Week 5 HW: Protein Design Part II
Part 1: SOD 1 Binder Peptide Design
Superoxide dismutase 1 sequence was retrieved from Uniprot database (P00441), this protein has a length of 154 amino acids.
SOD1 Sequence:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The mutated version of the human SOD1 caused by an A4V mutation was retrieved from the PDB database that contains a structure obtained from an X-Ray Diffraction study with a resolution of 1.90 Å (Hough et al., 2004)
1UXM_1 Superoxide Dismutase Mutated from Homo sapiens:
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
An alignment between normal SOD 1 and mutated SOD 1 was performed using Clustal Omega to corroborate the mutation at position four, an initial methionine was included into the mutated SOD 1 to have a protein of the same length. (Figure 1)

Small protein binders were generated using the PepMLM model made by Chen et al (2025). Four peptides were generated with a length of 12 amino acids and a Top K value of 3.
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WRYYAVVVAHKX | 12.802906286585648 |
| 1 | WHYGVVALAHKX | 7.909934706159041 |
| 2 | WLSYPAALRHKX | 11.125327842529979 |
| 3 | WRSPAAAVRWKE | 11.952399811426888 |
The four candidates have low pseudo perplexity values (< 20) indicating confidence from the model to the peptides designed (Chen et al. 2025). A fasta document was created including the four candidates with the mutated SOD sequence and SOD-1 binding peptide FLYRWLPSRRGG as a control. However Generated candidates contained an X amino acid coded that means an unknown residue.
These candidates were aligned with the original protein using Clustal Omega (Figure 2)

Part 2: AlphaFold 3 Binders
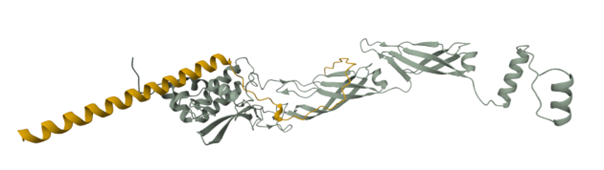
Peptide candidates were modeled using the AlphaFold Server together with the mutated SOD 1 sequence. The control peptide was also modeled and showed a close integration into the SOD 1 structure. Candidates 1, 2, and 3 haven’t shown an integration into the internal structure of SOD 1 (Figure 3)
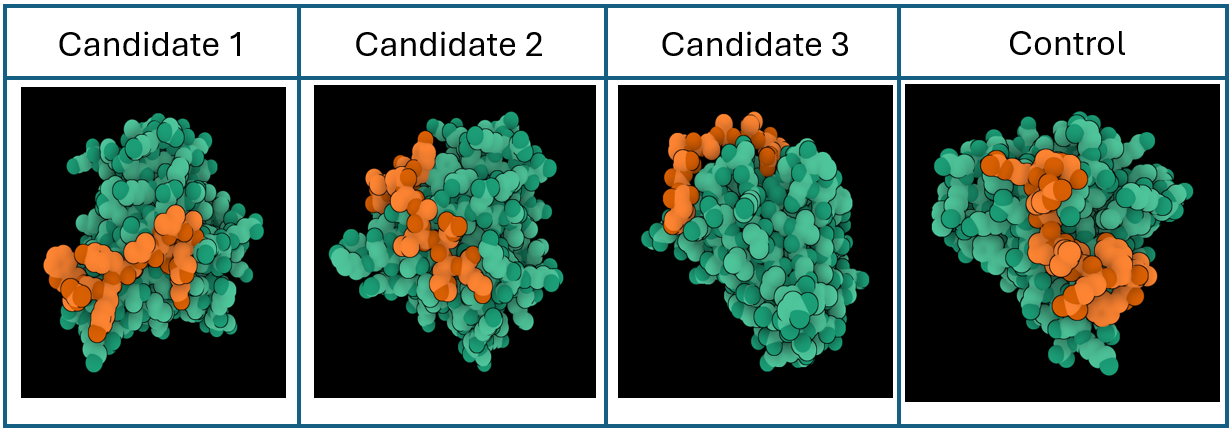
Confidence metrics are presented in the table below where pTM and ipTM scores are shown for each Candidate and the control. These scores measure the accuracy of the structures generated. For all candidates and the control, the pTM scores are more than 0.5, suggesting some confidence that the structure is like its true structure. On the other hand, ipTM value suggests poor confidence in the relative position of the subunits within the complex
| Peptide | ipTM | pTM |
|---|---|---|
| Control | 0.26 | 0.78 |
| Candidate 1 | 0.36 | 0.76 |
| Candidate 2 | 0.45 | 0.83 |
| Candidate 3 | 0.36 | 0.87 |
Part 3: PeptiVerse Evaluation
PeptiVerse was used to predict several characteristics that are required for proposing a binding peptide with therapeutical application.
