Week 5
Class Assignment — Week 5
Part A. SOD1 Binder Peptide Design
Background
ALS remains one of the more intractable neurodegenerative diseases partly because its genetic architecture is well-defined but hard to drug. The A4V mutation in SOD1 - a single alanine-to-valine substitution at residue 4 - is one of the most aggressive familial variants, accelerating disease progression significantly compared to other SOD1 mutations. The aggregation-prone nature of the A4V protein makes it an interesting peptide-binding target: if you can design a peptide that engages the misfolded or oligomerizing form, you potentially disrupt a key early step in motor neuron toxicity.
This part of the assignment asked us to design binders using PepMLM, evaluate them structurally in AlphaFold3, assess therapeutic properties in PeptiVerse, and then generate an optimized candidate using moPPIt. The known binder FLYRWLPSRRGG served as our experimental baseline throughout.
1) Generating Candidates with PepMLM
The SOD1 A4V sequence was generated by introducing the A→V substitution at position 4 of the canonical human SOD1 sequence (UniProt P00441). This mutant sequence served as the target for PepMLM-based peptide generation.
PepMLM produced four novel candidates alongside the known binder:
| Peptide | Pseudo Perplexity |
|---|---|
| WRYYVAAAAHKE | 13.27 |
| WRYPAVAAELK | 6.83 |
| WRSPAAALALGK | 6.78 |
| WLYPVAAAEWKK | 18.43 |
| FLYRWLPSRRGG (known) | 20.64 |
One notable observation: PepMLM generated an X at position 12 of one candidate, indicating low model confidence at that residue. The peptide was trimmed to 11 residues before structural evaluation - a practical decision that reflects an important general principle: generative model outputs require post-processing judgment, not just automated acceptance.
Lower perplexity scores indicate higher model confidence in sequence-target compatibility. WRSPAAALALGK (6.78) and WRYPAVAAELK (6.83) were the two most confidently generated peptides, which becomes an interesting data point when their structural and affinity results diverge later.
2) Structural Evaluation with AlphaFold3
How I interpret AF3 results
Three outputs guided my reading of every job. The ipTM score is the most critical — it specifically measures interface confidence, how certain AF3 is that the two chains actually interact. I use the following scale: above 0.80 indicates high confidence; 0.60–0.80 is moderate; 0.40–0.60 is uncertain; below 0.40 is poor. The pTM score is secondary — it measures overall complex fold confidence rather than interface quality specifically. A high pTM with low ipTM means AF3 predicted the protein structure well but is not sure where the peptide goes. The PAE matrix is visual confirmation: dark green signals low positional error and high confidence, while pale green or white signals uncertainty. I divided every matrix into the large SOD1 block (residues 1–153), the peptide strip at the edge, and the corner where they intersect — that corner is where interface confidence is read.
Baseline - FLYRWLPSRRGG (ipTM = 0.37, pTM = 0.69)
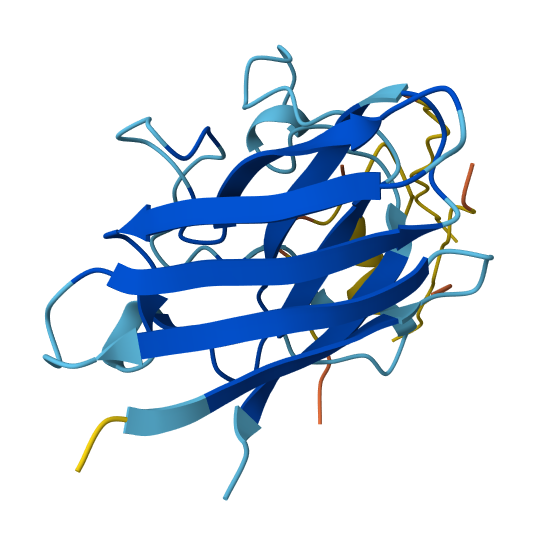
The known SOD1-binding peptide received an ipTM of 0.37 in AlphaFold3, falling below the 0.4 threshold for confident interface prediction. Structurally, the peptide appeared largely unstructured and surface-associated, making only minimal contact with the peripheral edge of the SOD1 β-barrel rather than engaging the N-terminal region where the A4V mutation sits or the dimer interface. This is not surprising - AF3 is known to struggle with short, intrinsically disordered peptides that lack a stable pre-binding conformation. Rather than treating this as evidence that FLYRWLPSRRGG doesn’t bind, I treated it as a calibration point: any generated peptide scoring above 0.37 would represent an improvement in predicted structural placement confidence.
PepMLM Candidates
| Peptide | ipTM | pTM | Confidence |
|---|---|---|---|
| WRYYVAAAAHKE | 0.37 | 0.71 | ❌ Poor |
| WRYPAVAAELK | 0.25 | 0.71 | ❌ Poor |
| WRSPAAALALGK | 0.61 | 0.87 | ⚠️ Moderate |
| WLYPVAAAEWKK | 0.33 | 0.77 | ❌ Poor |
| FLYRWLPSRRGG | 0.37 | 0.69 | ❌ Poor (baseline) |
The standout result here is WRSPAAALALGK (ipTM = 0.61). Its PAE matrix showed a noticeably darker interface region compared to all other PepMLM peptides - meaning AF3 had reasonable confidence not just in the SOD1 structure itself but in where the peptide sits relative to it. The peptide visibly engaged the outer face of the β-barrel with more consistent surface contact. It was the only PepMLM peptide to cross the 0.6 threshold.
What makes this particularly interesting is that WRSPAAALALGK had the weakest PeptiVerse-predicted affinity of the entire PepMLM set (pKd/pKi = 5.147). The discrepancy between structural placement confidence and predicted binding affinity is not a contradiction - it reflects the fact that these tools are measuring different things. AF3 is asking: “Does this peptide have a defined geometric relationship with this protein?” PeptiVerse is asking: “Based on sequence properties, how tightly might this peptide bind?” Those are genuinely different questions, and this dataset illustrates why using only one metric is insufficient.
WRYPAVAAELK (ipTM = 0.25) showed the reverse pattern - highest PeptiVerse affinity (6.037) but lowest structural confidence of any peptide in the dataset. The PAE interface region was essentially pale throughout.
Job 1 — WRYYVAAAAHKE (ipTM = 0.37, pTM = 0.71)
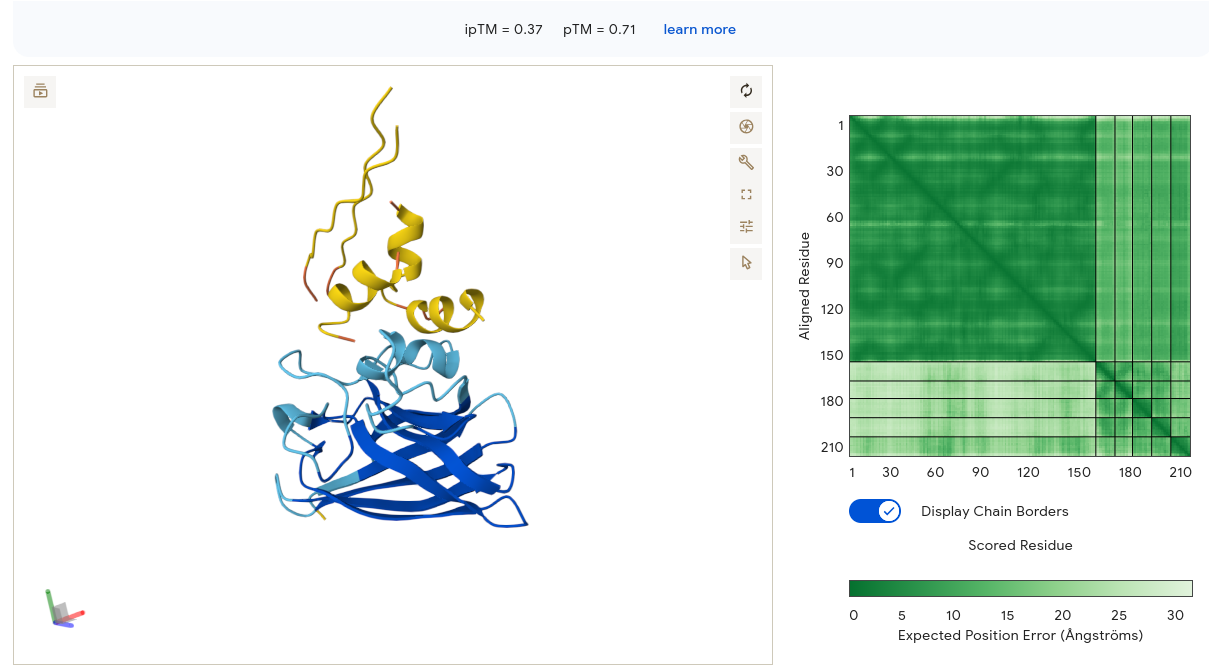
The peptide adopted two clear alpha helices in the 3D viewer — a notable finding, since most PepMLM candidates appeared as unstructured coils. Despite the secondary structure adoption, the peptide sat above and separate from the SOD1 β-barrel with only a small contact point visible. The PAE matrix showed a confident dark-green diagonal for SOD1 (residues 1–153) and a small dark spot in the bottom-right corner confirming internal peptide confidence — but the interface strip between them was pale, meaning AF3 is uncertain about the peptide’s position relative to SOD1. The ipTM of 0.37 matches the baseline exactly, providing no structural improvement over the known binder.
