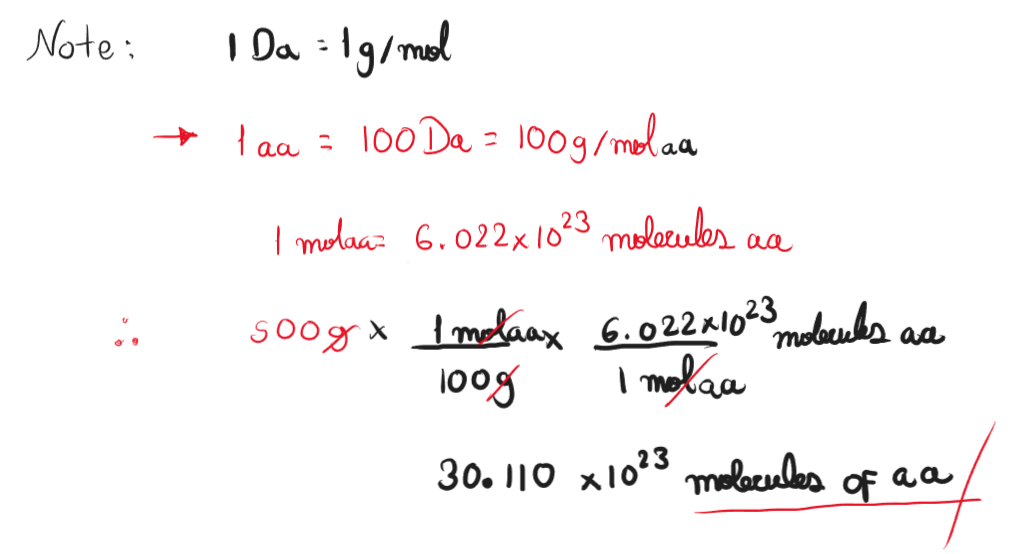B.Sc. Genetics and Biotechnology from the National University of San Marcos in Lima, Perú. My main experience is in snake-venoms protein research and antivenoms development. My insterest are protein research, designing tools and products that solve real-world problems, and learning new things every day.
I’m passionate about the development of new technologies and how they can shape the future, and I would like to contribute to their advancement. I also love computers and the many ways they can be used to create unique and meaningful experience.
As a professional, I have focused primarily on protein research. Using snake venoms, I purified various proteins and enzymes with promising applications. I conducted experiments to determine their biochemical characteristics and their roles in the envenomation process. Some proteins were selected for in vitro production through recombinant strategies, followed by further studies using X‑ray crystallography to understand the relationship between their catalytic function and structure.
I also carried out studies with antivenoms, evaluating their efficacy against different protein families. These studies not only revealed the biological effects of snake toxins on their prey but also provided insights into their evolution. Currently. I also have experience teaching science to students and designing interactive practices and lessons to introduce students to scientific topics.
Class Assignment Describe a biological engineering application or tool you want to develop and why. Aplication title: De novo design of proteins binders for neutralizing Bothrops venom toxins
Antivenoms are a mix of immunoglobulins produced traditionally by the hyperimmunization of large animals with crude venom obtained from clinically-relevant snakes (Ratanabangkoon, K., 2023). Novel alternatives have emerged to neutralize venom toxins without the use of animals. For example, Torres and collaborators (2025) designed proteins with high affinity for important regions of cytotoxins from the 3FTx family. These proteins showed great neutralizating capacity in vitro and great protective capacity in vivo .
Part 1: Benchling & In-silico Gel Art Lambda Sequence: Sequence from E.coli I cl857 S7 lambda bateriophage (Daniels, et al., 1983) available at New England Biolabs (N3011)
A digest simulation was performed using the lambda sequence and 7 different restriction enzyme (EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI). The range of fragments obtained from this simulation varies depending on the enzyme used.
Opentrons Artwork: Gel Designing Design: Snake Trimeresurus puniceus Inspired from a snake photo taken in the Oswaldo Meneses serpentarium, Lima, Peru. Art created Donovan’s Automation art interface
Python Script Design Opentrons script was created following the instructions and ideas offered by the HTGAA Opentrons Colab. To create the script first I created a pseudocode with the idea of how the robot will work
Part A: Conceptual Questions How many molecules of amino acids do you take with a 500 grams of meat? (on average an amino acid is ~100 Daltons) Assuming whole composition of meat is protein, the number of amino acids molecules in 500 grams is 3.011 x 1024 molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish? This is because our digestion breaks down macromolecules into their monomers. Proteins are broken down into amino acids that later are used for the biosynthesis of proteins. The phenotypic characteristic of an organism is defined in its principally by its genome, and it’s not affected by the food they consume.
