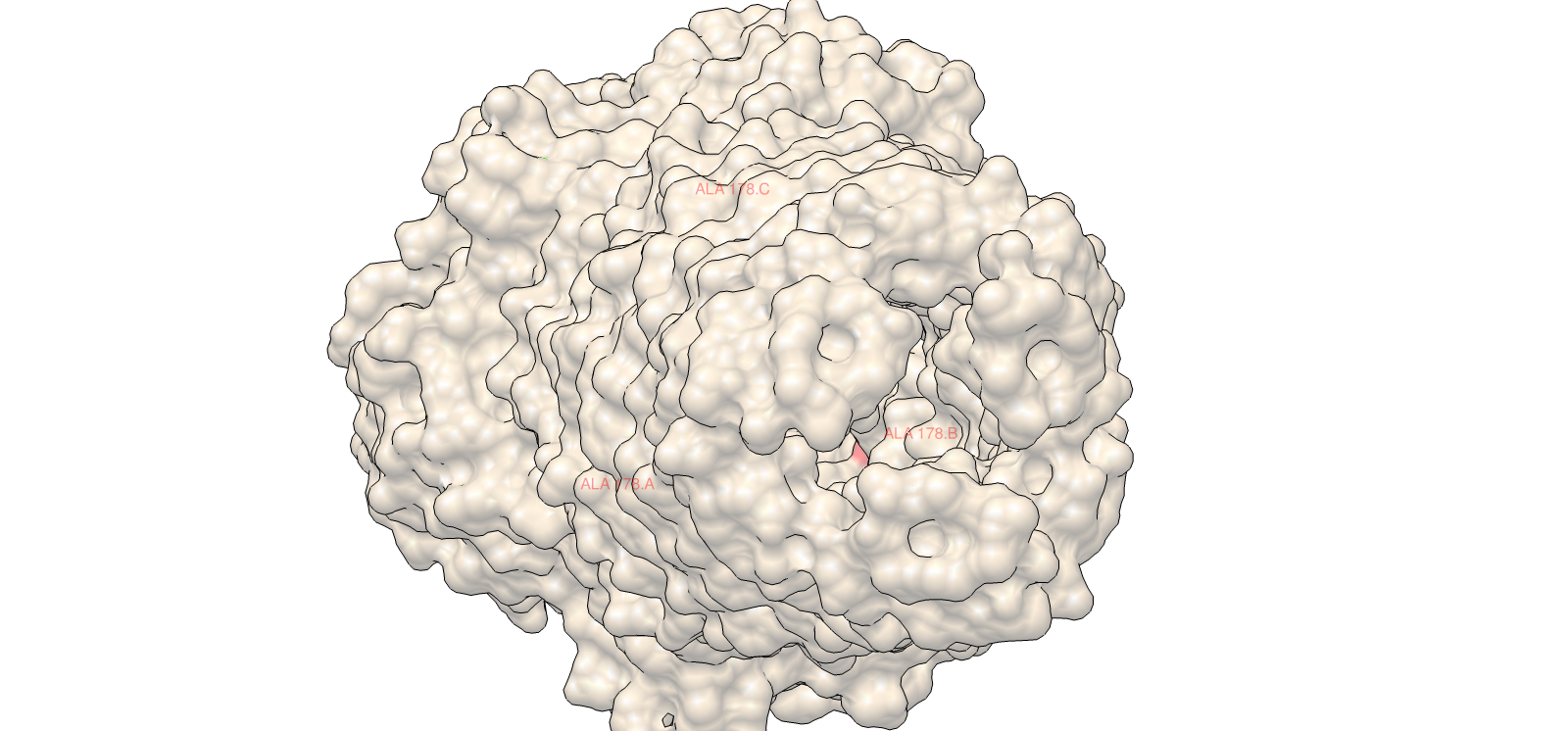
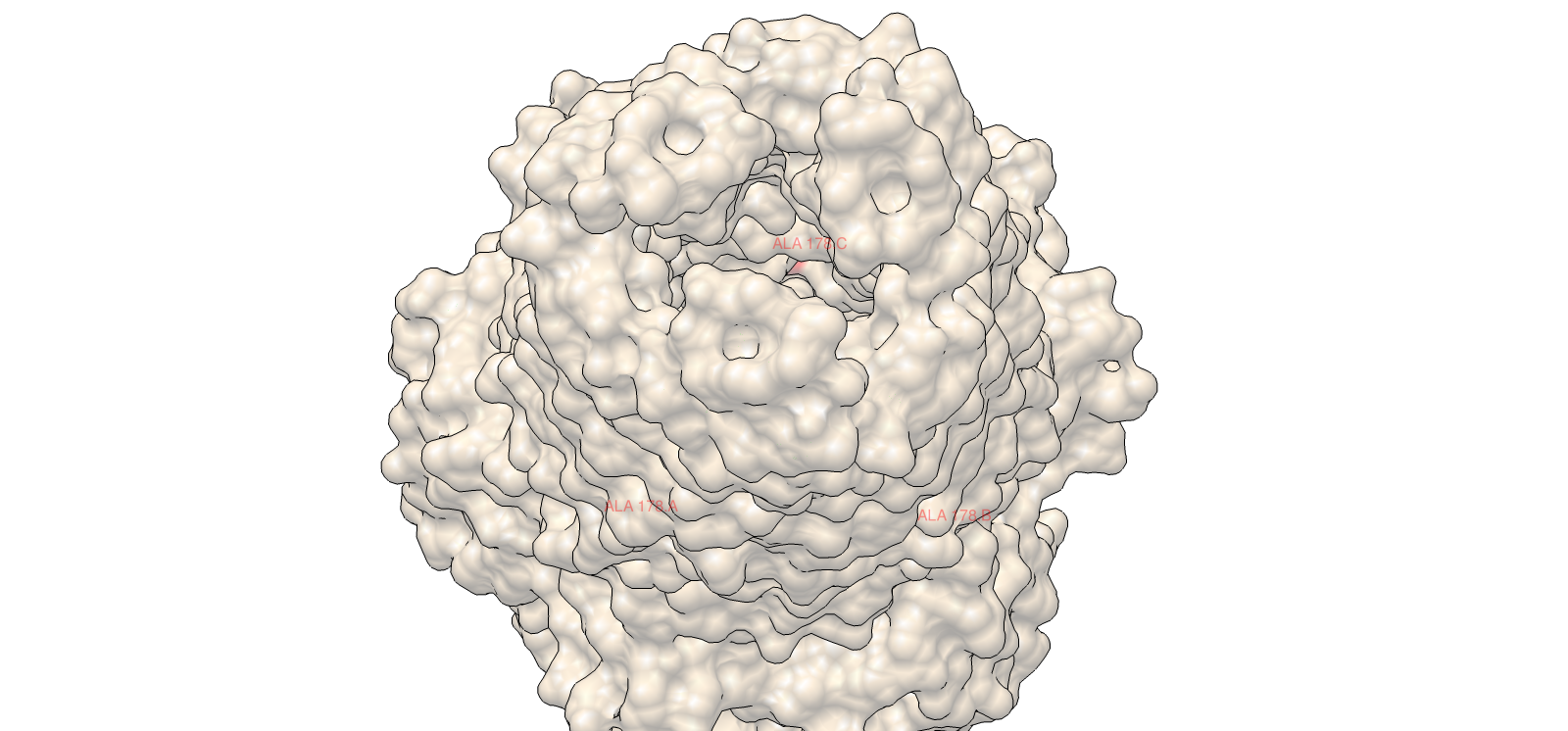
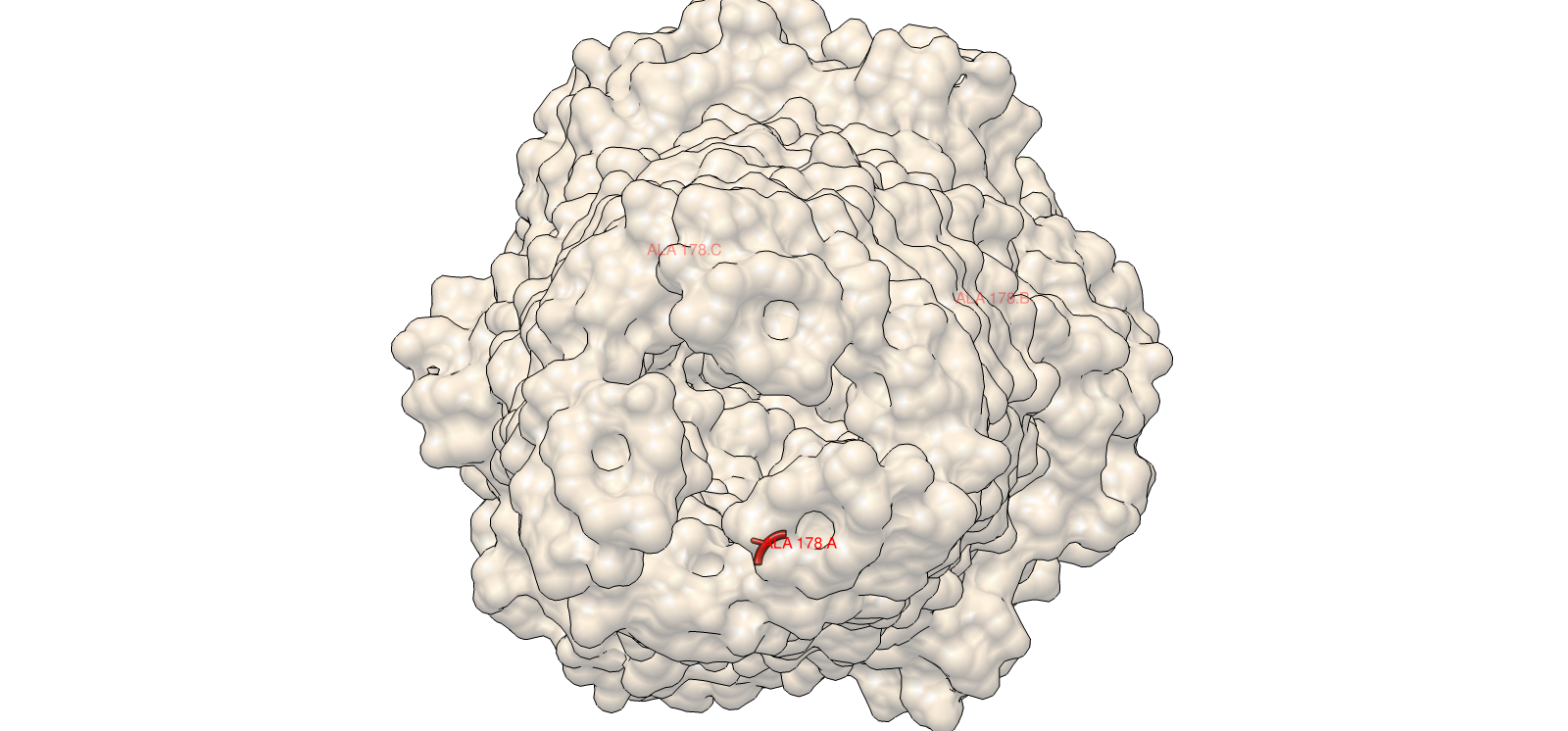
ESMFold inference (TolC chain)
Using the notebook workflow:
- Sequence length: 428
- Mode: mono
- Device: CUDA
- Prediction: pTM 0.858, mean pLDDT 90.2 (min 41.4, max 96.3)
- Outputs saved: PDB, PAE, pLDDT, contacts
- TolC_ChainA_ESMFold_ptm0.858_r3.pdb
- TolC_ChainA_ESMFold_ptm0.858_r3.pae.txt
- TolC_ChainA_ESMFold_ptm0.858_r3.plddt.txt
- TolC_ChainA_ESMFold_ptm0.858_r3.contacts.txt
This combination of language-model scoring and structural context gave a consistent interpretation of constraint and stability.