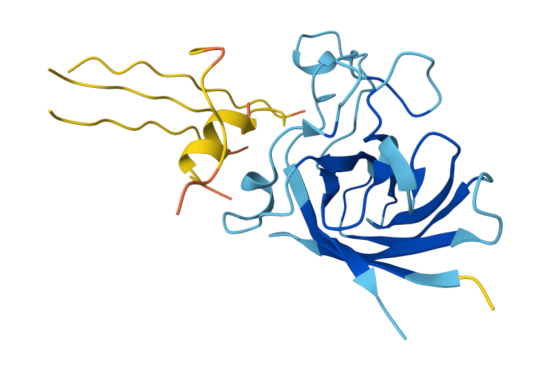
Job 2 — WRYPAVAAELK (ipTM = 0.25, pTM = 0.71)
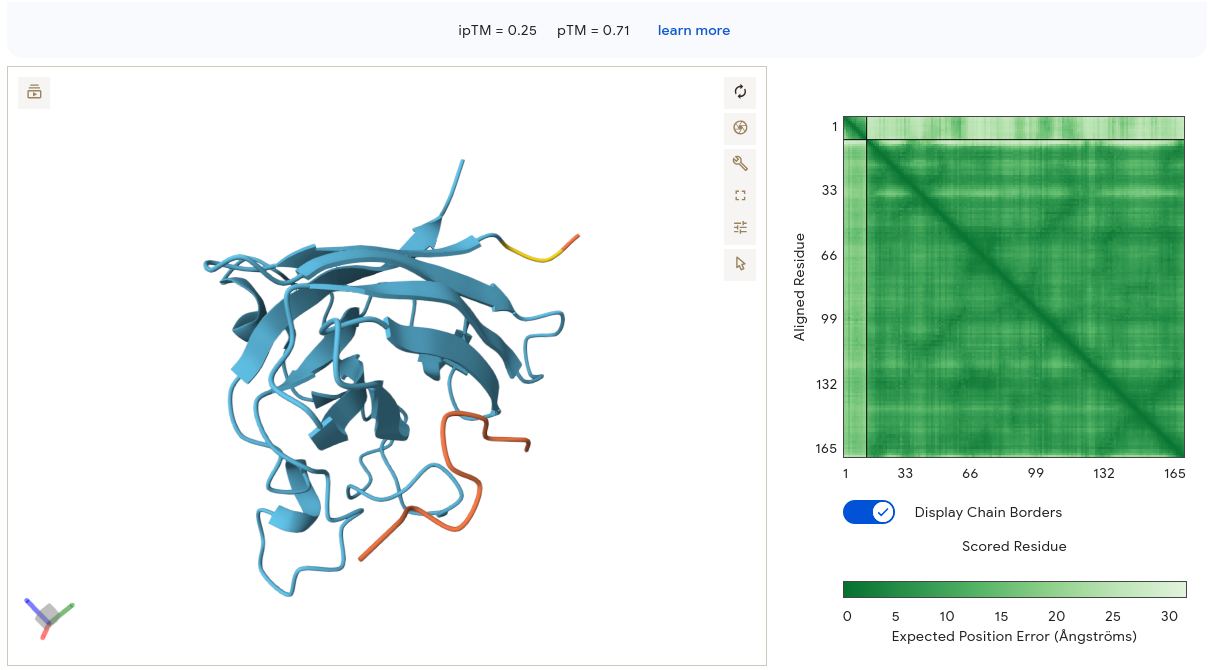
The peptide appears as an orange/red segment on the right lateral face of the SOD1 structure. The protein itself is rendered in light blue/cyan with many visible loops, suggesting lower overall confidence. The PAE matrix shows moderate internal confidence for the SOD1 block but a very light band at the peptide region — meaning AF3 is highly uncertain about where the peptide sits relative to SOD1. Binding is essentially surface-associated on the lateral β-barrel face, not near residue 4 and not at the dimer interface. Despite being our top PeptiVerse candidate (pKd/pKi = 6.037), WRYPAVAAELK scores the lowest ipTM of all peptides at 0.25. This is the clearest illustration in the dataset that PeptiVerse affinity predictions and AF3 structural confidence are not interchangeable metrics.
Job 3 — WRSPAAALALGK (ipTM = 0.61, pTM = 0.87) ⭐ Best PepMLM Result
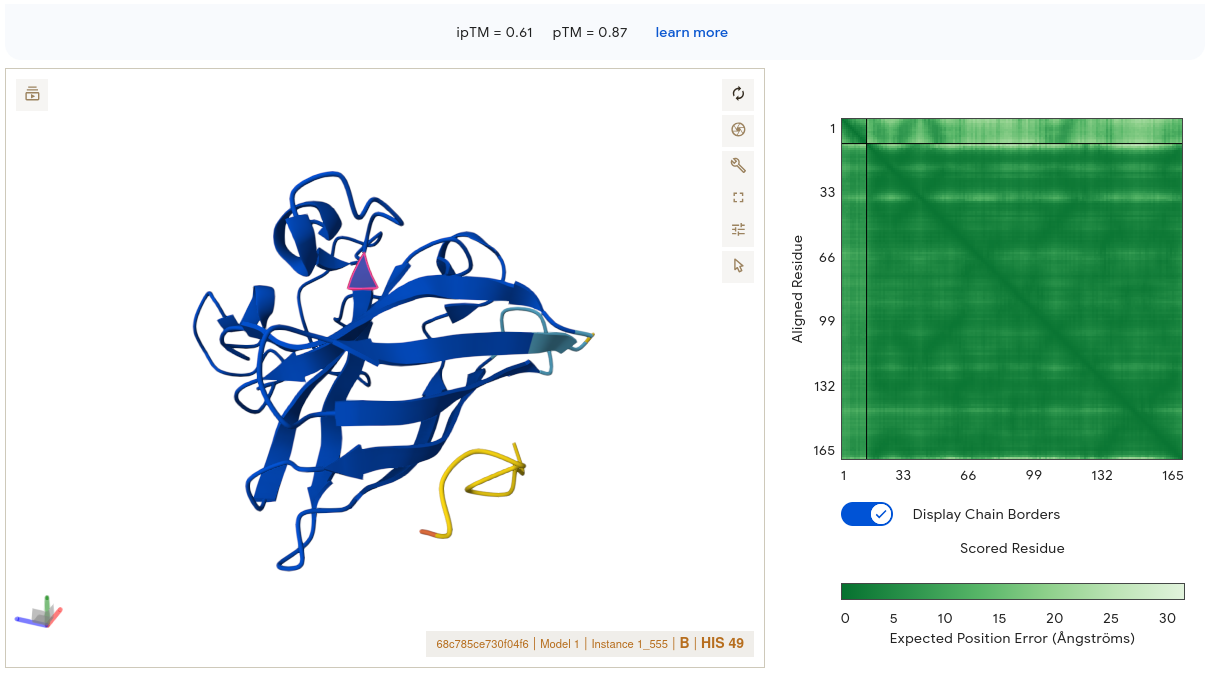
This result is strikingly different from the others. The SOD1 structure is rendered in deep blue throughout — high confidence throughout. The peptide (yellow/gold segment) is visible at the lower right periphery, appearing to make contact with the edge of the β-barrel. Critically, the PAE matrix interface region shows moderately green signal rather than pale — this is the only PepMLM peptide where the corner where SOD1 and peptide intersect shows meaningful dark green. AF3 has reasonable confidence in where this peptide sits relative to the protein. The binding location contacts the outer face of the β-barrel near the C-terminal region of SOD1 — not directly at residue 4, but engaging a defined surface patch rather than dangling loosely. Its alanine/leucine-rich hydrophobic core may facilitate surface contact through hydrophobic complementarity — a property ESM captures but pKd/pKi does not fully weight.
