Week 4 HW: Protein Design Part 1
Part A: Conceptual Questions
- How many molecules of amino acids do you take with a 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming whole composition of meat is protein, the number of amino acids molecules in 500 grams is 3.011 x 1024 molecules.
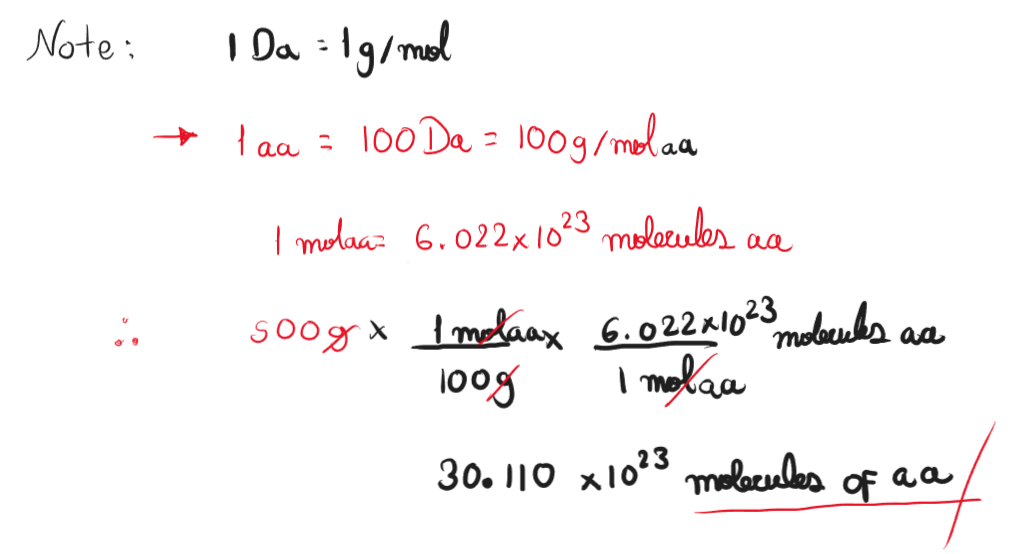
Why do humans eat beef but do not become a cow, eat fish but do not become fish? This is because our digestion breaks down macromolecules into their monomers. Proteins are broken down into amino acids that later are used for the biosynthesis of proteins. The phenotypic characteristic of an organism is defined in its principally by its genome, and it’s not affected by the food they consume.
Why are there only 20 natural amino acids? Amino acids origin is a complex field of study of many theories indicating why living organisms have conserved 20 amino acids. Crick for example mention the “Frozen accident theory” which states that amino acids have been kept the same as living beings’ complexity cross a limit where other amino acids cannot compete with the gold standard. On the other hand, there is a close relationship between these 20 amino acids and living being’s metabolism, suggesting that these amino acids are the result of metabolism optimization (Kirschning, 2022).
Can you make other non-natural amino acids? Design some new amino acids. Non-natural amino acids are modified amino acids that don’t belong to the traditional 20 amino acids, these amino acids can be produced by several organisms. Non-natural amino acids can be designed by modifying natural amino acids using techniques grouped in the field of Peptidomimetics.
Where did amino acids come from before enzymes that make them, and before life started? Several hypotheses have been proposed for the origin of amino acids, for example the “ARN World” hypothesis proposes the existence of a precursor to RNA whit the capability of processing information and catalyze chemical reactions. Evidence of this hypothesis is the presence of Ribozymes, RNA molecules with catalytic activity (Higgs & Lehman, 2014). Other theories propose other molecules like coenzymes or the association of metabolic pathways and amino acids for their production and the possible apparition of amino acids from meteorites (Kirschning, 2022)
If you make a -helix using D-amino acids, what handedness (right or left) would you expect? L- and D-Amino Acids are chiral molecules, meaning that L-Amino Acids are the reflection of D-Amino acids. Alpha-helix using L-amino Acids results in right handedness helix. D-Amino Acids Could result in left handedness in their alpha helix (Novotny & Kleywegt, 2005).
Why are most molecular helices right-handed? Most helices are right-handed because they are formed by L-amino Acids which are more stable at that orientation.