Additional outputs:
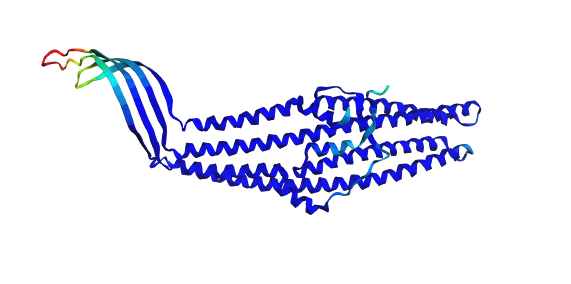
1B) Latent Space Analysis (ESM2 Embeddings)
Using ESM2 embeddings, protein sequences were projected into reduced-dimensional space using t-SNE. Each sequence was represented by the mean of its final hidden state embeddings, generating a fixed-length vector per protein. Dimensionality reduction to three components revealed structured clustering rather than random dispersion.
Proteins grouped into coherent neighborhoods, suggesting the embedding captures functional and structural similarity. When placing the TolC sequence into this latent map, it localized within a neighborhood consistent with outer membrane efflux proteins. Its nearest neighbors showed similar length profiles and domain architecture, supporting the idea that sequence-only embeddings can recover meaningful structural proximity.
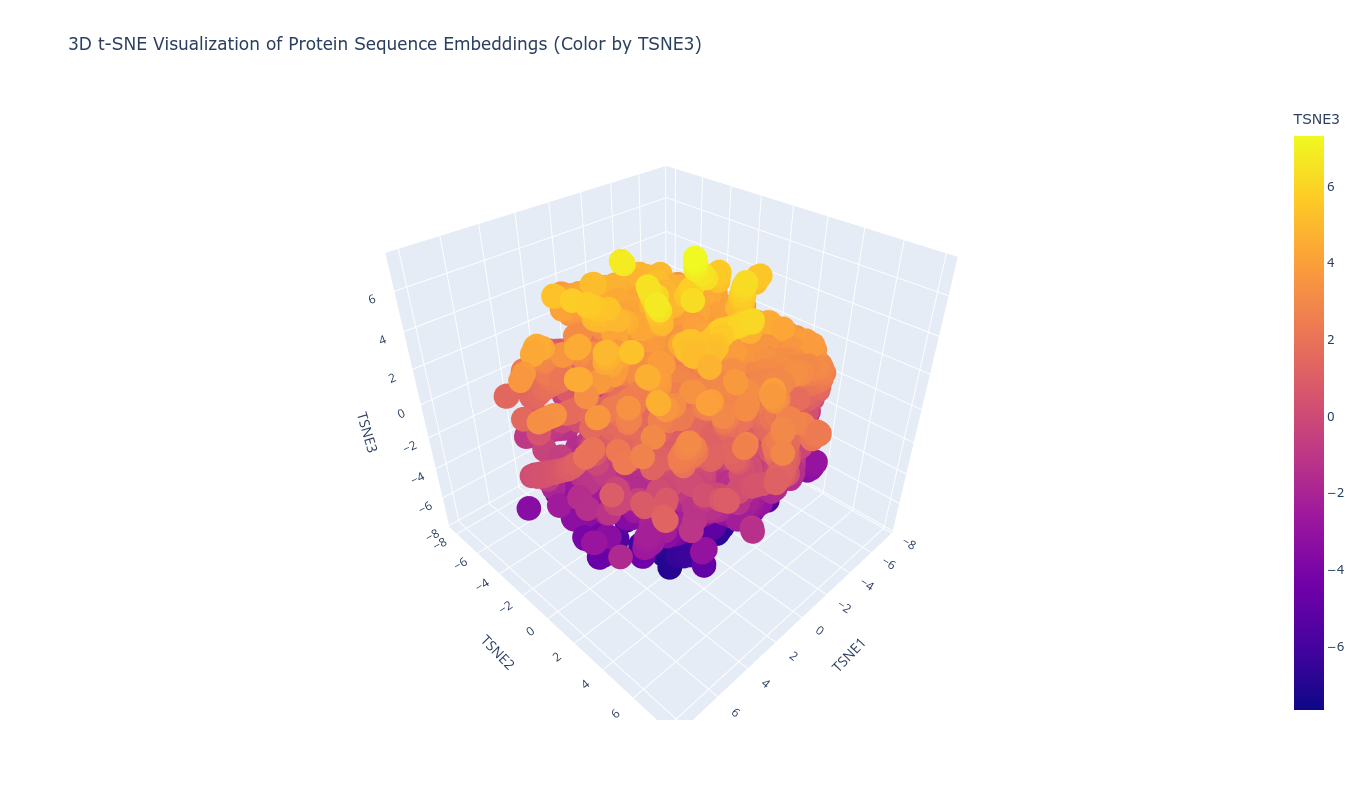
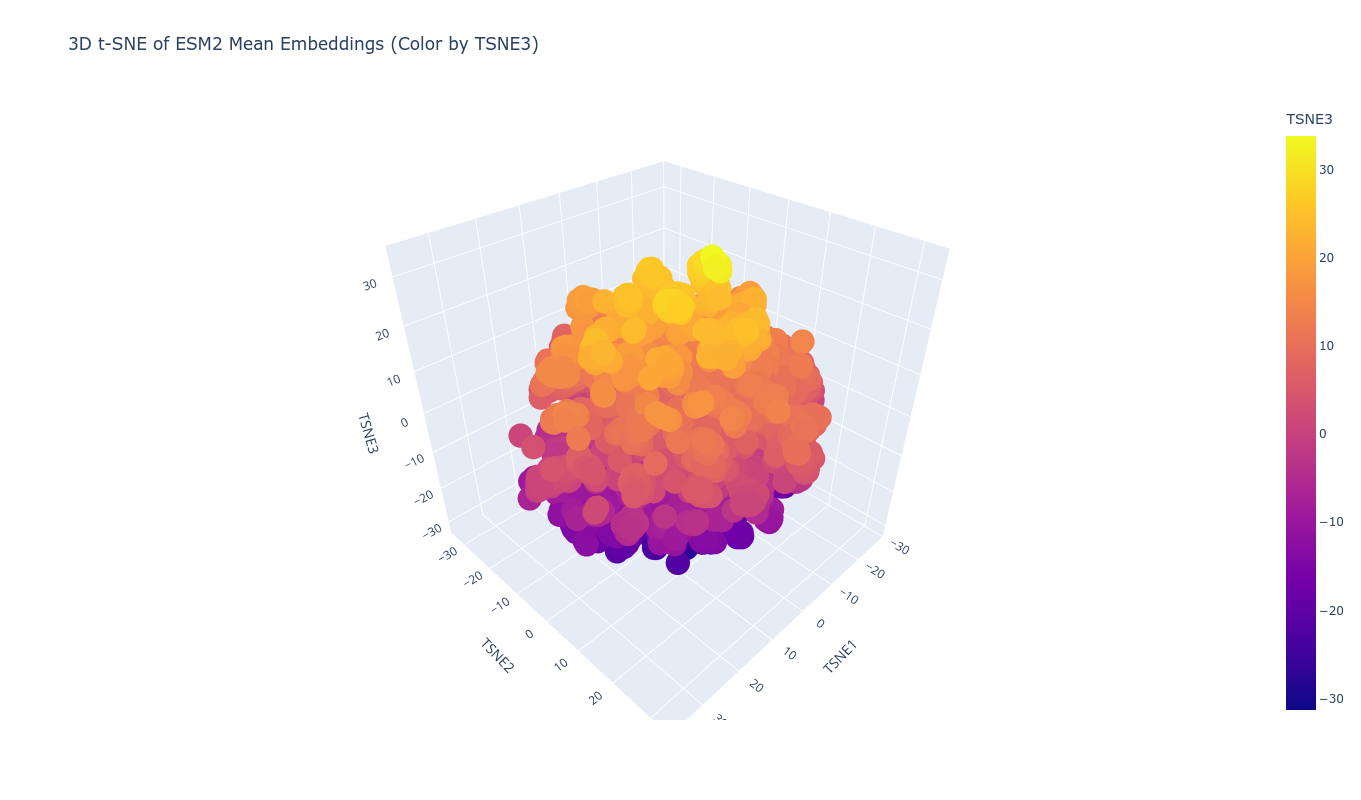
Top-10 nearest neighbors (cosine similarity):
- sim=0.6964 | d4nqra_ c.93.1.0 (A:) {Anabaena variabilis [TaxId: 240292]}
- sim=0.6958 | d3vvfa1 c.94.1.0 (A:1-236) {Thermus thermophilus [TaxId: 262724]}
- sim=0.6875 | d1tkja_ c.56.5.4 (A:) {Streptomyces griseus [TaxId: 1911]}
