Week 6
Node participant note: I am a remote Genspace node listener based in Nigeria without onsite lab access. The Week 6 Gibson Assembly lab was a wet-lab session at Genspace nodes. In lieu of physical bench access, I engaged with the assembly logic computationally: the primer design, overlap verification, and construct validation workflows documented in Parts A and B were completed in Benchling and represent my full remote engagement with the lab material.
Class Assignment — Week 6
Part A. DNA Assembly
1. Components of Phusion High-Fidelity PCR Master Mix
A) Phusion DNA Polymerase A DNA-binding protein subunit that ensures higher template processivity, speed, and accuracy/fidelity alongside 5´→3´ polymerase activity and 3´→5´ exonuclease activity for proofreading.
B) Phusion Reaction Buffer (HF or GC) An optimized buffer that provides high salt concentrations used to stabilize primer-template hybridization. HF Buffer is the default for high fidelity, while GC Buffer helps with GC-rich or difficult templates.
C) MgCl₂ Provides the necessary magnesium ions for Phusion DNA polymerase activity.
D) dNTPs Exist as Deoxynucleoside triphosphates in either dATP, dTTP, dGTP, or dCTP. They act as the building blocks for synthesizing the new DNA strand.
E) DMSO Dimethyl sulfoxide acts alongside the Phusion reaction buffer as a PCR additive to aid the denaturation of templates with high GC content or complex secondary structures.
F) Stabilizers Components that maintain the integrity and activity of the enzyme during storage and cycling, often including bovine serum albumin (BSA).
2. Factors Determining Primer Annealing Temperature During PCR
Primer annealing temperature in PCR is primarily determined by the melting temperature of the primer-template duplex, which represents the temperature at which 50% of the primers are bound to the template.
A) Primer Melting Temperature Directly related to primer annealing temperature.
B) Primer Length Directly related to primer annealing temperature; optimally 18–24 bp.
C) GC Content Total percentage of GC content is directly related to primer annealing temperature; usually optimal at 40–60%.
D) Ionic Strength Mg²⁺ concentration is directly related to primer annealing temperature.
E) Primer Concentration Directly related to binding probability and therefore to primer annealing temperature.
F) Presence of Additives DMSO, glycerol, or formamide presence is inversely related to primer annealing temperature.
G) Target DNA When the target contains GC-rich templates, a higher primer annealing temperature is often required — i.e. directly related.
3. PCR vs. Restriction Enzyme Digests: Comparison of Two Methods for Creating Linear DNA Fragments
Mechanism PCR uses a thermostable polymerase to exponentially amplify a target region using designed primers, starting from a tiny amount of template. It generates millions of identical copies through cycles of denaturation, annealing, and extension. A restriction enzyme (RE) digest, on the other hand, uses sequence-specific endonucleases that recognize short palindromic sequences (typically 4–8 bp) and cleave both strands at or near that site, producing non-identical fragments defined entirely by where those sites happen to fall in the existing DNA.
Ends Produced PCR with standard primers produces blunt-ended fragments, but with Gibson-specific primers the overhangs are built into the primer sequence itself, so the linear product has the exact 20–22 bp overlap sequence that is designed. REs typically leave either sticky ends (4 bp 5’ or 3’ overhangs) or blunt ends depending on the enzyme. These sticky ends can be directly ligated but are constrained by the availability of RE recognition sites in the template.
When Each Is Preferred PCR is the clear choice when there is a need to introduce mutations, when no convenient RE site flanks the insert, or when customized overhangs are needed especially for Gibson assembly. RE digests are preferred when working with a well-characterized vector/insert system that already has compatible sites, when high fidelity without PCR-introduced errors is required, or when performing directional cloning into a backbone pre-cut with two different enzymes.
Error Profile PCR can introduce point mutations at a rate that depends on polymerase fidelity. Phusion HF, used in this lab protocol, has an error rate approximately 50× lower than Taq, making it appropriate for mutagenesis work where only the intended changes should be introduced. RE digests introduce no sequence errors.
4. Ensuring DNA Sequences Are Appropriate for Gibson Cloning
A) Overlapping sequences must be present and correct Gibson exonuclease chews back 5’ ends to expose single-stranded tails that then anneal to complementary tails on the adjacent fragment. If PCR primers were designed with the correct 20–22 bp overhang matching the adjoining fragment, the overlap is automatically built in. For RE-digested fragments, it is important to confirm that the sticky ends of one fragment are complementary to those of the adjoining fragment, which typically means using compatible enzymes (e.g., BamHI + BglII both produce GATC overhangs).