Why do β-sheets tend to aggregate? Aggregation may be promoted by the presence of Aggregation Prone Regions (APRs) that are 5-15 residue long stretches in proteins. These APRs regions are usually buried inside the hydrophobic core of the protein. A protein with more APRs may aggregate because this rearing can link together through hydrogen bones which is also promoted by their hydrophobicity forming a “steric zipper” (Aggregation Prone Regions (APRs), n.d.).
a. What is the driving force for β-sheet aggregation? Β-sheet aggregation is promoted by intermolecular forces like hydrogen bonds and hydrophobic interaction between the Aggregation Prone RegionsCan you use amyloid β-sheets as materials? A study made by Cheng et al. (2012) proposes the use of amyloid β-sheets mimics (ABSMs) to antagonize the aggregation of amyloid proteins and reduce their toxic activity.
References
- Aggregation Prone Regions (APRs). (n.d.). VIB Switch Laboratory. https://switchlab.org/aprs
- Cheng, P., Liu, C., Zhao, M., Eisenberg, D., & Nowick, J. S. (2012). Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nature Chemistry, 4(11), 927–933. https://doi.org/10.1038/nchem.1433
- Higgs, P. G., & Lehman, N. (2014). The RNA World: molecular cooperation at the origins of life. Nature Reviews Genetics, 16(1), 7–17. https://doi.org/10.1038/nrg3841
- Kirschning, A. (2022). On the Evolutionary History of the Twenty Encoded Amino Acids. Chemistry - a European Journal, 28(55), e202201419. https://doi.org/10.1002/chem.202201419
- Novotny, M., & Kleywegt, G. J. (2005). A survey of left-handed helices in protein structures. Journal of Molecular Biology, 347(2), 231–241. https://doi.org/10.1016/j.jmb.2005.01.037
Protein Analysis and Visualization
- Protein Description: Bothrops atrox snake venom nerve growth factor (NGF) Nerve Growth Factor (NGF) is a protein involved in the process of neurite differentiation and growth through the activation of a receptor that promotes signaling cascades. Snake venoms possess NGFs that might contribute to the toxicity of the venom by activating pro-inflammatory signals that increase vascular permeability. Snake NGF has been proposed as an alternative for therapeutic strategies aimed at regenerating tissues.
- Amino acid sequence description The amino acid sequence was obtained from the Uniprot database (ID: A0A1L8D608) predicted at a transcriptomic level
- Protein Length: 241 amino acids
- Amino Acid Frequency
Amino Acid Count Frequency T: 22 (9.13%) N: 22 (9.13%) V: 19 (7.88%) S: 18 (7.47%) R: 16 (6.64%) A: 15 (6.22%) P: 14 (5.81%) K: 13 (5.39%) D: 13 (5.39%) L: 12 (4.98%) I: 12 (4.98%) E: 11 (4.56%) F: 10 (4.15%) G: 9 (3.73%) C: 8 (3.32%) Q: 8 (3.32%) Y: 6 (2.49%) H: 5 (2.07%) M: 4 (1.66%) W: 4 (1.66%) - Using Uniprot’s Blast tool I found that the protein has high homology with a broad number of NGFs from other species of snakes and also from other reptiles and fishes

- Uniprot’s database informs that this protein belongs to NGF-beta family and other related families according to different databases
- NFG structure page
- The B. atrox NGF doesn’t have a crystallyzed protein so its structure haven’t been resolved. To answer the protein structure questions another NGF from Mus musculus (ID: pdb_0001btg)
- This protein has been resolve with 2.50 A structure resolution. This makes the structure a good model to study
- This protein was resolved by X-RAY DIFFRACTION and consist in a Homo-3-mer and contains a ZN ligand (https://www.rcsb.org/structure/1BTG)
- Mus musculus Domains are part of the Neurotrophin family according to the Scop Database, each chain is linked with the 1BET A: 10-116 Domain (SCOP ID: 8026411)
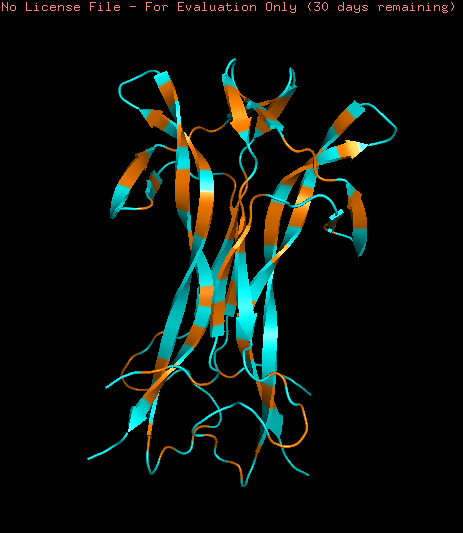
- Protein Visualization For the visualization of the B. atrox NGF a model was created using the SWISS-MODEL tool to generate an predicted structure base on homology (A0A1L8D608)
Protein Visualization was performed using Pymol Software
NGF model was visualized as ribbons, cartoon and sticks. This model is a homo-2-mer


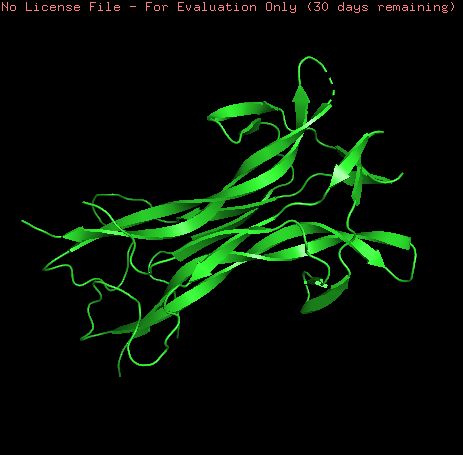
Coloring the protein by secondary structure shows that the protein lacks of alpha helix structures and is mainly formed by b-sheets structures and loops.