- sim=0.6858 | d1lu4a_ c.47.1.10 (A:) MPT53 {Mycobacterium tuberculosis [TaxId: 1773]}
- sim=0.6855 | d2w7qa_ b.125.1.0 (A:) {Pseudomonas aeruginosa PA01 [TaxId: 208964]}
- sim=0.6783 | d3jzja_ c.94.1.0 (A:) {Streptomyces glaucescens [TaxId: 1907]}
- sim=0.6747 | d4a82a1 f.37.1.1 (A:1-323) SAV1866 {Homo sapiens [TaxId: 9606]}
- sim=0.6687 | d5tfqa_ e.3.1.0 (A:) {Bacteroides cellulosilyticus [TaxId: 537012]}
- sim=0.6686 | d1xoca1 c.94.1.1 (A:17-520) OppA {Bacillus subtilis [TaxId: 1423]}
- sim=0.6658 | d3kcma1 c.47.1.0 (A:28-165) {Geobacter metallireducens [TaxId: 269799]}
Overall, the clustering behavior was consistent with the embedding reflecting shared fold-level or domain-level properties, rather than superficial sequence identity alone.
2A) Folding the Protein with ESMFold
The TolC sequence (length 428 residues) was folded using ESMFold with three recycles.
- Predicted pTM: 0.858
- Mean pLDDT: 90.2 (min 41.4, max 96.3)
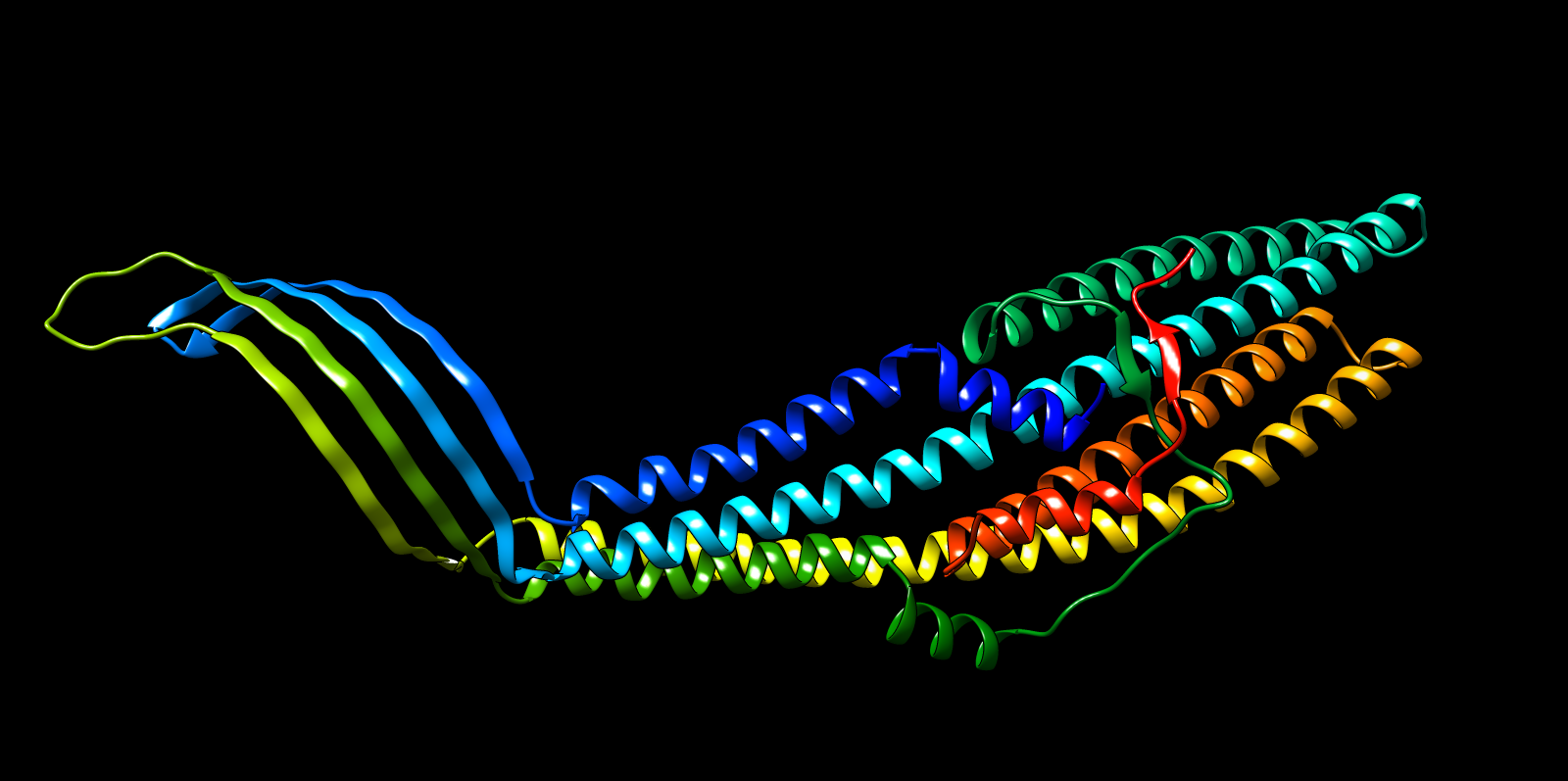
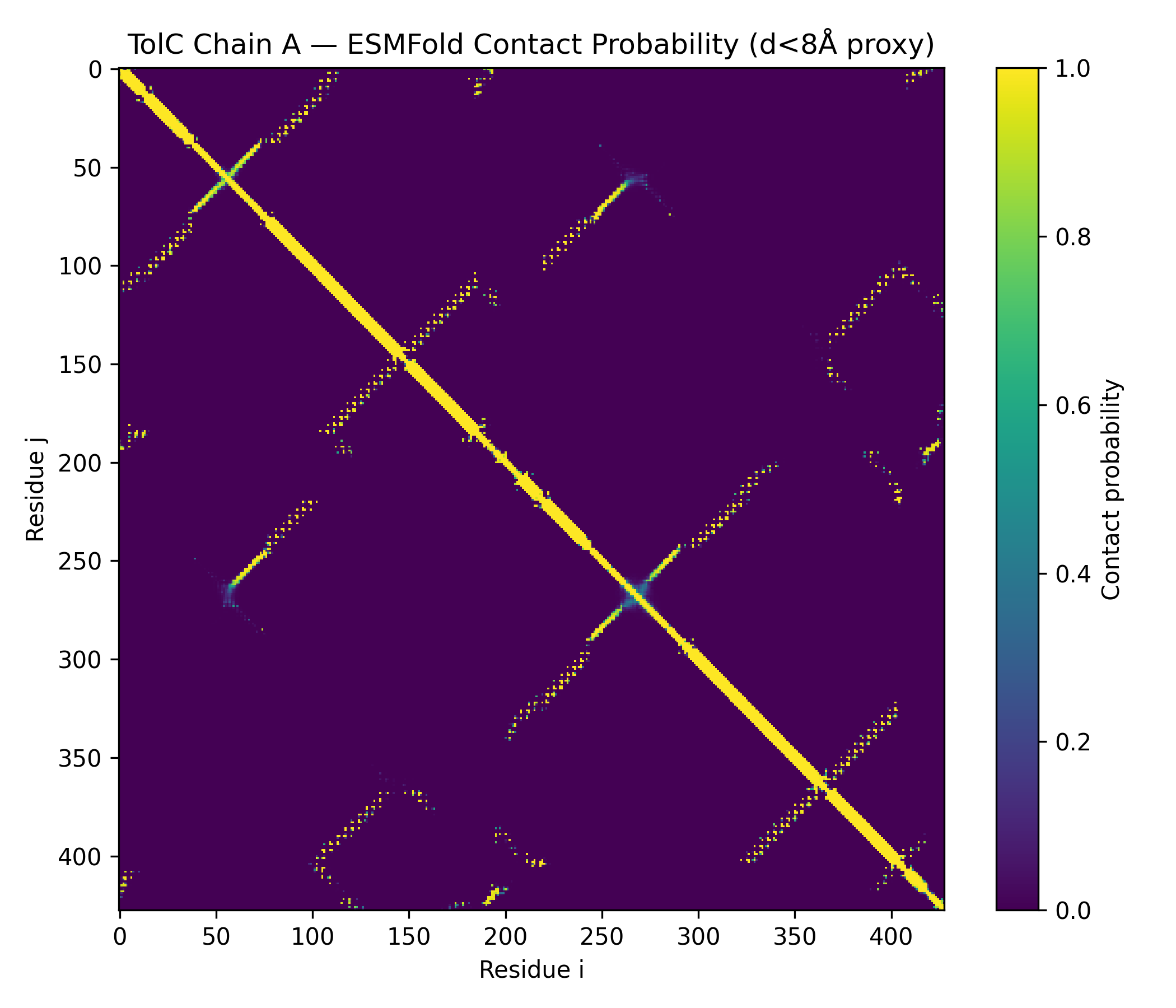
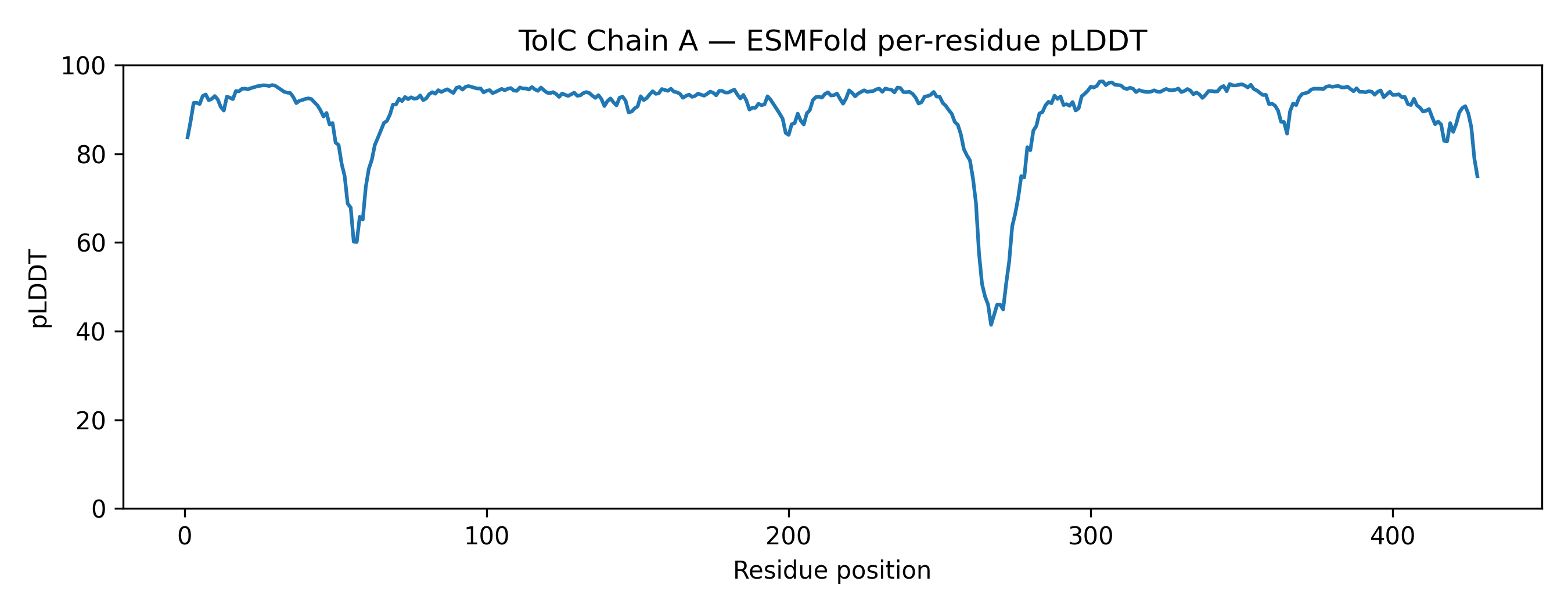