B) Fragment orientation must be correct (5’→3’) Each primer and fragment sequence should be verified in Benchling or SnapGene to confirm that directionality is preserved. A reversed insert is the most common and often the most costly error.
C) Fragment length and concentration must be within working range After gel electrophoresis, bands must appear at the expected sizes — backbone at approximately 3 kb and insert at approximately 300 bp as expected from the mUAV plasmid. Nanodrop concentration should exceed approximately 30 ng/µL.
5. How Plasmid DNA Enters E. coli Cells During Transformation
The process involves heat-shock transformation with chemically competent DH5α cells. Competent cells are pre-treated with divalent cations (typically CaCl₂), which partially neutralize the negative charge of the cell membrane’s lipopolysaccharide layer and the DNA backbone, reducing electrostatic repulsion. When the 42°C heat shock is applied for exactly 45 seconds, it creates a transient thermal imbalance that temporarily disrupts the membrane, creating pores or channels through which the plasmid can enter by diffusion. The cells are immediately transferred back to ice to reseal the membrane. Recovery in SOC media (Super Optimal broth with Catabolite repression) for 60 minutes at 37°C allows cells to repair the membrane, express the chloramphenicol resistance gene from the newly acquired plasmid, and begin dividing so that when plated on selective media, only transformants survive. Alternatively, electroporation works more definitively by using a brief high-voltage pulse to create quantifiable electropores, which generally yields higher efficiency than heat shock.
6. Alternative Assembly Method: Golden Gate Assembly
Overview
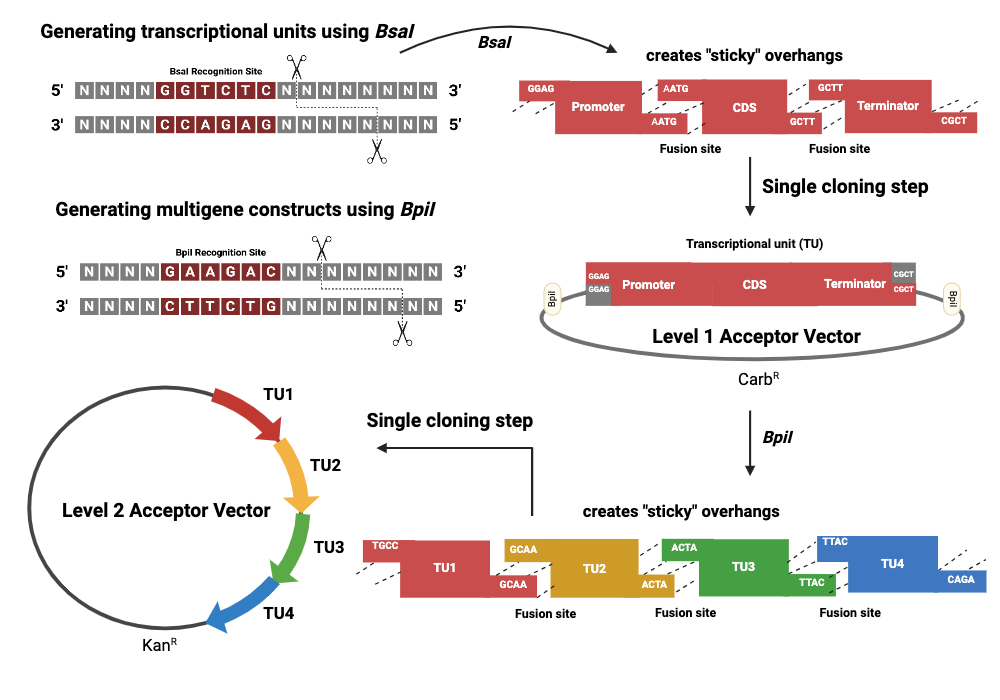
Golden Gate Assembly is a DNA assembly method that leverages Type IIS restriction enzymes — most commonly BsaI or Esp3I — which cut outside their recognition sequence at a defined offset, generating customizable 4 bp overhangs. Unlike conventional REs, which leave their recognition site in the product, the Type IIS enzyme cuts away from itself so that the recognition site is excised along with the surrounding primer sequence, leaving a scar-free junction. Each fragment is PCR-amplified with primers that embed the BsaI site facing outward, followed by the desired 4 bp overhang unique to that junction. The enzyme cuts all fragments simultaneously, exposing these complementary 4 bp tails, which then direct fragment annealing in the correct order — because only perfectly complementary overhangs will anneal stably. T4 DNA ligase seals the nicks in the same reaction tube. The reaction cycles between the cutting temperature (~37°C) and ligation temperature (~16°C) repeatedly, driving the equilibrium toward a fully assembled, circularized product. Golden Gate can assemble up to approximately 10 fragments simultaneously with high efficiency and directional fidelity, making it especially powerful for large combinatorial pathway assembly such as building multi-part biosynthetic operons, where Gibson’s exonuclease-dependent overlap system becomes less efficient.