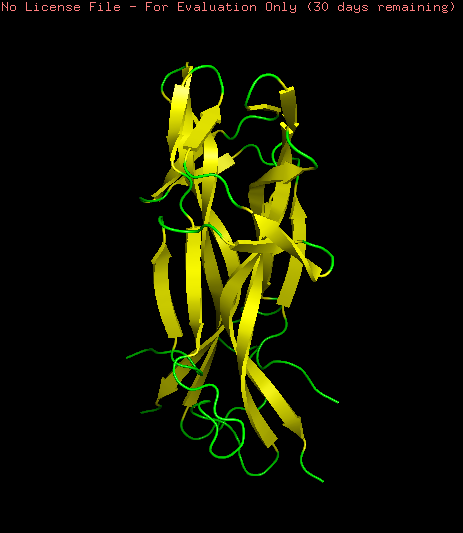
Hydrophobic (orange) and Hydrophilic (skyblue) residues were colored in the model. The model contains a homogeneous distribution of these residues along the beta sheets structure while the loops are mainly hydrophobic.
Part C1: ML-Based Protein Design Tools
The protein selected is the crystal structure of beta nerve growth factor at 2.5 A resolution from Mus musculus (ID: pdb_00001btg).
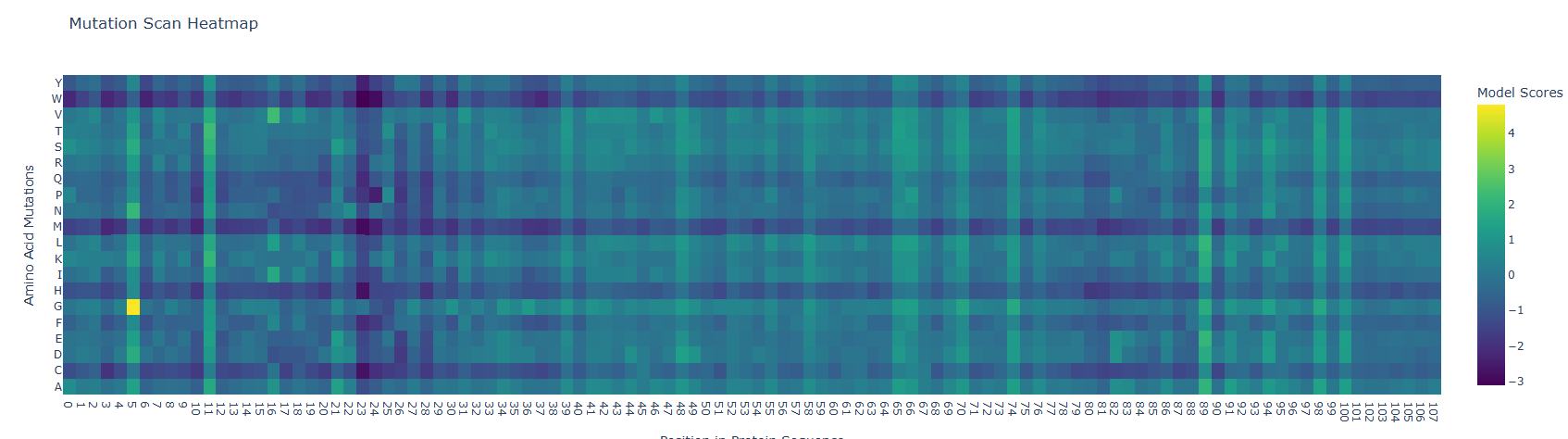
Deep Mutational Scans: Mutation Scan Heatmap results show low scores along the protein sequence as can be observed below. However the fifth residue (S) showed a high mutational score with the glycine amino acid. Low scores in the mutation heatmap may be related with their protein conservation. A study from Barker et al. (2020) states that Mammalian nerve growth factors (NGF) are conserved, this could be related with a low mutational capacity. To corroborate this hypothesis I suggest comparing mutational scores of NGF of mammals and other animal groups.