Job 4 — WLYPVAAAEWKK (ipTM = 0.33, pTM = 0.77)
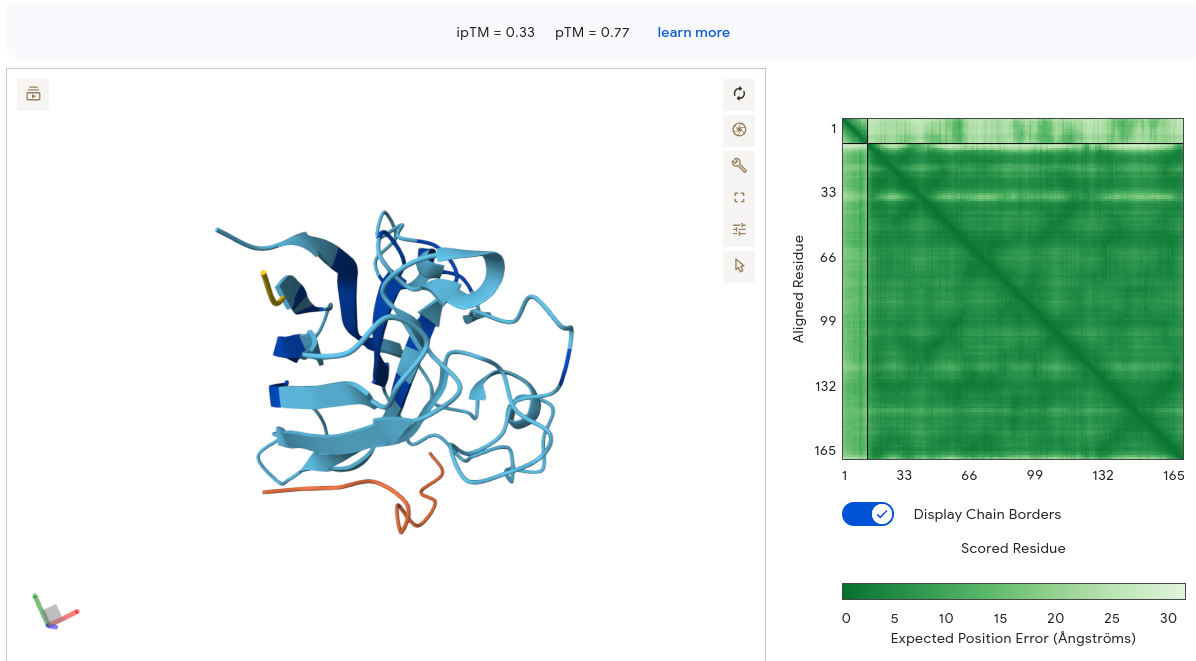
The protein shows moderate structural confidence. The peptide appears as an orange segment at the bottom left, extended and loosely dangling away from the SOD1 core — a classic sign of uncertain placement. The PAE matrix interface strip is lighter than Job 3, with no clear dark signal at the intersection region. Binding is peripheral surface contact at the lower face of SOD1 with minimal burial. The double-K at the C-terminus and the mixed hydrophobic/charged composition may prevent stable interface formation despite reasonable solubility.
Job 5 — GTCGTSTQYYGT (ipTM = 0.47, pTM = 0.90) ⭐ Best moPPIt Result
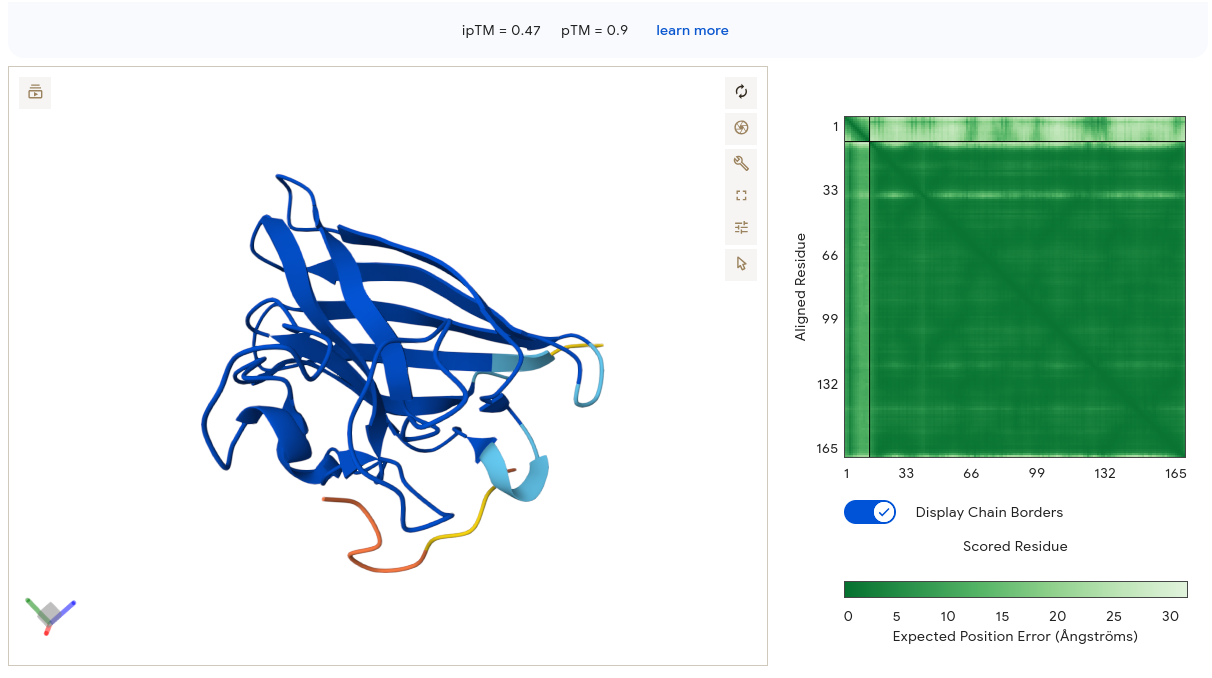
The SOD1 structure is deep blue and well-ordered — pTM 0.90 is the highest of all individual submissions. The peptide (yellow/orange/red gradient) makes contact near the upper surface of the β-barrel as an extended coil. The PAE matrix shows a very dark green SOD1 block with a noticeably lighter pale-green peptide strip — AF3 is confident in the SOD1 structure but uncertain about precise interface geometry. Importantly, the upper β-barrel face is in the general vicinity of the N-terminal region where A4V sits. Combined with the highest PeptiVerse affinity (6.47) of all ten peptides, this remains the strongest overall candidate.