| Candidate | Solubility | Hemolysis | Binding Activity | pH | Length | Molecular Weight |
|---|---|---|---|---|---|---|
| Candidate 1 | Soluble | Non-Hemolytic | Weak | 9.70 | 12 | 1373.7 Da |
| Candidate 2 | Soluble | Non-Hemolytic | Weak | 9.99 | 12 | 1323.8 Da |
| Candidate 3 | Soluble | Non-Hemolytic | Weak | 10.84 | 12 | 1456.7 Da |
| Control | Soluble | Non-Hemolytic | Weak | 11.71 | 12 | 1507.7 Da |
Candidates 1, 2, and 3 showed high solubility and low hemolytic probability, indicating their possible expression and use. However, pHs obtained a highly basic making it difficult to keep their structure in blood. Predicted Binding activities suggest that the candidates would have a weak interaction with their target. This result is also supported by the ipTM values gotten indicating that these candidates could not be able of binding to the target.
Part 4: Optimized Peptides Generation with moPPIt
Peptide binders were produced using the moPPIt using the mutated SOD1 N-terminal as target region. I propose that these candidates would bind to the mutated region and prevent the aggregation by stabilization of the structure. Peptides were generated considering as objectives and weights their Hemolysis probability, Solubility, Affinity and Specificity. A total of 4 candidates who were generated have low pseudo-perplexity values indicating low uncertainty for the model to the predicted sequence (OFS Pseudo-perplexity for Protein Fitness, n.d.)
| Candidates | Sequence | Pseudo-Perplexity |
|---|---|---|
| Candidate 1 | WRYYAVVVAHKX | 12.80 |
| Candidate 2 | WHYGVVALAHKX | 7.90 |
| Candidate 3 | WLSYPAALRHKX | 11.12 |
| Candidate 4 | WRSPAAAVRWKE | 11.95 |
A Clustal Omega alignment was performed for all the candidates generated by moPPIt and PEPMLM showing close similarities in their sequences (Figure 4)

moPPIt candidates were evaluated using the PeptiVerse programs to evaluate their main characteristics and therapeutical applicability.
| Candidate | Solubility | Hemolysis | Binding Activity | pH | Length | Molecular Weight |
|---|---|---|---|---|---|---|
| Candidate 1 | Soluble | Non-Hemolytic | Weak | 9.70 | 12 | 1373.7 Da |
| Candidate 2 | Soluble | Non-Hemolytic | Weak | 8.61 | 12 | 1262.7 Da |
| Candidate 3 | Soluble | Non-Hemolytic | Weak | 9.99 | 12 | 1323.8 Da |
| Candidate 4 | Soluble | Non-Hemolytic | Weak | 10.84 | 12 | 1456.7 Da |
All candidates were predicted with weak affinity and presented a pH superior to 7 making them difficult to use directly in a human.
References
- Hough, M. A., Grossmann, J. G., Antonyuk, S. V., Strange, R. W., Doucette, P. A., Rodriguez, J. A., … & Hasnain, S. S. (2004). Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proceedings of the National Academy of Sciences, 101(16), 5976-5981.
- Chen, L. T., Quinn, Z., Dumas, M., Peng, C., Hong, L., Lopez-Gonzalez, M., … & Chatterjee, P. (2025). Target sequence-conditioned design of peptide binders using masked language modeling. Nature Biotechnology, 1-9.
- Zhang, Y., Tang, S., Chen, T., Mahood, E., Vincoff, S., & Chatterjee, P. (2026). PeptiVerse: A Unified Platform for Therapeutic Peptide Property Prediction. bioRxiv, 2025-12.
- OFS Pseudo-perplexity for protein fitness. (n.d.). https://www.emergentmind.com/topics/one-fell-swoop-ofs-pseudo-perplexity
Part C: L-protein mutants
The MS2 lysis proteins is a small protein resposible for host cell lysis during bacteriphage infection and can be used as antimicrobial candidates. The aim of this project evaluate mutants of the MS2 lysis protein to improve the stability of the lysis protein and its interaction of de DnaJ protein. The following protein sequences were used:
MS2 lysis protein sequence:
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DnaJ chaperone protein:
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Stage 1: Prediction of potential mutation sites
MS2 lysis sequence was analyzed using Blatp tool most related sequences were extracted. Clustal Omega tool was used to align the lysis protein with their most related sequences to identify possible conserved sites (Figure 1).