Latent Space Analysis The figure below shows the latent space result after introducing the NGF sequence.
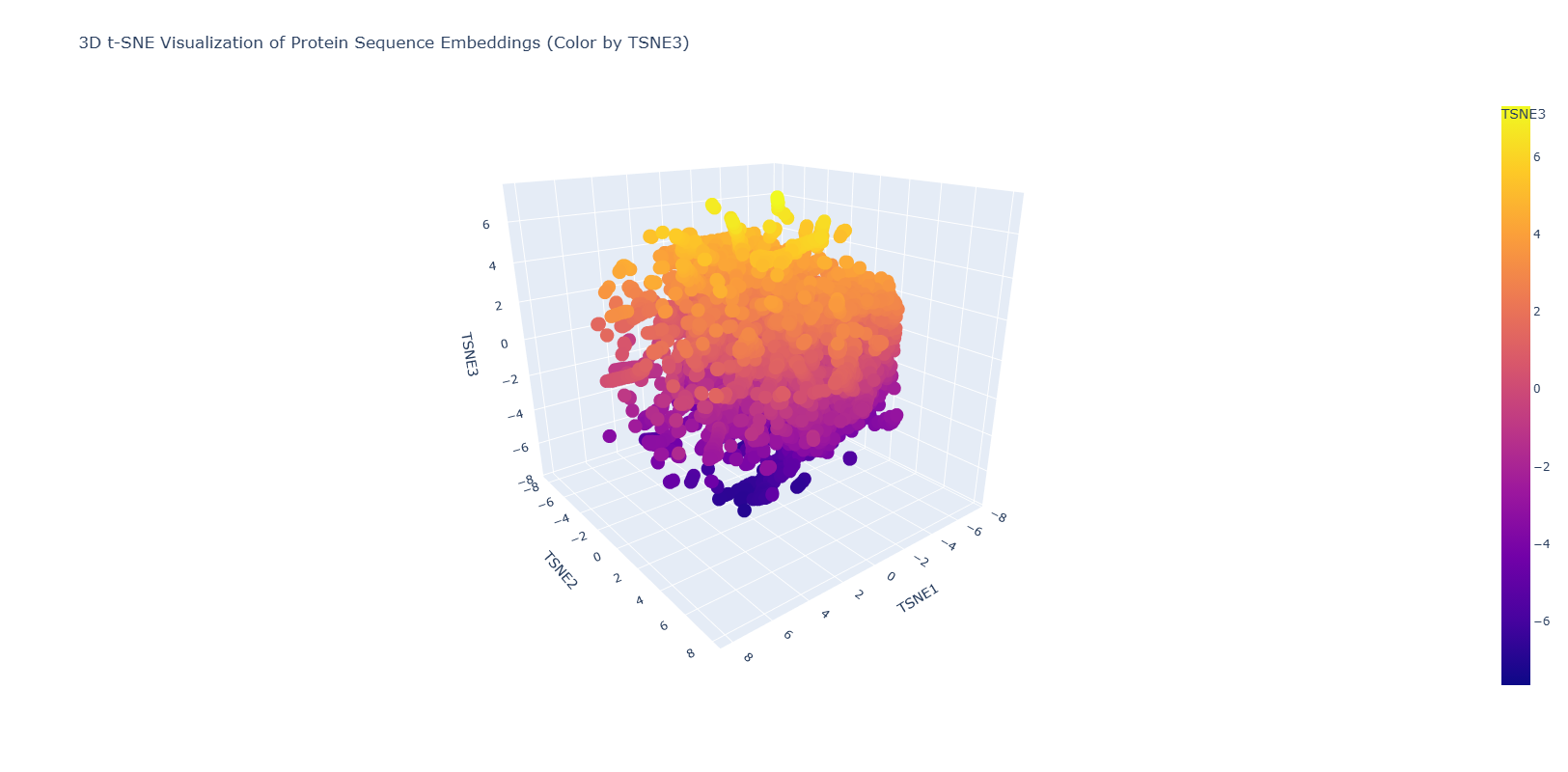
The resulting figure shows a homogeneous distribution of the proteins in the database with small dispersed regions. This could suggest a gradual evolution of the proteins used in this database. Dispersed regions are diverse in origin and also comprises automated matches.