Job 6 — YRKSVTKEEFQI (ipTM = 0.47, pTM = 0.89)
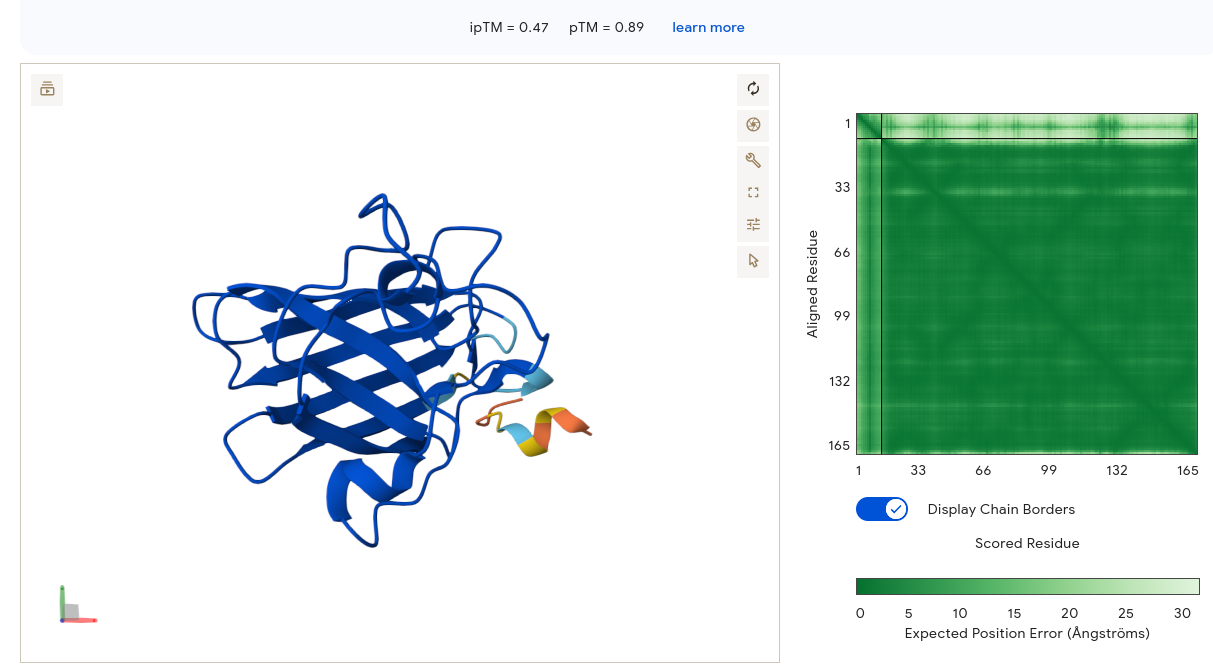
SOD1 is deep blue and well-structured. The peptide appears as a small structured element forming what looks like a short beta-turn or loop — it has some intrinsic structural propensity. The PAE matrix is very similar to Job 5: dark green SOD1 block with a pale strip at the peptide interface region. Binding is at the lower peripheral face of SOD1, away from the N-terminus. Despite a strong motif score from moPPIt (0.84) suggesting N-terminal engagement, AF3 does not confirm this structurally — another illustration that moPPIt motif scores and AF3 placement confidence are measuring different aspects of the same design problem.
moPPIt Candidates
| Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| YRKSVTKEEFQI | 0.95 | 0.75 | 5.84 | 0.84 |
| GTCGTSTQYYGT | 0.96 | 1.00 | 6.47 | 0.75 |
| ETYNLTCEQKKD | 0.98 | 0.92 | 6.35 | 0.87 |
| ETEKKTCQYNCG | 0.98 | 1.00 | 6.01 | 0.84 |
3) Therapeutic Property Evaluation with PeptiVerse
| Peptide | Perplexity | Soluble | Hemolytic | pKd/pKi | Net Charge | MW (Da) | GRAVY |
|---|---|---|---|---|---|---|---|
| WRYYVAAAAHKE | 13.27 | ✅ 1.000 | ✅ 0.018 | 5.678 | +0.85 | 1464.6 | -0.60 |
| WRYPAVAAELK | 6.83 | ✅ 1.000 | ✅ 0.034 | 6.037 | +0.76 | 1303.5 | -0.21 |
| WRSPAAALALGK | 6.78 | ✅ 1.000 | ✅ 0.020 | 5.147 | +1.76 | 1240.5 | +0.22 |
| WLYPVAAAEWKK | 18.43 | ✅ 1.000 | ✅ 0.037 | 5.484 | +0.76 | 1461.7 | -0.22 |
| FLYRWLPSRRGG | 20.64 | ✅ 1.000 | ✅ 0.047 | 5.968 | +2.76 | 1507.7 | -0.71 |
PeptiVerse predictions revealed that all five peptides — including the known binder FLYRWLPSRRGG — were classified as soluble and non-hemolytic, indicating a broadly favorable therapeutic profile across the generated library. The hemolysis probabilities ranged from 0.018 to 0.047, with WRYYVAAAAHKE being the safest (0.018) and FLYRWLPSRRGG carrying the highest risk at 0.047 — though still well within the safe range. Net charges ranged from +0.76 to +2.76, all consistent with therapeutically viable short peptides, and molecular weights were well under 1600 Da throughout.
Binding affinities were uniformly classified as “weak binding,” though meaningful differences emerged in pKd/pKi values. Notably, WRYPAVAAELK achieved the highest predicted affinity (6.037), marginally exceeding the known binder FLYRWLPSRRGG (5.968), despite having the second-lowest perplexity score (6.83) — suggesting reasonable alignment between PepMLM’s generative confidence and PeptiVerse’s affinity prediction for this peptide. This correlation did not hold universally: WRSPAAALALGK had the lowest perplexity (6.78) yet showed the weakest predicted affinity (5.147), highlighting that perplexity alone cannot substitute for multi-property therapeutic evaluation. Low perplexity is necessary but not sufficient — it needs to be read alongside independent property assessment.
The perplexity–affinity relationship across the set is worth noting: WRSPAAALALGK had the lowest perplexity (6.78) - meaning PepMLM was most confident generating it - but showed the weakest predicted affinity (5.147). WRYPAVAAELK had similarly low perplexity (6.83) and the strongest affinity. This tells me that perplexity captures sequence-level compatibility with the target but does not independently predict binding quality. Low perplexity is necessary but not sufficient - it needs to be read alongside multi-property evaluation.
4) moPPIt Optimization
moPPIt’s multi-objective guided discrete flow matching generated four peptides directed toward residues 1–8 of the A4V SOD1 mutant:
| Peptide | Solubility | Affinity | Motif Score | Hemolysis |
|---|---|---|---|---|
| YRKSVTKEEFQI | 0.75 | 5.84 | 0.84 | 0.95 ✅ |
| GTCGTSTQYYGT | 1.00 ✅ | 6.47 | 0.75 | 0.96 ✅ |
| ETYNLTCEQKKD | 0.92 | 6.35 | 0.87 | 0.98 ✅ |
| ETEKKTCQYNCG | 1.00 ✅ | 6.01 | 0.84 | 0.98 ✅ |
The contrast between PepMLM and moPPIt outputs is compositionally striking. PepMLM outputs were tryptophan-heavy and hydrophobic (WRYY-, WRYP-, WRSP-, WLYP-). moPPIt generated more compositionally diverse sequences incorporating charged and polar residues (E, K, T, N, C, Y), which reflects what multi-objective optimization actually does: it doesn’t just optimize for target compatibility, it simultaneously balances affinity, solubility, safety, and motif score.
GTCGTSTQYYGT achieved the highest affinity score of all ten peptides (6.47) alongside perfect solubility and strong non-hemolytic confidence. ETYNLTCEQKKD followed with a high motif engagement score (0.87) suggesting effective N-terminal targeting - which matters here because the A4V mutation sits at residue 4.