The predicted structure displayed a clear alpha-helical barrel architecture consistent with known TolC topology. Confidence was highest across the helical core and reduced mainly in flexible loop regions and termini, which is typical for long membrane-associated channels.
A structural check against experimental PDB 1EK9 showed strong global agreement in fold topology. The helical bundle organization was preserved, supporting the reliability of the prediction for this fold class.
2B) Structural Resilience to Mutation
Single mutation: W178D

Residue W178, identified as buried within the trimeric core, was mutated to aspartate (W178D). This substitution replaces a large hydrophobic aromatic residue with a charged polar residue.
ESMFold outputs:
- TolC_W178D_ESMFold pTM: 0.859, mean pLDDT: 90.3 (min 41.3, max 96.4)
- TolC_W178D_ESMFold_ptm0.859_r3.pdb
- TolC_W178D_ESMFold_ptm0.859_r3.plddt.txt
Interpretation: the mutant maintained high overall confidence and preserved the global helical barrel architecture. The expected effect is primarily local disruption around the buried site, consistent with the ESM2 penalty, rather than a full fold collapse.
Segment mutation: alanine window (173–182)
A short segment around position 178 was mutated to alanine residues to test fold robustness under broader perturbation.
- TolC_AlaWindow_173_182_ESMFold pTM: 0.845, mean pLDDT: 89.8 (min 42.7, max 96.4)
- TolC_AlaWindow_173_182_ESMFold_ptm0.845_r1.pdb
- TolC_AlaWindow_173_182_ESMFold_ptm0.845_r1.plddt.txt
Interpretation: compared to the single-site mutation, the alanine window produced a slightly lower confidence score and broader local destabilization, but the overall topology remained recognizable. This supports that TolC’s fold stability is distributed across the structure rather than being dominated by one residue.
3A) Inverse Folding with ProteinMPNN
Using the backbone coordinates of PDB 1EK9, ProteinMPNN generated alternative sequences compatible with the fixed TolC structure.
Run details captured in output:
- Model: v_48_020
- Edges: 48
- Noise: 0.2 Å
- Designed chains: A, B, C
- Sampling temperature: 0.1
- Native score (lower is better): 1.6983
- Best design score reported: 0.8601 (sample=2)
High-level pattern: the designed sequences remained strongly alpha-helix compatible, with many alanine, leucine, and lysine residues, consistent with maintaining a stable helical barrel scaffold.
FASTA output (ProteinMPNN_designs.fasta) was generated and evaluated for structural compatibility.