Part C2: Protein Folding
NGF folding obtained using the ESMFold program was created using the sequence from the Mus musculus NGF (ID: pdb_00001btg). The sequence was used creating 3 copies since the original PDB sequence presents a homotrimeric structure. The resulting prediction (Yellow) was compared with the original structure (Multicolor) using the Pymol software
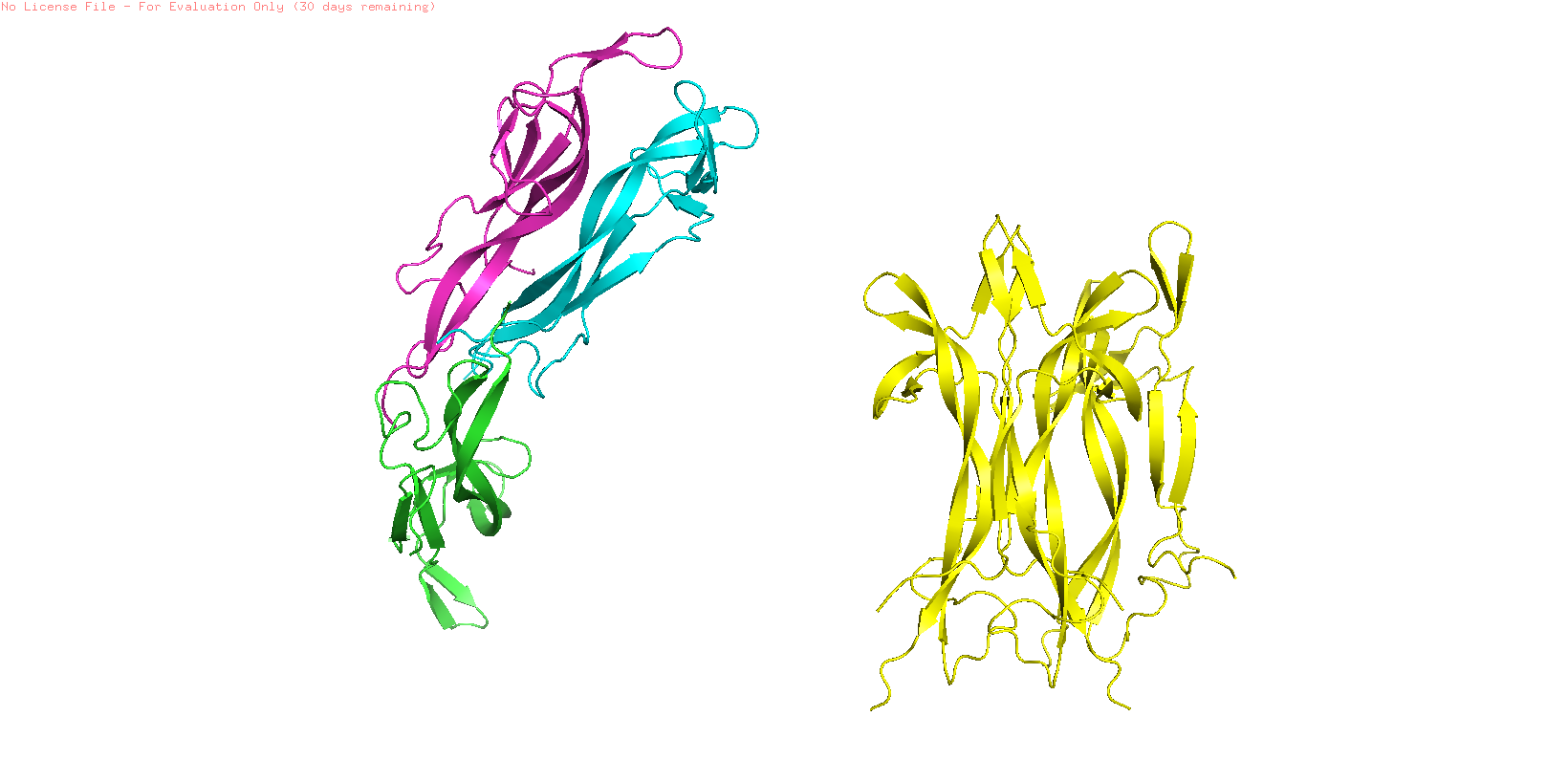
To study the quality of the protein folding the predicted region was aligned with the original structure