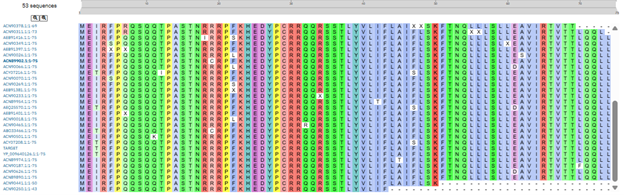
Clustal alignment showed a strong conserved N-terminal region with a conserved hydrophobin core, however some variable residues were found in the region 23-29 and 44-49.
Using the google collab offered in the activity a mutation heatmap was produced to see the mutational scores of the MS2 protein (Figure 2)
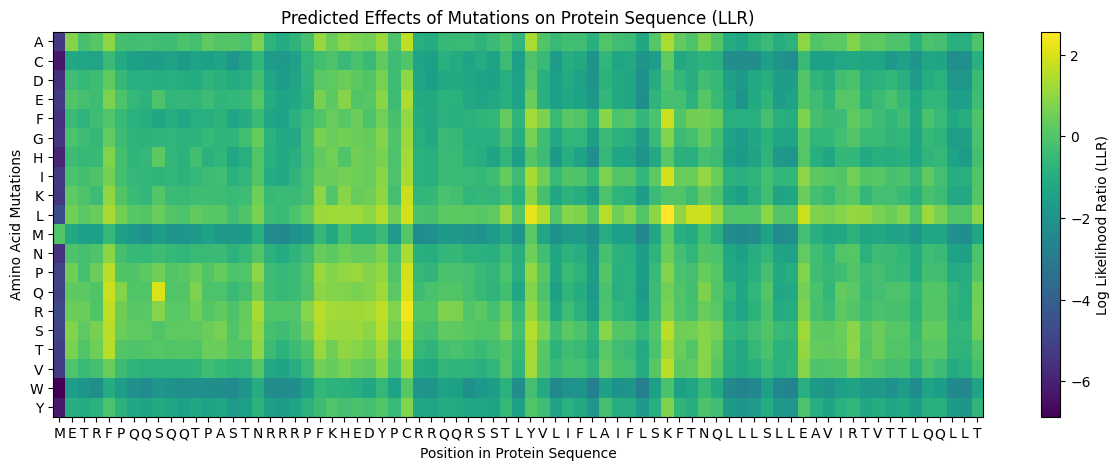
Heatmap results show lower mutation scores in predicted conserved sites in Clustal Omega suggesting that these conserved regions could have important functions in the protein. Comparing multiple alignment with the heatmap suggest that residues 5, 24-29, 38, 44-49 may be mutated.
These regions were analyzed by comparing their function with Chamakura et al (2017) that suggests that regions close to the C-terminal region like the LS motif can affect the integration of the protein to the membrane, for that reason these region were avoided. Additionally, residues 44-49 have different effect in the activity of the protein and these residues were also considered as potential mutation sites.
By comparing Clustal aligment, mutational heatmap and experimental values I selected the following residues to generate mutants
- Residues 23 - 29 based on conserved and mutational sites
- Variable residues 44, 47, 49 based on experimental studies
To determine the type of mutation, experimental results found in the cvs document were use to determine the types of subtitutions that doesn’t affect the activity of the protein lysis. Mutated sequence is shown below:
Mutated MS2 Lysis Protein (K23E, C29R, L44P, F47Y, S49L)
METRFPQQSQQTPASTNRRRPFEHEDYPRRRQQRSSTLYVLIFPAIYLLKFTNQLLLSLL EAVIRTVTTLQQLLT

Stage 2: Interaction Analysis using Boltz
Boltz interactions were produced between the native and mutated lysis proteins and DnaJ proteins as a target. Streptococcus pneumonia DnaJ pdb was used based on similarity search in the PDB database (ID: 6JZB)
Figure 4 shows the interaction of the mutated protein (green) and native protein (red). Mutated lysis protein showed a loop around the DnaJ protein, but this wasn’t observed in the native protein, however binding confidence values in both structures were low indicating that these results aren’t conclusive.
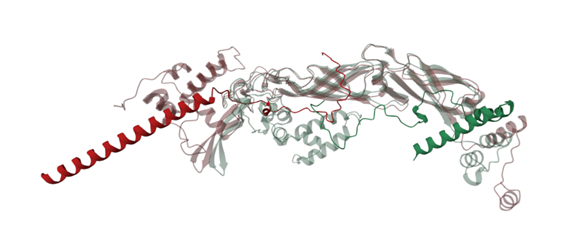
Modification of residue 49 seems to be related with the change of the folding, to corroborate this the mutation of the residue 49 was reverted (Figure 5) showing that the protein have lost the loop that generates it folding around the DnaJ protein, suggesting that residue 49 may have an important structural role in the protein, however further studies are required.