3B) Folding Designed Sequences with ESMFold
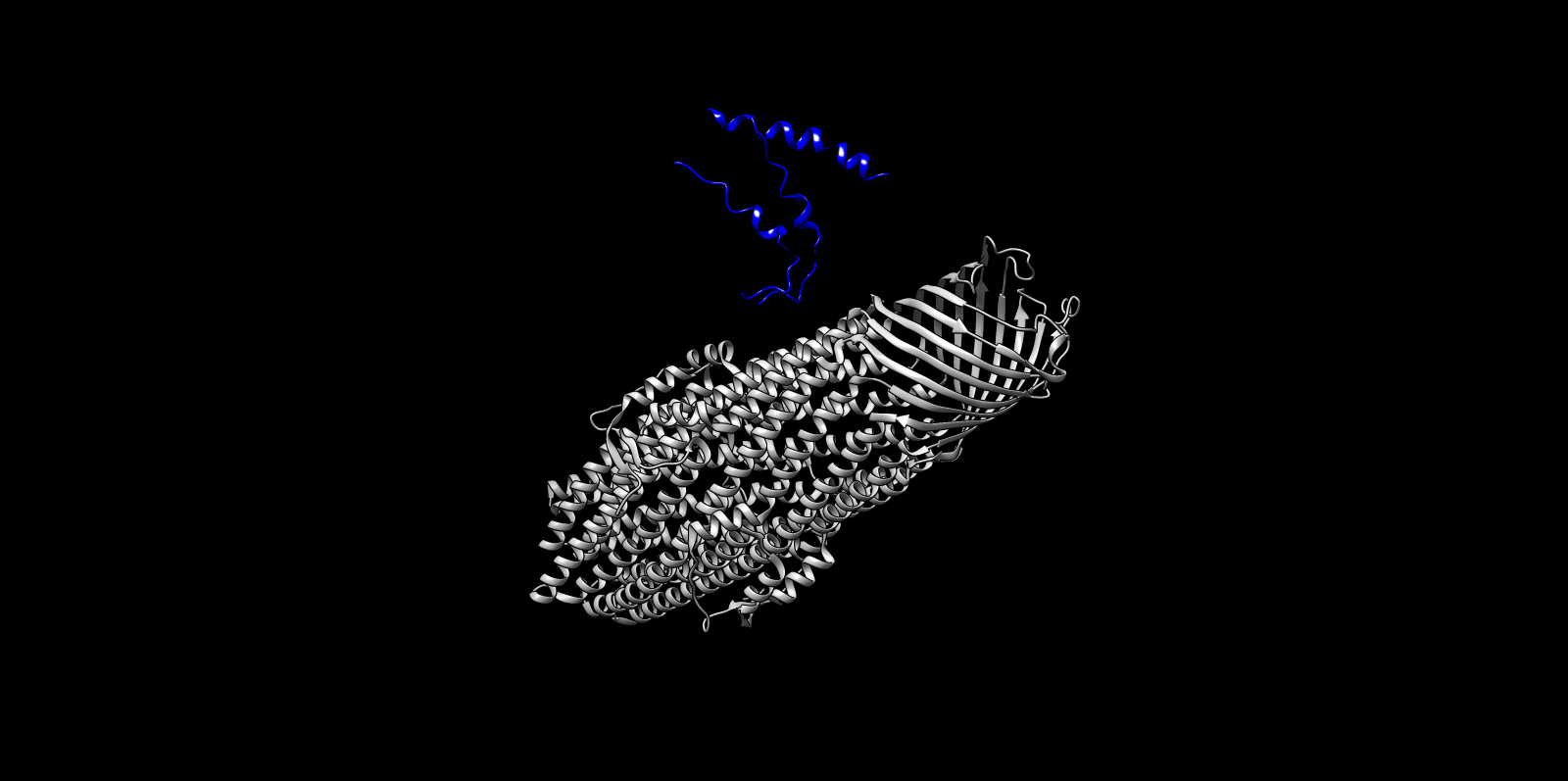
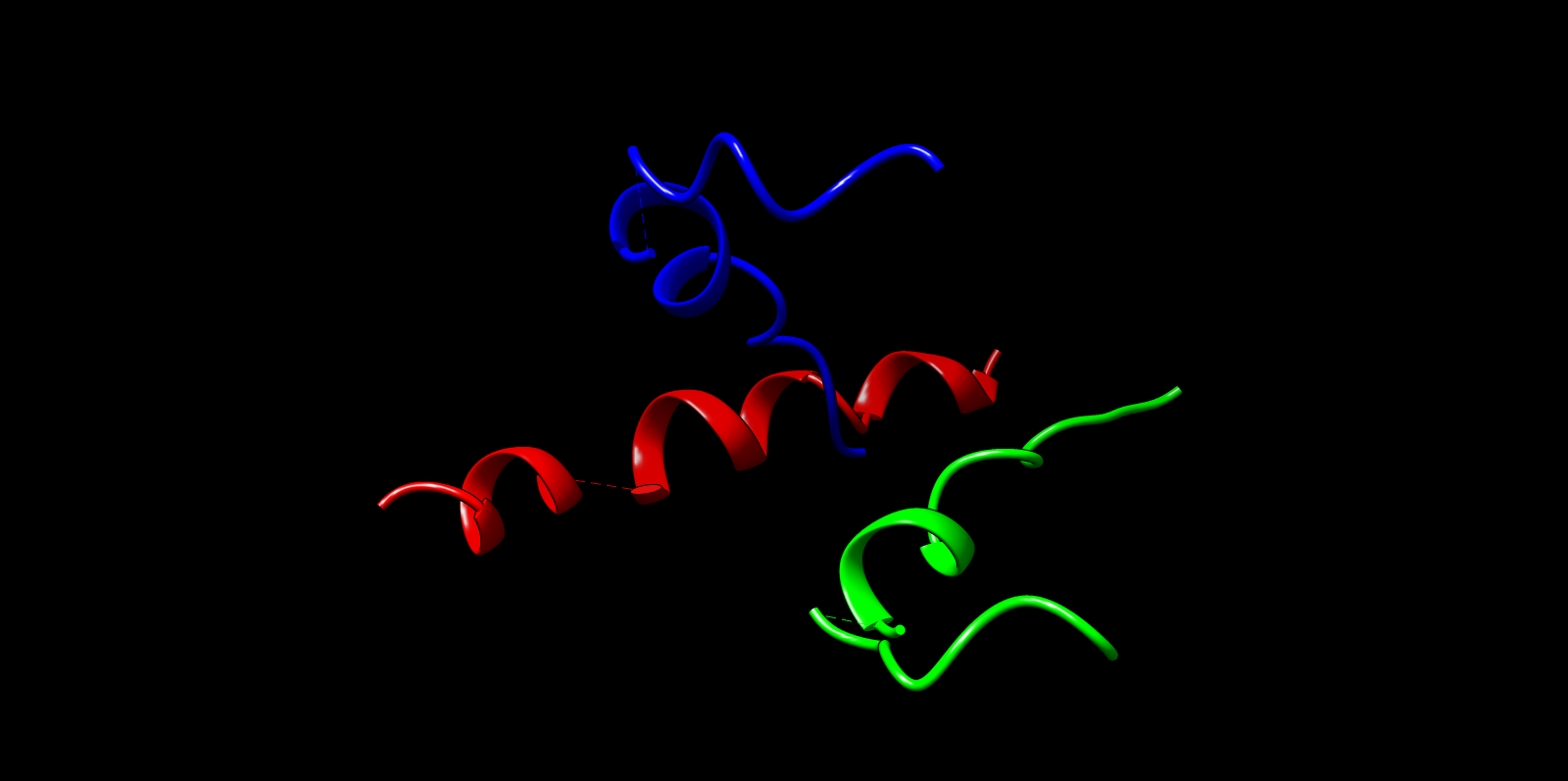
The top ProteinMPNN-designed sequence was refolded using ESMFold to assess structural compatibility. The predicted fold preserved the alpha-helical barrel topology. Differences were mainly confined to loop regions, while the core architecture remained consistent with the TolC backbone. This supports that ProteinMPNN successfully proposed sequences structurally compatible with the TolC fold.