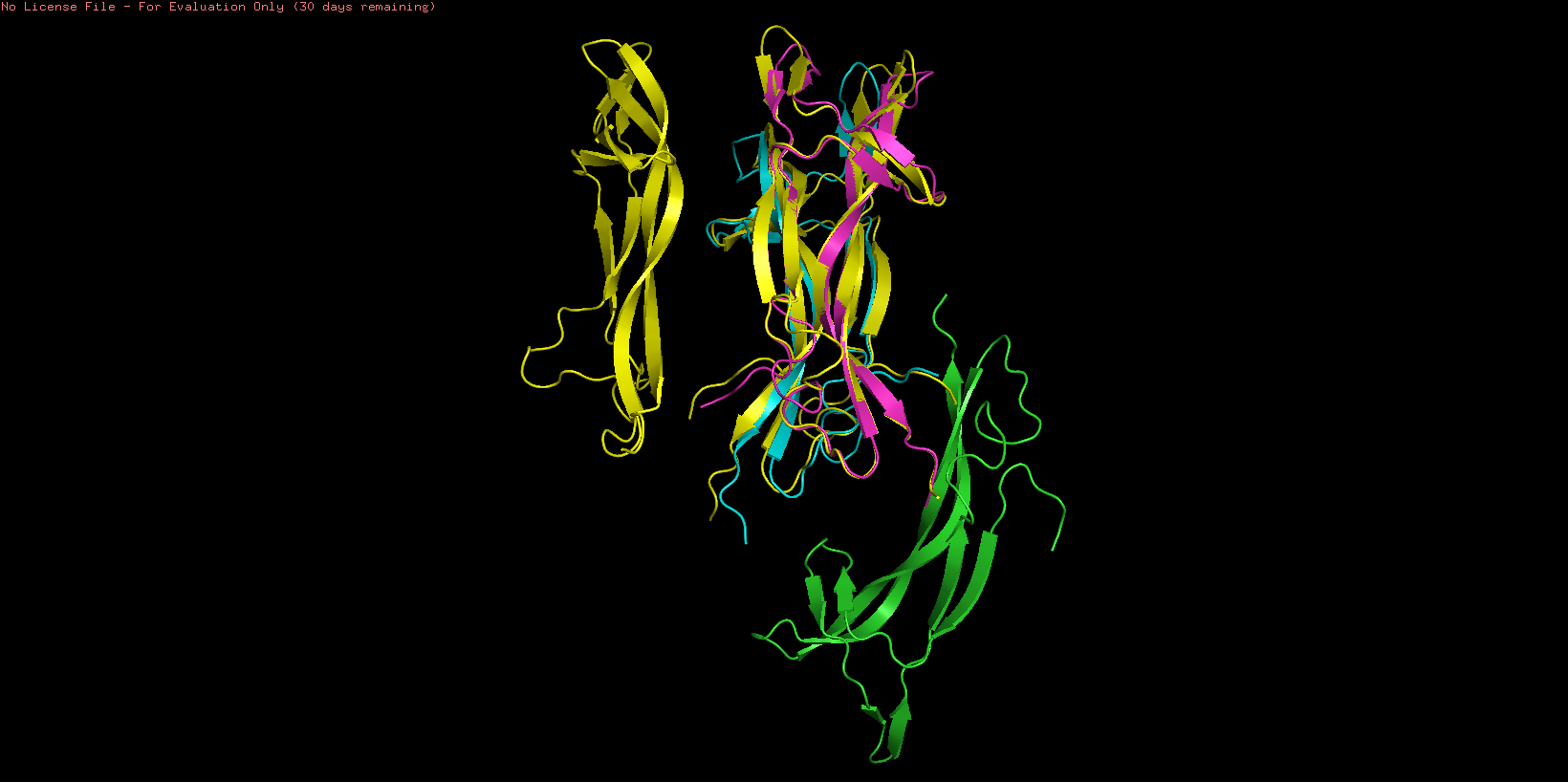
As showed in the image above, an aligment was performed using only two chains of both structures (B and C) resulting an RMSD = 0.487 indicating a close alignment between the predicted structure and it’s original model. Similar result can be observed comparing the resulting structures, observing similarities en their secondary structure. This result suggests the correct application of the ESMFold language and indicates that the predicted protein can be used for other structural studies.