Part 1: SOD 1 Binder Peptide Design Superoxide dismutase 1 sequence was retrieved from Uniprot database (P00441), this protein has a length of 154 amino acids.
SOD1 Sequence: sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
The mutated version of the human SOD1 caused by an A4V mutation was retrieved from the PDB database that contains a structure obtained from an X-Ray Diffraction study with a resolution of 1.90 Å (Hough et al., 2004)
Part 1: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fifelity PCR Master Mix offered by New England Biolabs is a product that contains a DNA polymerase with high fidelity useful for cloning and amplification of difficult amplicons. Phusion DNA polymerase contains proofreading activity (3’ -> 5’ exonuclease) and a higher fidelity 50X greater that Taq polymerase. Thermo Fisher Scientific Phusion High-Fidelity PCR Marter Mix is composed of a HF Buffer or GC Buffer; both buffers are used to reduce the error rate of the DNA polymerase. HF Buffer contains a lower error rate (4.4 x 107) than GC Buffer (9.5 x 107), however GC buffer can improve the performance of the polymerase on some difficult or long templates with high GC-rich templates or with secondary structures. The master mix is also provided with a optimized concentration of MgCl2 which is an essential cofactor that stabilizes the DNA double helix and facilitates primer annealing. High Fidelity PCR reaction can also include DMSO that is used to reduce the melting temperature through its association with Cytosine residues that changes the conformation of the DNA template. What are some factors that determine primer annealing temperature during PCR? Primer annealing temperature is affected by the Primer melting temperature, annealing temperature is usually 3-5 °C below the primer Tm this temperatura depends on the GC content of the primers or primer length and the prescense of secondary structures. Another factor that can modify the Ta is the MgCl2 concentration indicating the higher concentrations can increase the Ta. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR is a laboratory method often used to increase the amount of a desired DNA sequences, it uses primer to determine the boundaries of the fragment and uses a DNA polymerase that can be genetically engineered to get different results. PCR is often used to amplify sequences for other analysis like sequencing or cloning. Resctriction enzyme digest used restriction endonucleases that recognize specific palindromic sequences and cuts these sites, it doesn’t not amplify the target sequence and is often used for its integration in a cloning vector for recombinant technologies. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? To ensure that the DNA sequences will be appropiate for Gibson cloning the regions must have overlaping regions that can be used to connect correctly with the vector. The digested vector mus be linear and it must be purified to prevent re-ligation with the vector. How does the plasmid DNA enter the E. coli cells during transformation? The era multiple transfection methods use to insert a target DNA sequence inside the E. coli cells different methods can be used, one method called electroporation uses an electric shock to disturb the membrane and insert the DNA vector, another method uses a heat shock to disturb and insert the desired vector. Describe another assembly method in detail (such as Golden Gate Assembly) Golde Gate Assembly uses Type IIS restriction enzyme and T4 DNA ligase that cuts outside their recognition sequence, this allows to desing overhangs that can be ligated later, later this recognition sites are removed during assembly. Golden gate method allows assembling up to nine fragments at a time in a recipient plasmid. This can be done in a single tube that contains the donors, the recipient vector, a type IIS restriction enzyme and ligase (Engler & Marillonnet,. 2013) Some advantages of the Golden Gate Assembly are:
Genetic Circuits Part II: Neuromorphic Circuits Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? One characteristic of IANNs is that they can process continuous inputs rather than only discrete boolean data, this resembles with the natural process of a living system that is composed of a complex network of molecular interactions. Boolean login can simplify complex living activities making it difficult to process and engineer systems with this logic but IANNs uses complex analysis that can be used to reproduce complex process in living beings that uses millions of parameters. By training an IANN is possible to simulate complex genetic activities that resemble more to the living processes than only using boolean functions (Nilsson et al, 2022) Because IANNs works with continous data they can work better where a system have more molecular noise and context effect thant Boolean switches making them useful in therapetic context where there is a high amount of molecular noise that must be processed to produce a suitable answer (Müller et al., 2025)
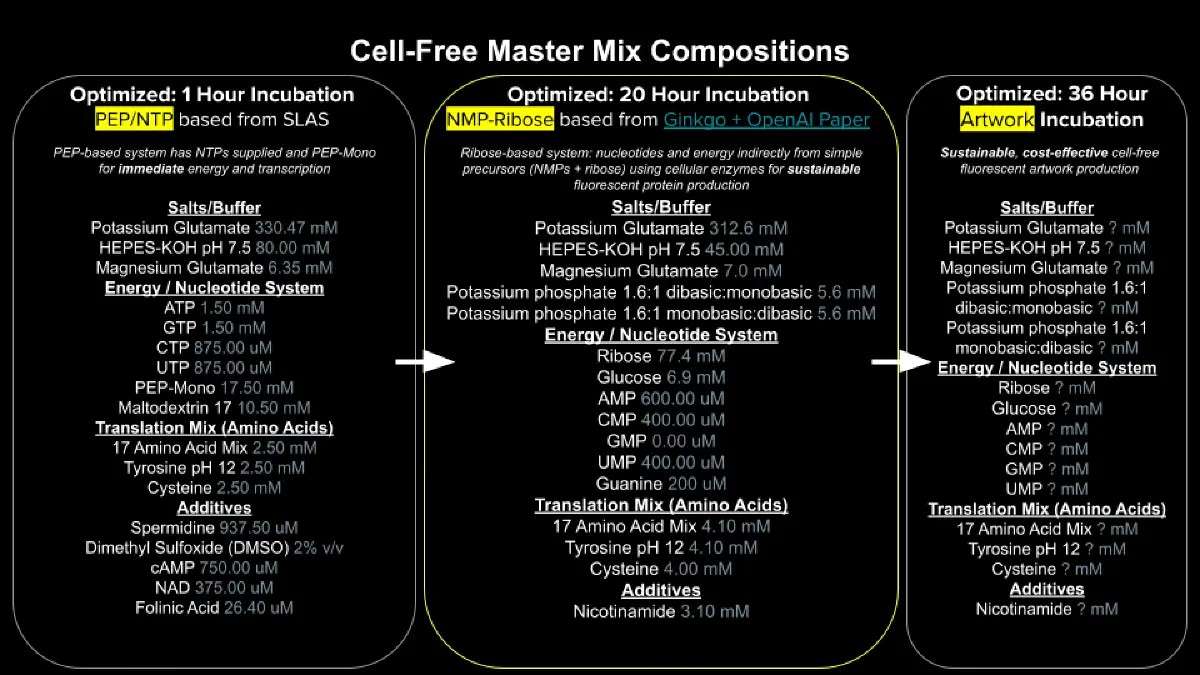
Part A: General and Lecturer-Specific Questions General Homework Questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production: Cell-free protein synthesis (CFPS) has greater flexibiltiy and experimental control that recombinar expression systems because the entire reaction happens outside a living organism. Without the use of a cell system to express the protein we can control and optimize every component of the system, making them scalable a can be applied in low amount making them suitable to analyze multiple protein candidates in parallel.
Homework: Final Project Figure 1 below presents some key aspects of my final project that require experimental testing and quantitative evaluation. These aspects refer to the expression of the protein binders generated using a Deep Learning Model and selected after in silico prediction of their therapeutic characteristics.
Figure 1: Experimental aspects of the final project: AI-driven Antivenom: A Generative Pipeline for De novo Neutralizing Peptides Against Snake Toxins. Image generated using: Copilot AI Aspect 1: Cell-Free Expression System (CFS) Cell-Free Expression System (CFS) enables rapid expression of multiple protein variants in parallel. Since this project aims to predict and express different protein binders, CFS provides a scalable and automatable platform that avoids cell culture and allows preliminary functional testing without full purification (Cui et al., 2022) To ensure reproducibility, the CFS workflow must be standarized and quantitatively monitored. Expression efficiency will be measures by using detection tags like biotinylated lysine or His-tag into the peptide of interest, enabling detection through SDS-PAGE followed by Western Blotting. A colorimetric readout using biotin-binding secondary antibody and chromogenic substrate will aloow quantification of expression levels (Hunt et al., 2024)
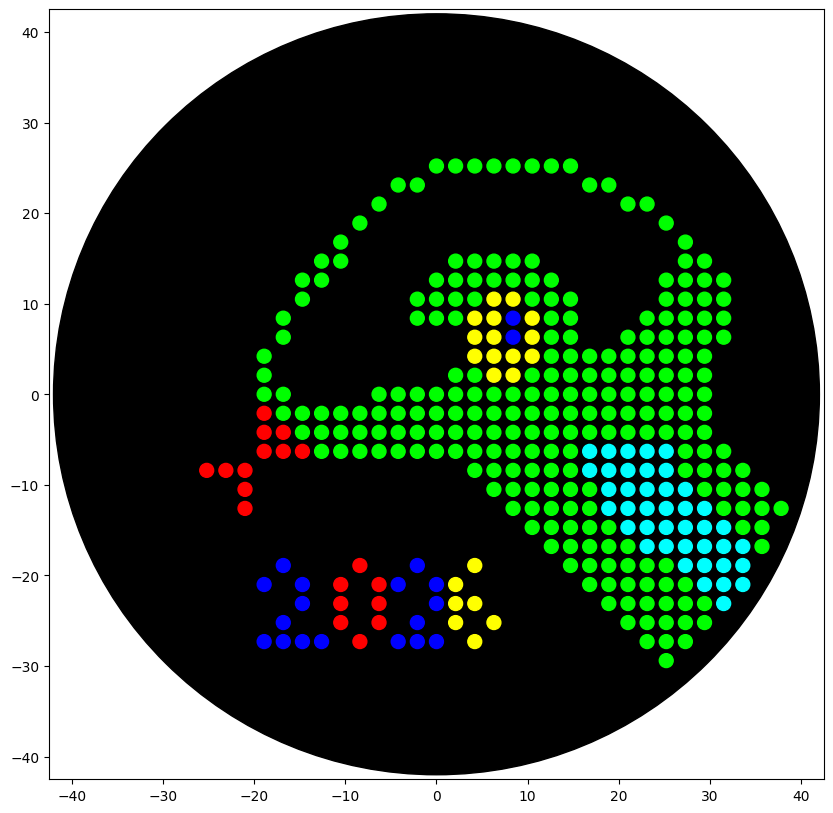