Notebook note: the 3-chain complex folding run saved a PDB file:
- TolC_3chain_ESMFold_len69_r0.pdb
3C) Structural Alignment Interpretation
| Metric | Value | Meaning |
|---|---|---|
| Aligned residues | 22 | Only a small fragment of the full TolC structure was compared |
| RMSD | 2.49 Å | Shows reasonable backbone structural similarity within the fragment |
| Sequence identity | 4.5% | Very low sequence similarity |
| TM-score (normalized by reference structure) | 0.047 | Low because fragment is tiny relative to the full protein |
Why the TM-score is Low but RMSD is Informative
The TM-score appears low (0.047) because it is normalized by the length of the full TolC protein (423 residues). The designed model represents only 22 residues, so TM penalizes the short fragment. In contrast, RMSD is calculated over the aligned residues only, reflecting how well the fragment overlaps structurally with the native region. An RMSD of 2.49 Å indicates that the backbone conformation of the designed fragment reasonably resembles the native TolC fold.
Structural alignment between the designed TolC fragment and the native TolC structure (PDB: 1EK9) yielded an RMSD of 2.49 Å across 22 aligned residues, demonstrating moderate backbone similarity. The TM-score (0.047) is artificially low due to normalization against the full TolC protein (423 residues). Despite very low sequence identity (4.5%), the RMSD indicates that the designed fragment adopts a backbone conformation consistent with the corresponding native region.
Overall Conclusion
Across embedding analysis, forward folding, mutational perturbation, and inverse design, TolC shows:
- strong structural determinism captured by sequence models
- robustness of the global fold to a single-site perturbation (W178D)
- broader but still localized destabilization under a short alanine-window mutation
- backbone-constrained sequence flexibility under inverse folding, with high compatibility upon refolding
Overall, the results support that protein language models encode structural priors that transfer across mutation scanning, folding, and inverse design tasks.
Process Reflections
This assignment forced me to move beyond simply “running models” into understanding how each computational layer interacts with biological structure. I began with deep mutational scanning using ESM2, where selecting W178D and confirming its buried structural context in Chimera made the relationship between sequence, structure, and stability concrete rather than abstract. That step shifted my thinking from score interpretation to spatial reasoning.
In latent space analysis, I learned the importance of runtime management and reproducibility, especially when Colab resets interrupted long embedding jobs. Rebuilding Step 2 to function independently reinforced modular workflow design. ProteinMPNN inverse folding introduced another layer: generating sequences under structural constraints while interpreting native scores and recovery metrics carefully.
The most instructive challenge was ESMFold memory failure when attempting to fold the trimer as a single concatenated chain. Debugging GPU out-of-memory errors clarified how sequence length scales computational complexity. Representing the trimer properly and adjusting chunk size, precision, and recycles emphasized computational discipline.
Overall, this process strengthened my systems thinking: model outputs are not endpoints but components within an engineered pipeline requiring structural awareness, resource management, and iterative refinement
Works Cited
Works Cited
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., Smetanin, N., Verkuil, R., Kabeli, O., Shmueli, Y., dos Santos Costa, A., Fazel-Zarandi, M., Sercu, T., Candido, S., & Rives, A. (2023). Evolutionary-scale prediction of atomic-level protein structure with ESMFold. Science, 379(6637), 1123–1130. https://doi.org/10.1126/science.ade2574
Rives, A., Meier, J., Sercu, T., Goyal, S., Lin, Z., Liu, J., Guo, D., Ott, M., Zitnick, C. L., Ma, J., & Fergus, R. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. PNAS, 118(15), e2016239118. https://doi.org/10.1073/pnas.2016239118
Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas, R. J., Bethel, N., Leung, P. J. Y., Huddy, T. F., Pellock, S., Tischer, D., Chan, F., Koepnick, B., Nguyen, H., Kang, A., Sankaran, B., … Baker, D. (2022). Robust deep learning-based protein sequence design using ProteinMPNN. Science, 378(6615), 49–56. https://doi.org/10.1126/science.add2187
Koronakis, V., Sharff, A., Koronakis, E., Luisi, B., & Hughes, C. (2000). Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature, 405(6789), 914–919. https://doi.org/10.1038/35016007
National Center for Biotechnology Information. (2024). GenBank accession CAM8152351.1, Microcin M precursor [Escherichia coli]. https://www.ncbi.nlm.nih.gov/protein/CAM8152351.1
RCSB Protein Data Bank. (2000). PDB ID: 1EK9. https://www.rcsb.org/structure/1EK9
AI Prompts Employed (Claude AI)
- Why is ESMFold running out of GPU memory, and what does sequence length do to memory
- How do I represent a 3-chain complex properly in ESMFold without concatenating chains
- Rewrite the inverse folding protein process to minimise memory usage (half precision, chunking, fewer recycles)
- Add a safe CPU fallback that still saves the PDB cleanly
- Explain why TM-score can appear low while RMSD is still informative