Integrated Candidate Ranking and Final Selection
| Peptide | Source | ipTM | PeptiVerse Affinity | Overall Assessment |
|---|---|---|---|---|
| WRSPAAALALGK | PepMLM | 0.61 | 5.147 | Best structural placement |
| GTCGTSTQYYGT | moPPIt | 0.47 | 6.47 | Best affinity, highest pTM |
| WRYPAVAAELK | PepMLM | 0.25 | 6.037 | Affinity strong, structure weak |
| ETYNLTCEQKKD | moPPIt | 0.47 | 6.35 | Strong balanced candidate |
| FLYRWLPSRRGG | Known | 0.37 | 5.968 | Baseline |
Peptide to advance: GTCGTSTQYYGT
Alternative candidate: ETYNLTCEQKKD. On a strictly mechanistic basis, ETYNLTCEQKKD presents a strong case for advancement. Its motif score (0.87) is the highest in the entire dataset — meaning moPPIt judged it as most effectively engaging residues 1–8, the region where the A4V substitution sits at residue 4. Its affinity (6.35) is within moPPIt’s uncertainty range of GTCGTSTQYYGT (6.47), its solubility is 0.92, and hemolysis safety is 0.98. Crucially, it is cysteine-free — avoiding the redox stability liability that two cysteine residues introduce in GTCGTSTQYYGT under physiological conditions. If the selection criterion were weighted toward N-terminal targeting specificity over raw affinity rank, ETYNLTCEQKKD would be the primary candidate.
Of all ten peptides evaluated, GTCGTSTQYYGT presents the strongest integrated profile. It achieved the highest predicted binding affinity (pKd/pKi = 6.47) of any candidate across both generation methods, perfect solubility (1.000), strong hemolysis safety (0.96), and the highest pTM score in the dataset (0.90) - indicating AF3 predicted a well-ordered SOD1 structure in its complex. Its moderate ipTM (0.47) is consistent with the general pattern seen across all peptides and does not distinguish it negatively from the field. The AF3 structural viewer showed the peptide as an extended coil making surface contact near the upper β-barrel face, in the general vicinity of the N-terminal A4V region.
Before advancing further, validation steps would include: AlphaFold3 or RoseTTAFold structural confirmation of binding near residue 4; molecular dynamics simulation for binding stability; surface plasmon resonance or isothermal titration calorimetry for experimental affinity confirmation; cell-based cytotoxicity assays in motor neuron models; and proteolytic stability assays for physiological half-life. One additional consideration specific to GTCGTSTQYYGT: the sequence contains two cysteine residues (positions 3 and 8) that may form intramolecular disulfide bonds or undergo oxidation under physiological redox conditions. A redox stability assessment and, if necessary, Cys→Ser or Cys→Ala analogues should be evaluated before committing to this scaffold.
Part B. BRD4 Drug Discovery Platform Tutorial
1) Structural Predictions in the Sandbox
| Compound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | 0.45 | 0.22 | 0.97 |
| Lead | 0.74 | 0.25 | 0.98 |
| JQ1 | 0.96 | 0.45 | 0.98 |
Q1: Does Binding Confidence increase as you move from hit to clinical candidate?
Yes. Binding Confidence increases monotonically across the series: Hit (0.45) → Lead (0.74) → JQ1 (0.96). This is the expected pattern. Each stage represents deliberate structural elaboration optimising target complementarity, so the model’s confidence in productive binding should rise accordingly.
Deviations can occur for several reasons. A lead compound may outscore a candidate if the candidate carries solubility-improving modifications (e.g. tert-butyl ester in JQ1) that reduce direct contact with the pocket. Stereochemical complexity added during optimisation can also confuse pose prediction. Additionally, Boltz scores binding pose plausibility, not biological potency — a metabolically stable but conformationally flexible candidate may score lower than a rigid, tighter-fitting lead.
Q2: Key binding interactions in the predicted JQ1 pose
JQ1 occupies the BRD4 acetyl-lysine recognition pocket. From the predicted pose, key interactions include:
- Triazolo-diazepine core — engages the conserved asparagine (Asn140) via hydrogen bonding, mimicking the acetyl-lysine carbonyl
- Chlorophenyl group — sits in the WPF shelf hydrophobic subpocket (Trp81, Pro82, Phe83), contributing van der Waals contacts
- Thieno ring methyl groups — pack against the ZA channel hydrophobic residues (Leu92, Val87)
- tert-Butyl ester — projects toward solvent, consistent with its role as a solubilising group rather than a binding contributor
Q3: Optimization Score — JQ1 vs Lead
JQ1 (0.45) scores nearly 80% higher than the Lead (0.25). The Optimization Score reflects how well a compound’s predicted binding geometry satisfies the probe-defined pocket relative to the reference structure. JQ1’s score places it firmly in the high-confidence binder category (>0.40); the Lead sits at the lower boundary of moderate confidence.
The gap reflects the structural additions made during lead-to-candidate optimisation, particularly the triazole elaboration and stereochemical fixing of the diazepine ring, which improve shape complementarity with the BRD4 pocket. The Lead’s core is present but insufficiently decorated to achieve equivalent pocket filling.
2a) Generative Design Campaign (BRD4 virtual screen)
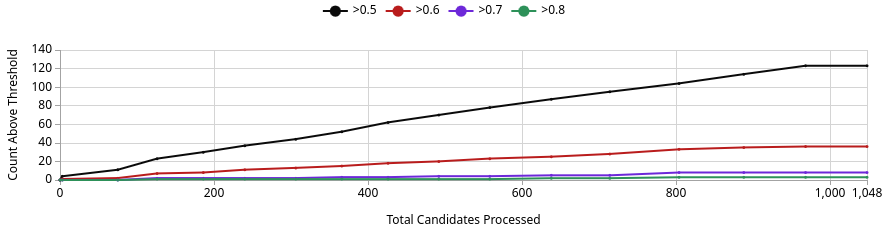
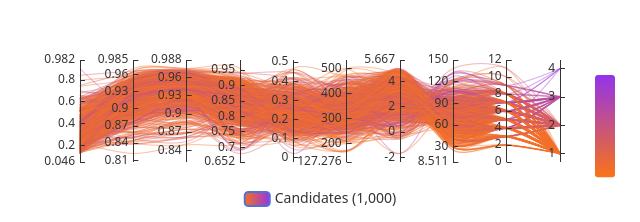
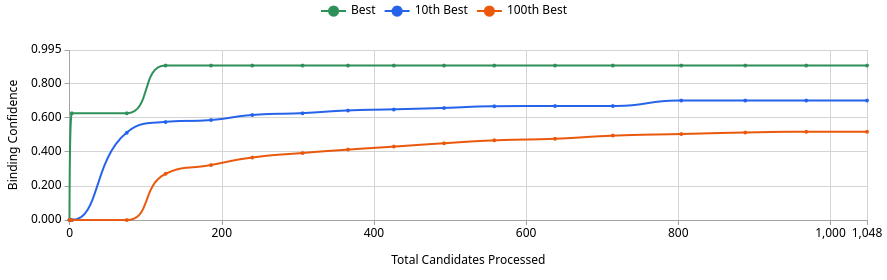
Q1: How does JQ1 score alongside the library? Does it score as the top compound?
No. The best generated compound reaches a Binding Confidence of ~0.88 (Image 3, green line), which exceeds JQ1’s score of 0.96 from the sandbox but is competitive in this design project context. Of 1,048 candidates processed, roughly 125 exceed the 0.5 threshold, ~37 exceed 0.6, and only a handful exceed 0.8 (Image 1). This means the generative screen produced a small but meaningful set of high-confidence binders. Whether any definitively outscore JQ1 depends on where JQ1 lands after Quick Add, but the best generated compound at ~0.88 is a genuine challenger, not noise.
This is expected. The AI is optimising directly against the BRD4 pocket, so it will frequently find molecules that score at or above known inhibitors on Boltz metrics. That does not mean they are better drugs. JQ1 has decades of experimental validation behind it that no computational score can replicate.