Part C3: Reverse Folding
The Mus musculus NGF was used with the Protein MPNN tool to predic the protein sequence based on the structural information. Using the default parameters of the tool a new sequence was obtained:
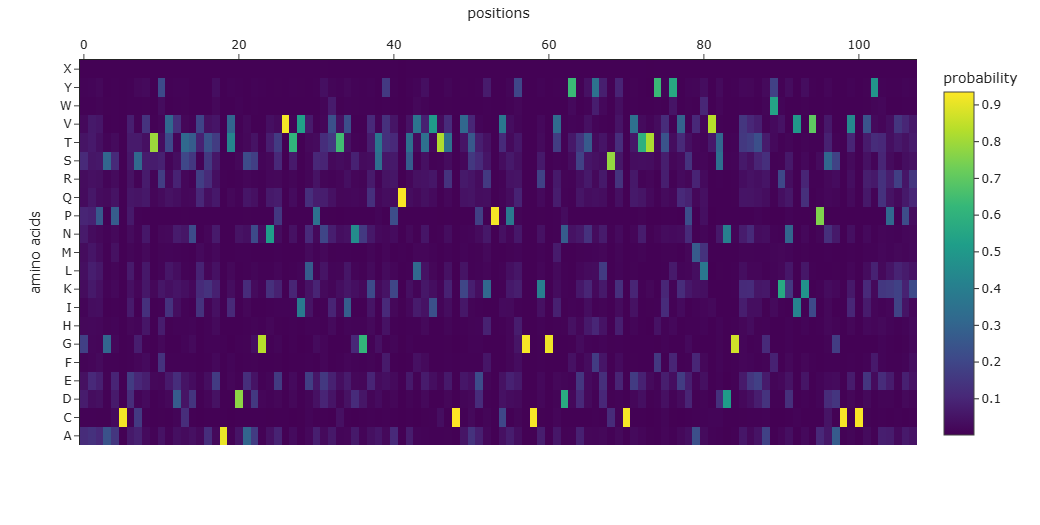
The resulting sequence presents a recovery score of 0.3519 wich suggest a low recovery percentage of the original sequence. Similar result can be observed when aligning both original and predicted sequence using Clustal Omega and through the probability graph of every aminoacid:

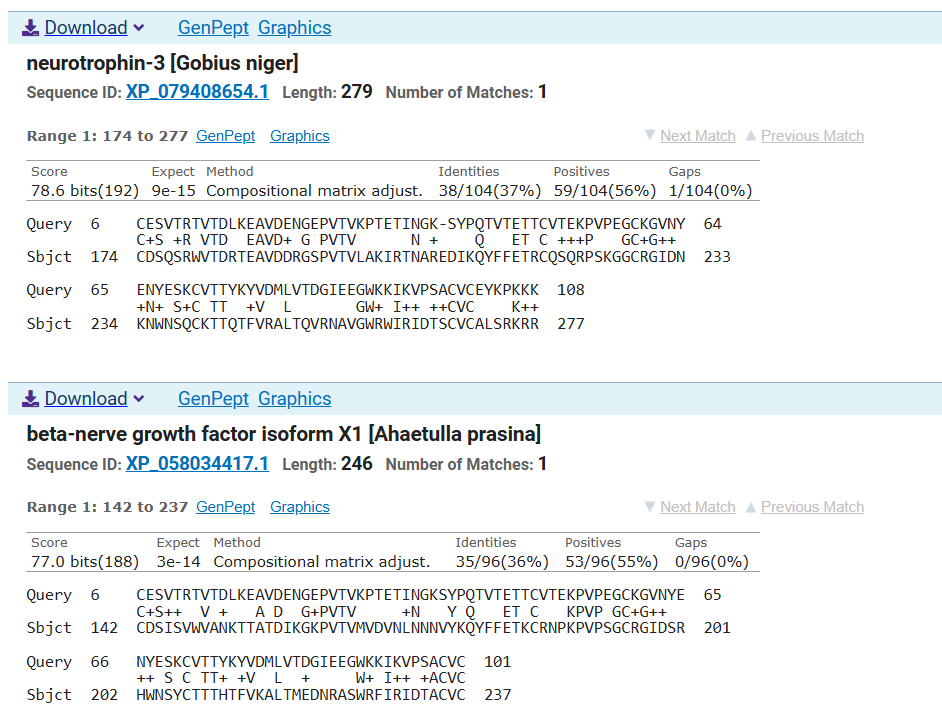
However Blast analysis predicted a close relationship of the predicted sequence with other NGFs of different species and other neurotrophins which could be supported with their evolutionary conservation of this family of proteins
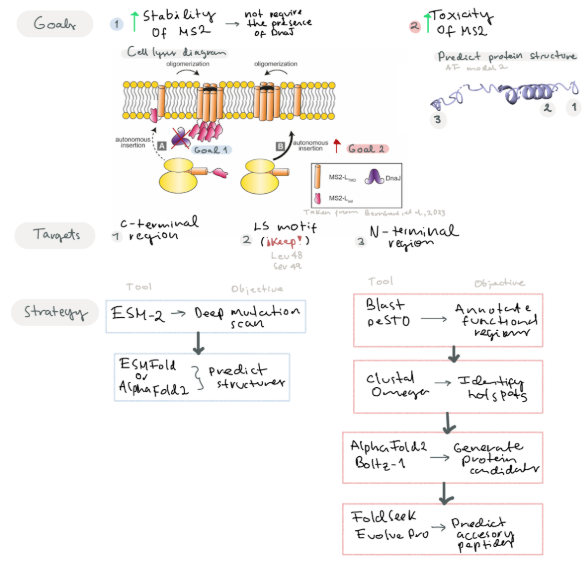
Part D. Group Brainstorm on Bacteriophage Engineering
- Main Goals:
- Goal 1: Increase the stability of the MS2 lysis protein by predicting mutations of residues near the C-terminal region and surrounding the LS motif
- Goal 2: Improve the N-terminal region by modifying residues to contribute to its toxic activity or add new functional regions that may increase its toxicity.
Improving MS2 lysis protein by modifying regions not related with the Leu48 and Ser49 (LS motif) and surrounding to improve protein toxicity (Chamakura et al, 2017). Predict mutations that may improve its stability. We suggest that by increasing protein stability, the protein would not require the presence of the DnaJ for its action.
Another goal is to design new accessories to the N-terminal region to improve lysis toxicity. Berkhout et al, 1985 suggest C-terminal region is key for protein activity, so taking this in consideration we can try to modify the N-terminal region to improve protein stability or add a new characteristics that may improve the toxicity of the protein
- Strategy
Given our two main goals, we propose different strategies to address each objective
For the first goal, we propose using a protein language model such as ESM-2 to perform in silico deep mutational scan that evaluates the plausibility of all possible single-point mutations in the MS2 L protein. Subsequently, we will employ ESMFold or AlphaFold2 to predict the resulting 3D structural variations.