Golden Gate vs. Gibson Assembly

Gibson uses a 5’ exonuclease to chew back fragments and generate long (20–40 bp) single-stranded overhangs for annealing, which then require a polymerase to fill gaps and a ligase to seal them. Golden Gate uses short 4 bp Type IIS-generated overhangs and no exonuclease — simpler biochemistry, but the overhangs are shorter and specificity depends entirely on the 4 bp sequence design. Ligation of wrong-order fragments can occur if overhang sets are not carefully designed to be unique. Gibson is more forgiving for large fragments; Golden Gate is faster and more multiplexable for modular, repetitive assemblies.
| Feature | Gibson Assembly | Golden Gate Assembly |
|---|---|---|
| Enzyme type | 5’ exonuclease + polymerase + ligase | Type IIS RE + T4 ligase |
| Overlap length | 20–40 bp | 4 bp |
| Scars left | None | None (RE site excised) |
| Max fragments | 5–6 efficiently | Up to 10+ |
| Best for | Large fragments, flexible design | Modular, combinatorial assemblies |
| Error risk | PCR errors at junctions | Wrong-order ligation if overhangs not unique |
Benchling Model
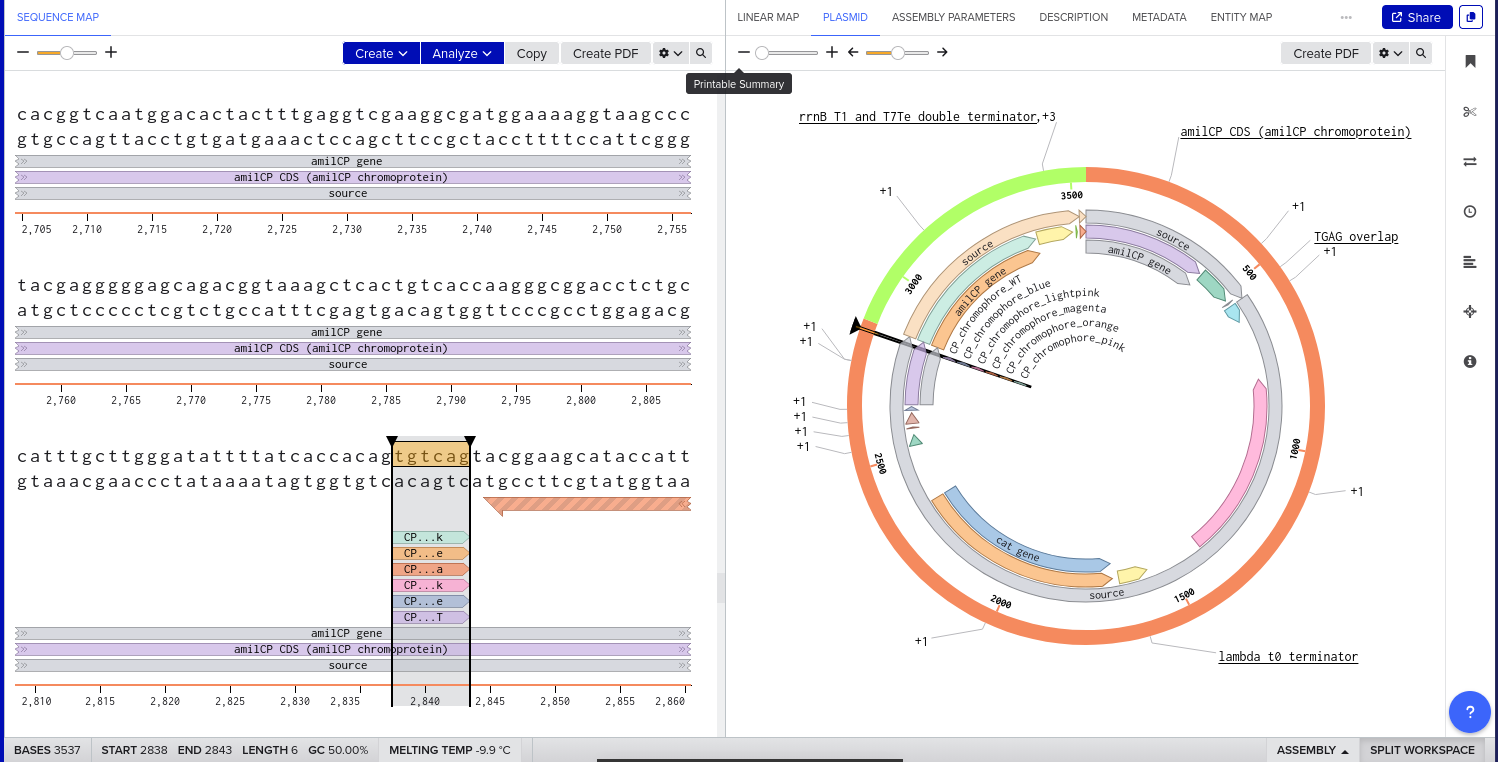
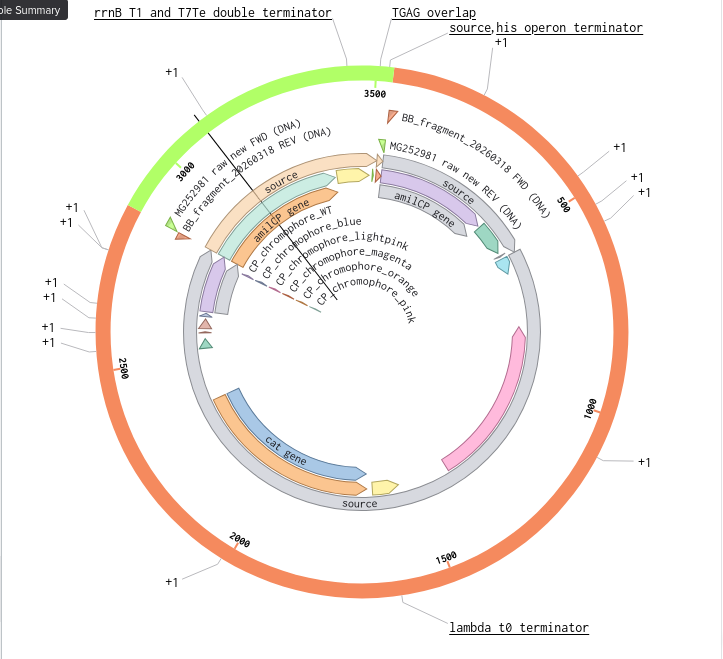
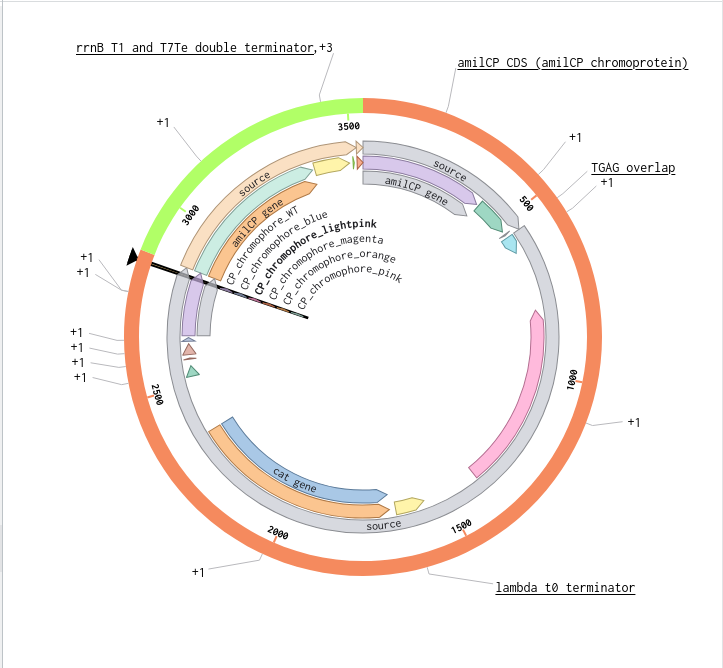
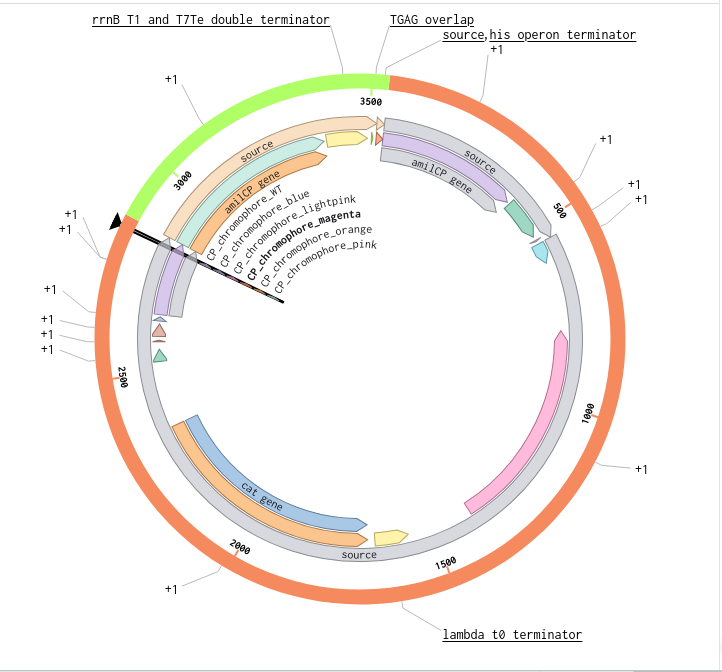
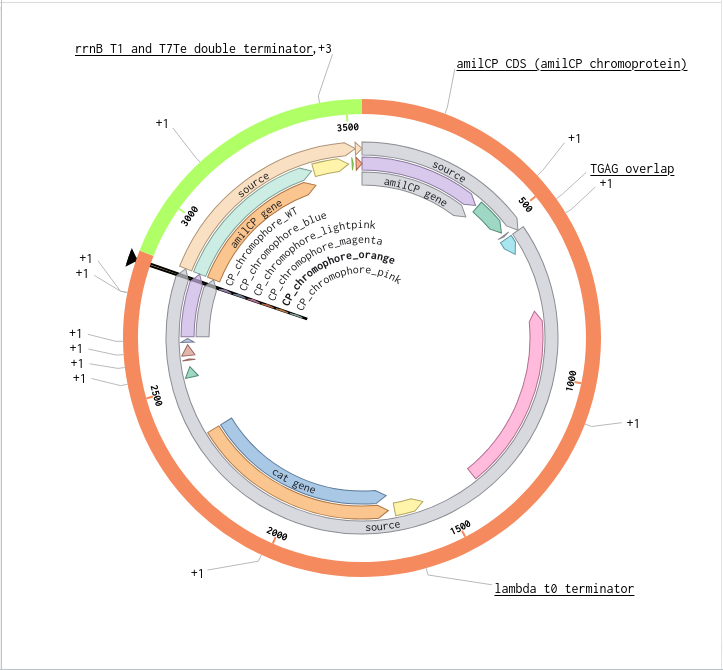
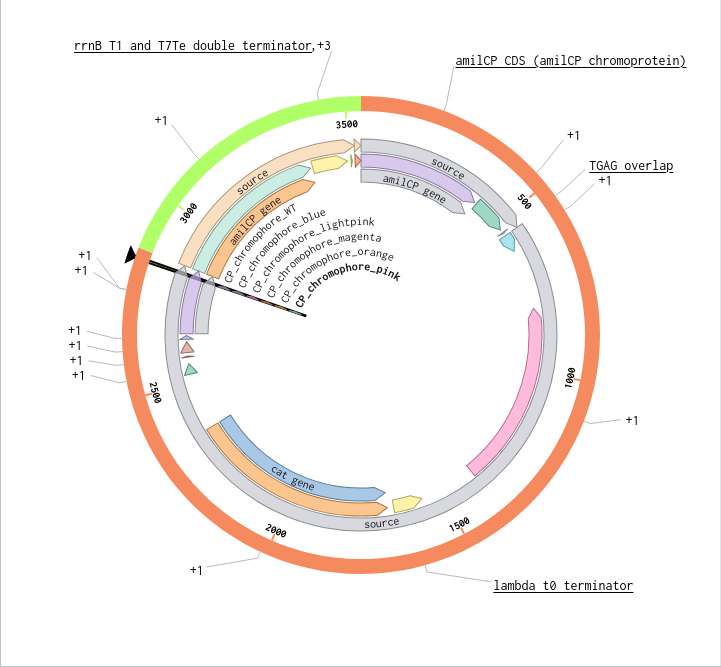
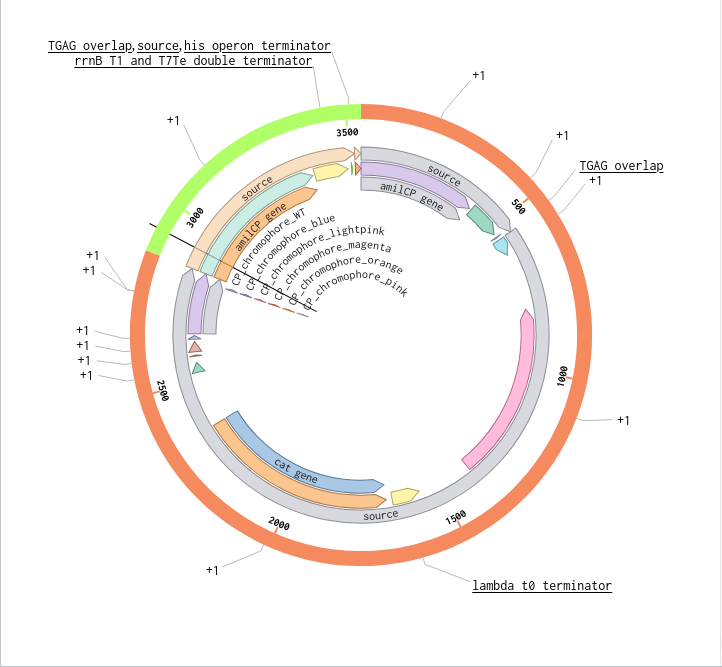
Part B. Asimov Kernel
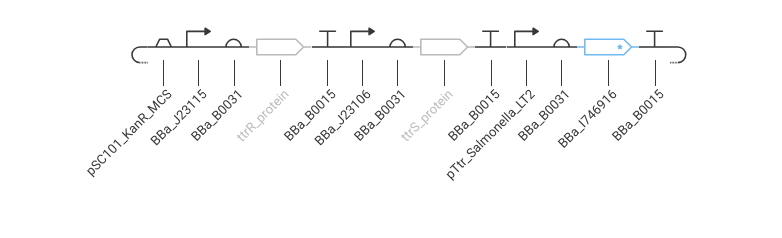
Folder: John_Adeyemo_Adedeji_Genspace (Benchling workspace)
The construct I designed in the Asimov Kernel exercise is a minimal tetrathionate-responsive MccH47 expression cassette for E. coli Nissle 1917 (EcN). The design logic follows directly from the ÌṢỌ project architecture.
Process Reflections
What struck me most this week was how much assembly method choice is actually a design decision rather than a technical one. The distinction between Gibson and Golden Gate is not simply about what enzymes you use, it is about what failure modes