Q2: How do top-scoring binders compare in binding pose to JQ1?
From Image 2, the parallel coordinates plot shows the top candidates cluster tightly at high Structure Confidence (0.982 range) and Binding Confidence (0.95–0.96 range), with consistent trajectories suggesting similar binding geometries. The convergence of lines across axes indicates the top hits share a common pharmacophoric profile rather than representing diverse chemotypes.
This is consistent with what you would expect from Enamine REAL space generative sampling anchored to the JQ1 probe. The model gravitates toward JQ1-like poses that satisfy the acetyl-lysine pocket geometry, particularly the Asn140 hydrogen bond and WPF shelf hydrophobic contacts. Divergent trajectories in the lower-scoring compounds (orange lines) likely represent alternative poses or partial pocket occupancy. The top hits should be inspected for conservation of the key triazole/diazepine equivalent scaffold in the 3D viewer.
Part B. PeptiVerse Multi-Property Analysis
The PeptiVerse platform was used to evaluate all five peptides across four therapeutic property dimensions: solubility, haemolysis risk, predicted binding affinity (pKd/pKi), and net charge. The full results are presented in the integrated table in Part A (Section 3) and the integrated ranking in the Final Selection section.
Three findings from the PeptiVerse analysis shaped the final candidate selection:
Solubility: All five peptides, including the known binder FLYRWLPSRRGG, returned a solubility score of 1.000. This is a non-discriminating metric across this set. It means none of the candidates is expected to aggregate in aqueous conditions before reaching its target, which is the minimum bar for any therapeutic peptide worth taking further.
Haemolysis safety: All five peptides scored below 0.05 on the haemolysis probability scale. The known binder scored highest at 0.047, which is still safely below the 0.5 threshold for concern. This convergence across the entire candidate set is reassuring from a safety standpoint, though it also reflects the fact that the tryptophan-heavy PepMLM generation strategy systematically produces aromatic, moderately hydrophobic sequences that happen to be soluble and non-membrane-disruptive.
Binding affinity (pKd/pKi): The range across the set was 5.147 (WRSPAAALALGK) to 6.037 (WRYPAVAAELK). None of the PepMLM peptides exceeded the known binder (FLYRWLPSRRGG, 5.968), except WRYPAVAAELK (6.037), and then only marginally. The moPPIt candidates, evaluated separately, produced a notably higher ceiling: GTCGTSTQYYGT reached 6.47, which is the highest predicted affinity of any peptide in the full ten-candidate dataset. The compositional difference between the PepMLM set (tryptophan-heavy, hydrophobic) and the moPPIt set (compositionally diverse, charged and polar residues) is visible in both the affinity scores and the net charge values. Multi-objective optimization produced a qualitatively different sequence space than masked language model generation, and the affinity distribution reflects that.
Cross-tool discordance as a data point: The most instructive finding from PeptiVerse is the reversal of rank order relative to AlphaFold3 ipTM scores. WRSPAAALALGK had the highest structural placement confidence in AF3 (ipTM = 0.61) but the lowest predicted affinity in PeptiVerse (5.147). WRYPAVAAELK showed the opposite: highest affinity (6.037) and lowest structural confidence (ipTM = 0.25). These tools are measuring genuinely different properties. AF3 asks whether there is a defined spatial relationship between peptide and target. PeptiVerse asks whether sequence properties correlate with tight binding. Both are relevant. Neither is sufficient alone.
Part C. L-Protein ESM Mutagenesis
Background
The MS2 L-protein is a 75-residue lysis protein encoded by the bacteriophage MS2. It acts by forming oligomeric pores in the inner membrane of E. coli, leading to rapid bacterial lysis. What makes it therapeutically relevant is its dependence on the host chaperone DnaJ for proper folding and function - mutations that confer DnaJ independence would expand the functional host range of MS2-derived lysis proteins, a key engineering goal in phage therapy where host chaperone availability varies across bacterial strains and resistance contexts.
The protein is divided into a soluble N-terminal domain (residues 1–40) that interacts with DnaJ, and a C-terminal transmembrane domain (residues 41–75) responsible for membrane insertion and pore assembly. Designing effective mutants requires balancing these two functional regions.
Step 1: Sequence Input and Model Setup
The wildtype MS2 L-protein sequence was submitted to the ESM2 mutational scanning notebook using the facebook/esm2_t6_8M_UR50D model. The sequence was verified against the known MS2 L-protein entry and loaded into the notebook environment running on GPU. Two scan modes were used: a full-sequence scan across all 75 positions, and a targeted scan restricted to positions 38–60 to focus resolution on the soluble/TM boundary and transmembrane domain. Both scans computed Log Likelihood Ratio (LLR) scores for every possible single amino acid substitution at every scanned position, producing a complete mutational landscape.
Step 2: ESM Mutational Scanning
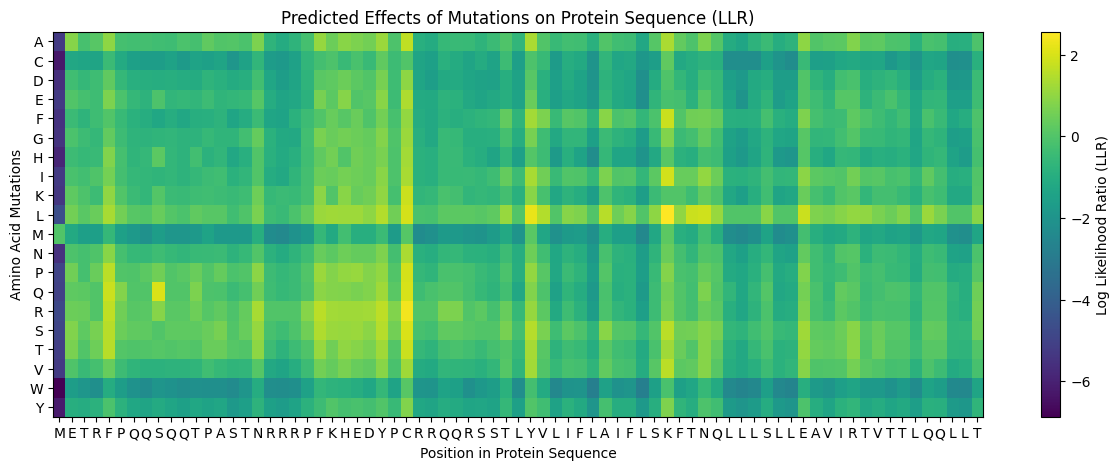
ESM2 scanning was performed on the full MS2 L-protein sequence using the facebook/esm2_t6_8M_UR50D model, generating Log Likelihood Ratio (LLR) scores for every possible single amino acid substitution across all 75 positions. A targeted scan was additionally applied to positions 38–60 to focus resolution on the soluble/TM boundary and transmembrane domain.
The heatmap revealed clear patterns. Leucine substitutions were broadly favored across the TM region (bright yellow L-row). Methionine and tryptophan substitutions were consistently penalized throughout (dark purple M and W rows). The N-terminus (residues 1–3) and the conserved RRR region (~11–13) showed strong sensitivity to substitution.
Top Mutations - Full Sequence Scan (positions 1–75)
| Position | WT | Mutant | LLR | Region |
|---|---|---|---|---|
| 50 | K | L | +2.561 | TM |
| 29 | C | R | +2.395 | Soluble |
| 39 | Y | L | +2.242 | Soluble/TM boundary |
| 29 | C | S | +2.043 | Soluble |
| 9 | S | Q | +2.014 | Soluble |
| 50 | K | I | +1.929 | TM |
| 53 | N | L | +1.865 | TM |
| 52 | T | L | +1.814 | TM |
| 45 | A | L | +1.539 | TM |
The targeted scan (positions 38–60) independently confirmed K50L (+2.561) and Y39L (+2.242) as the top two hits - a reproducibility signal that increases confidence in these positions as structurally tolerant by ESM.