For the second goal:
- Step 1: Identify and Annotate key functional regions near the C-terminal motif and LS motif
- Software: Blast (For conserved domains), PeSTO (Functional motifs) Predict mutations near the N-terminal and C-terminal site that may improve protein stability
- Software: Clustal Omega (To identify hotspots for mutations)
- Generate different protein candidates with mutations and evaluate their stability
- Software: Alpha-Fold Multimer, Boltz-1 We propose using Alpha-Fold with a specific training set for bacteriophages
- Predict accessory peptide sequences to insert in their N-terminal region and improve its toxicity
- Software: FoldSeek (To find remote sequences with similar folding), EvolvePro (To suggest optimized N-terminar sequences)
- Test suitability of these protein candidates by performing docking essays with a bacterial membrane model, etc.
- Suggested Pipeline
Strategy/Software Core Limitation Risks Structural prediction & design (AlphaFold, FoldSeek, EvolvePro, Boltz-1) The model can predict structures that look stable and coherent, but it does not measure real folding energy, membrane insertion, or toxicity. “Looks good” in silico ≠ “works better” in vivo. Selection of variants that appear structurally improved but do not increase stability or toxicity — or even reduce lytic activity. Phage-specific training / limited viral datasets If the model is trained or fine-tuned on a small, biased set of phage proteins, it may learn dataset-specific patterns instead of general biological rules (overfitting). Mutations may seem optimized in the model but fail outside that narrow dataset. Reduced generalizability and misleading predictions.
References:
- Chamakura, K. R., Edwards, G. B., & Young, R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology, 163(7), 961–969. https://doi.org/10.1099/mic.0.000485
- Berkhout, B., De Smit, M., Spanjaard, R., Blom, T., & Van Duin, J. (1985). The amino terminal half of the MS2-coded lysis protein is dispensable for function: implications for our understanding of coding region overlaps. The EMBO Journal, 4(12), 3315–3320. https://doi.org/10.1002/j.1460-2075.1985.tb04082.x