you are willing to accept and what flexibility you need downstream. Gibson forgives imprecise fragments but penalises you on multiplexability. Golden Gate rewards modular combinatorial thinking but demands that you get the 4-bp overhang design exactly right, every time.
The deeper insight was about error propagation. In a sequential biological engineering pipeline, a mistake at the assembly stage is not recoverable at the sequencing stage, it shows up as a wrong construct that passes gel verification but fails functional testing. Designing assembly from the perspective of what can go wrong, rather than what should go right, shifted how I think about planning synthesis-to-expression workflows for ÌṢỌ.
The Asimov kernel exercise reinforced that genetic circuit design has a grammar, not just a vocabulary. Parts have semantics. Composability is a property you engineer for, not something you assume.
Works Cited
Gibson, D. G., Young, L., Chuang, R.-Y., Venter, J. C., Hutchison, C. A., & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345. https://doi.org/10.1038/nmeth.1318
Engler, C., Kandzia, R., & Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS ONE, 3(11), e3647. https://doi.org/10.1371/journal.pone.0003647
Palmer, J. D., Piattelli, E., McCormick, B. A., Silby, M. W., Brigham, C. J., & Bucci, V. (2017). Engineered probiotic for the inhibition of Salmonella via tetrathionate-induced production of Microcin H47. ACS Infectious Diseases, 4(1), 39–45. https://doi.org/10.1021/acsinfecdis.7b00114
Benchling, Inc. (2024). Molecular biology platform. https://benchling.com
AI Prompts Employed (Claude AI)
- What are the actual failure modes of Gibson assembly versus Golden Gate, not just the standard advantages
- Explain what Type IIS restriction enzymes are doing differently from conventional enzymes
- Why does Golden Gate have a higher error rate when overhang uniqueness is not enforced
- Walk me through what an Asimov kernel construct definition looks like for a biosensor circuit