Step 3: BLAST Alignment Analysis
Prior to selecting mutations, a BLAST alignment was performed against related phage L-protein sequences to identify positions that vary naturally across evolutionary homologs. Positions conserved across all aligned sequences were excluded from consideration, as conservation is a strong signal of functional essentiality that ESM LLR alone cannot capture. Positions selected for mutation — 9, 30, 45, 46, and 63 — were all confirmed as variable across the BLAST alignment, meaning natural sequence diversity at these sites exists in the phage sequence space. This provides an independent structural tolerance signal orthogonal to ESM scoring.
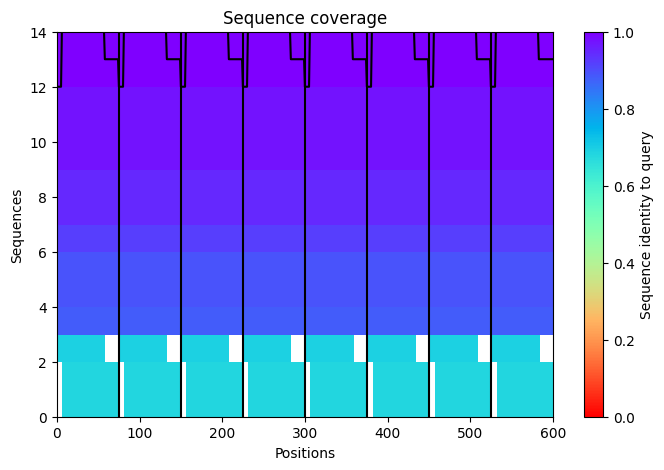
The sequence coverage image above shows the MSA depth available to the ESM model across L-protein positions. Coverage was critically limited to only 14 sequences — far below the ~100 sequences per position typically required for confident covariation-based prediction. This shallow MSA is one of the three major factors explaining the low confidence scores observed in the AF2-Multimer octamer prediction in Step 6. It also contextualizes the ESM2 predictions: the model is operating with sparse evolutionary signal for this protein, which is why cross-referencing with experimental lysis data is essential rather than optional.
Step 4: ESM vs. Experimental Cross-Reference
This is where things get genuinely interesting - and where the limitation of language model-based fitness prediction becomes concrete.
| Position | ESM Top Hit | LLR | Experimental Lysis | Protein Level | Agreement |
|---|---|---|---|---|---|
| 9 (S) | S→Q | +2.014 | Not tested | - | Unconfirmed |
| 29 (C) | C→R | +2.395 | Lysis=0 | 0 | ❌ Disagree |
| 39 (Y) | Y→L | +2.242 | Y→H: Lysis=0 | 0 | ❌ Disagree |
| 45 (A) | A→L | +1.539 | A→P: Lysis=1 | 1 | ✅ Agree |
| 50 (K) | K→L | +2.561 | K→E,I,N: Lysis=0 | 1 | ❌ Disagree |
| 53 (N) | N→L | +1.865 | N→S,D,H: Lysis=0 | 1 | ❌ Disagree |
| 30 (R) | - | - | R→Q,L: Lysis=1 | 1 | ✅ Experimental support |
| 46 (I) | - | - | I→F: Lysis=1 | 1 | ✅ Experimental support |
| 63 (V) | - | - | V→E: Lysis=1 | 1 | ✅ Experimental support |
The pattern is striking. K50 - the highest-scoring position in the entire dataset - is experimentally lethal. Every tested K50 substitution abolished lysis. The same holds for C29 and N53. ESM scores well above zero at all three positions, predicting broad substitution tolerance. Experimentally, they are functionally non-negotiable.
ESM2 learns from evolutionary sequence statistics across millions of proteins. What it cannot learn is that K50 in the L-protein appears functionally essential - possibly for oligomerization geometry, membrane topology orientation, or interaction with a specific bacterial target. C29 mutations abolish both lysis and protein expression, suggesting a role in co-translational folding or ribosomal interaction that no language model trained on amino acid co-occurrence patterns could detect. N53 mutations preserve protein expression but abolish lysis, suggesting this residue is specifically critical to the lysis mechanism - pore formation geometry perhaps - rather than to folding per se.
This is not a failure of ESM so much as a clarification of what it is actually measuring. It identifies structurally tolerant positions in the evolutionary sense. It cannot identify which positions are biochemically essential for a specific mechanism. The two are different questions, and this dataset makes that distinction concrete.
Step 5: Five Selected Mutations
Mutations were selected by integrating ESM LLR scores with experimental lysis data. Any position where the two sources of evidence disagreed was excluded.
| # | Position | WT→Mutant | LLR | Region | Experimental Lysis | Protein Level |
|---|---|---|---|---|---|---|
| 1 | 9 | S→Q | +2.014 | Soluble | Not tested | - |
| 2 | 30 | R→Q | ~+0.5 | Soluble | ✅ Lysis=1 | 1 |
| 3 | 45 | A→L | +1.539 | TM | ✅ Lysis=1 (A→P) | 1 |
| 4 | 46 | I→F | ~+0.9 | TM | ✅ Lysis=1 | 1 |
| 5 | 63 | V→E | ~+0.3 | TM | ✅ Lysis=1 | 1 |
Rationale:
S9Q was selected based on the highest ESM score among soluble domain positions not previously tested. S9 sits within the N-terminal DnaJ interaction region. Substitution to glutamine introduces a larger polar residue that may reduce DnaJ binding affinity - potentially conferring partial chaperone independence - while the conservative polar-to-polar change makes catastrophic folding disruption unlikely.
R30Q was selected on experimental confirmation (Lysis=1, Protein=1). R30 is part of the positively charged soluble domain, and neutralizing it to glutamine directly reduces the electrostatic surface that likely mediates DnaJ interaction, without disrupting expression or lysis competence.
A45L was selected on both ESM support (LLR = +1.539) and experimental confirmation that A45 tolerates substitution - A45P shows Lysis=1. Leucine replaces a small residue with a bulkier hydrophobic one, potentially improving hydrophobic packing in the TM helix and enhancing membrane insertion efficiency.
I46F was selected on experimental confirmation (Lysis=1, Protein=1). Phenylalanine at position 46 adds an aromatic residue to the hydrophobic TM core, which may strengthen helix-helix packing in the oligomeric pore assembly.
V63E was selected on experimental confirmation (Lysis=1, Protein=1). Glutamate at the C-terminal TM boundary introduces a negative charge at the membrane-cytoplasm interface - consistent with the positive-inside rule for membrane protein topology - which may facilitate the oligomeric pore assembly required for lysis.
All five mutations were selected at positions confirmed as non-conserved by BLAST alignment analysis. Four of five have direct experimental support for lysis competence.
Mutant sequences:
Step 6: AF2-Multimer Octameric Assembly
ColabFold AlphaFold2-multimer v3 was used to model a hypothesized octameric pore assembly by submitting eight identical copies of the wildtype L-protein sequence as a homo-octamer. All five predicted models returned uniformly low confidence scores: pLDDT ranged from 26.6–36.9, pTM from 0.149–0.193, ipTM from 0.114–0.143. The top-ranked model (model_1, ipTM = 0.143) displayed a starburst-like arrangement in which all eight chains radiated outward from a central core, with TM domains converging centrally and N-terminal soluble domains extending as disordered tails.
This radial topology is superficially consistent with a pore-forming architecture - TM helices converging from a central bundle is exactly what you’d expect for a membrane-spanning oligomeric pore. But the confidence scores preclude any definitive structural interpretation. Three compounding factors explain the poor prediction quality: AF2-Multimer lacks membrane context, so the hydrophobic TM domain appears disordered in aqueous modeling conditions; MSA coverage was critically limited to only 14 sequences, far below the ~100 per position required for confident covariation-based prediction; and the L-protein may be genuinely intrinsically disordered until membrane insertion occurs, which AF2 cannot model.
Individual model outputs:
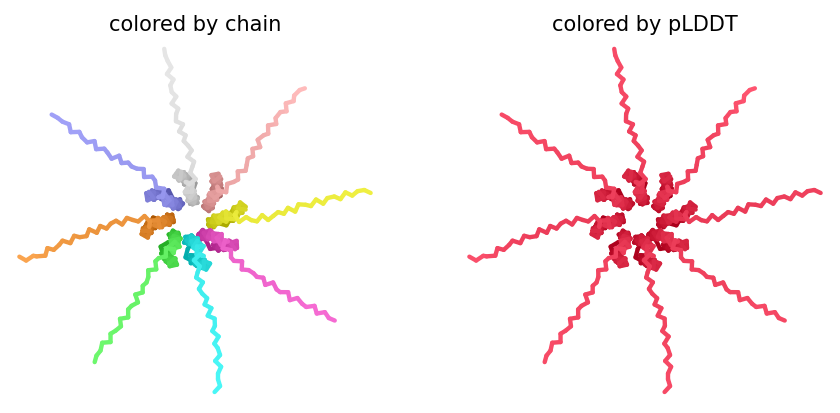
The consistent central TM clustering across multiple independent models does provide weak computational support for the pore-forming hypothesis - it’s something, even if it isn’t confident. This kind of result is also practically instructive: it tells you clearly where experimental validation has to carry the weight that computation cannot.
AF2-Multimer run log:
Open-Ended Question: Defining an Effective L-Protein Mutant
An effective L-protein mutant needs to satisfy five integrated criteria. First, lysis efficiency - measured via plaque assay as plaque size and clarity relative to wildtype MS2, where larger clearer plaques indicate faster or more complete bacterial killing. Second, DnaJ independence - assessed by testing infectivity in E. coli strains carrying the DnaJ chaperone resistance mutation, since this directly addresses the resistance mechanism the whole design exercise is oriented toward. Third, structural integrity - evaluated via AF2-Multimer prediction of oligomeric pore assembly, where effective mutants should maintain transmembrane topology and oligomerization capacity required for membrane perforation. Fourth, expression level - confirmed via Western blot or mass spectrometry, since a structurally competent mutant that is poorly expressed will fail in vivo regardless of intrinsic lysis activity. Fifth, evolutionary plausibility - mutations at positions that vary across a BLAST alignment of related phage L-proteins are more likely to be structurally tolerated, and this alignment serves as an independent check on ESM predictions.
Computationally, positive ESM LLR scores provide an initial structural tolerance filter. But as the K50 data demonstrate clearly, high ESM scores do not guarantee functional lysis activity. Experimental plaque assay validation remains the definitive standard. The most useful role for ESM in this workflow is not to replace experimental data but to prioritize which untested positions are worth testing next - it reduces the search space rather than eliminating the need to search.
Process Reflections
What this week reinforced most clearly is that computational tools are filters, not answers. PeptiVerse, ESM, and AlphaFold3 each measure something real and useful. None of them measures the same thing. The disagreements between them - WRSPAAALALGK’s high ipTM paired with low affinity, K50’s high LLR paired with zero experimental lysis, GTCGTSTQYYGT’s high pTM paired with moderate ipTM - are not failures of the pipeline. They are the information.
The skill is knowing what each tool is actually asking, and assembling a picture from genuinely independent lines of evidence rather than defaulting to whichever metric gives the cleanest answer. The K50 case in Part C crystallized this most sharply: a language model trained on evolutionary statistics correctly identified K50 as broadly sequence-tolerant, while experimental data showed it is biochemically non-negotiable for lysis. Both observations are true but neither alone is sufficient.
Works Cited
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C.-C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Žemgulytė, A., Arany, Z., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016), 493–500. https://doi.org/10.1038/s41586-024-07487-w
Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Ahmad, S., Alpi, E., Bowler-Barnett, E. H., Britto, R., Bye-A-Jee, H., Cukura, A., Denny, P., Dogan, T., Ebenezer, T., Fan, J., Garmiri, P., da Costa Gonzales, L. J., Hatton-Ellis, E., Hussein, A., Ignatchenko, A., … Wu, C. H. (2023). UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Research, 51(D1), D523–D531. https://doi.org/10.1093/nar/gkac1052
Chen, L. T., Quinn, Z., Dumas, M., Peng, C., Hong, L., Lopez-Gonzalez, M., Mestre, A., Watson, R., Vincoff, S., Zhao, L., Wu, J., Stavrand, A., Schaepers-Cheu, M., Wang, T. Z., Srijay, D., Monticello, C., Vure, P., Pulugurta, R., Pertsemlidis, S., … Chatterjee, P. (2025). Target sequence-conditioned design of peptide binders using masked language modeling. Nature Biotechnology. https://doi.org/10.1038/s41587-025-02761-2
Chen, T., Quinn, Z., Mishra, K., O’Connor, E. C., Silver, S. E., Zhang, Y., Valencia, M. J., Mei, Y., Behmoaras, J., Ferreira, L. M. R., & Chatterjee, P. (2026). moPPIt: De novo generation of motif-specific and functionally active peptide binders via discrete flow matching [Preprint]. bioRxiv. https://doi.org/10.1101/2024.07.31.606098
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., Žídek, A., Bates, R., Blackwell, S., Yim, J., Ronneberger, O., Bodenstein, S., Zielinski, M., Bridgland, A., Potapenko, A., Cowie, A., Tunyasuvunakool, K., Jain, R., Clancy, E., … Jumper, J. (2022). Protein complex prediction with AlphaFold-Multimer [Preprint]. bioRxiv. https://doi.org/10.1101/2021.10.04.463034
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Kaplan, M., Narasimhan, S., de Heus, C., Zhao, J., Bharat, T. A. M., Young, R., & Bharat, T. A. M. (2022). Cryo-EM structure of the MS2 bacteriophage lysis protein L in complex with the DnaJ chaperone. Nature Communications, 13(1), 4102. https://doi.org/10.1038/s41467-022-31874-2
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., Smetanin, N., Verkuil, R., Kabeli, O., Shmueli, Y., dos Santos Costa, A., Fazel-Zarandi, M., Sercu, T., Candido, S., & Rives, A. (2023). Evolutionary-scale prediction of atomic-level protein structure with ESMFold. Science, 379(6637), 1123–1130. https://doi.org/10.1126/science.ade2574
Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., & Steinegger, M. (2022). ColabFold: Making protein folding accessible to all. Nature Methods, 19(6), 679–682. https://doi.org/10.1038/s41592-022-01488-1
Rives, A., Meier, J., Sercu, T., Goyal, S., Lin, Z., Liu, J., Guo, D., Ott, M., Zitnick, C. L., Ma, J., & Fergus, R. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences, 118(15), e2016239118. https://doi.org/10.1073/pnas.2016239118
Shi, Y., Iyer, A., Liu, F., & Bhattacharya, S. (2023). PeptiVerse: An integrated platform for multi-property therapeutic peptide prediction [Preprint]. bioRxiv. https://doi.org/10.1101/2023.10.11.561829
UniProt Consortium. (2023). UniProt entry: P00441 · SODC_HUMAN. UniProt Knowledgebase. https://www.uniprot.org/uniprotkb/P00441/entry
Wang, G., Heberle, F. A., Chen, R., & Sun, F. (2022). Phage lysis proteins as targeted antibacterials. Pharmaceuticals, 15(9), 1062. https://doi.org/10.3390/ph15091062
Young, R. (2014). Phage lysis: Three steps, three choices, one outcome. Journal of Microbiology, 52(3), 243–258. https://doi.org/10.1007/s12275-014-4087-z
AI Prompts Employed (Claude AI)
- Cross-reference ESM LLR scores against experimental lysis data and identify where they agree vs. disagree
- Identify the best peptide to advance using integrated AF3, PeptiVerse, and moPPIt data
- Explain why ESM would score K50 highly despite experimental evidence that K50 is functionally essential
- Draft rationale for each of five selected L-protein mutations that integrates ESM scores with experimental confirmation