Part A: Artwork Canvas Contibution: I contribute with 52 dots making the “Love” Desing at the bottom left plate. I liked the collaborative working of the canvas and the constant organic evolution of the design that resembles very well with the development of synthetic biology. For a next collaborative art experiment I could propose an experiment that serves as a competence of different groups, or a collaborative project that shows thet location of the participant in the world.
Subsections of Homework
Week 1 HW: Principles and Practices
Class Assignment
Describe a biological engineering application or tool you want to develop and why.
Aplication title:De novo design of proteins binders for neutralizing Bothrops venom toxins
Antivenoms are a mix of immunoglobulins produced traditionally by the hyperimmunization of large animals with crude venom obtained from clinically-relevant snakes (Ratanabangkoon, K., 2023). Novel alternatives have emerged to neutralize venom toxins without the use of animals. For example, Torres and collaborators (2025) designed proteins with high affinity for important regions of cytotoxins from the 3FTx family. These proteins showed great neutralizating capacity in vitro and great protective capacity in vivo .
“Omics” strategies applied to snake venoms have been developed as Venomics, these strategies allows the characterization of whole venoms building protein profiles. Similar estrategies can also be used for studying antibody-toxin complexes as Antivenomics (Lomonte, 2017). The information obtained through venomics and antivenomics give us the ability to build databases with structural and functional information of snake toxins; this information is important for disigning de novo proteins
Bothrops genus is one of the most relevant in South America and has been studied broadly. Many venomics and antivenomics studies has been developed giving high amounts of information that can be used for the design of proteins with high affinity to their important regions.
The following proposal aims to create a system that involves venomics and antivenomics estudies of Bothrops venoms to create a database that can be used to identify key regions with high impact in their toxic activities. Through these regions I propose the design of proteins using artificial intelligence. These candidates could be useful to explore the possibility of designing synthetic antivenoms that don’t depend on the use of animals for their production (Figure 1)
Figure 1: Schematic representation of de novo desing workflow
Justification: Snakebites are classified as Neglected Tropical Diseases by the World Health Organization (WHO) affecting low-and middle-income countries from Africa, Asia, and South America (World Health Organization, 2019).
Antivenoms are the only approved treatment against snakebites. Antivenoms show several limitations in their efficacy and production. Snake venoms present variability in their composition. This could lead to antivenoms with different efficacies depending on the venom used for their production. Additionally, antibodies present in the antivenom can cause adverse reactions when administered to the patient.
Antivenom production is technologically complex with high costs, resulting in a limitation for low-income countries (Alangode et al., 2020). Antivenoms not only present but also numerous challenges in their production but also in their requisites at different levels to be used safely (Figure 2, Potet et al, 2021)
Figure 2: Access antivenom barriers at different levels from global to local. Figure obtained from Potet et al, 2021
The limitations observed in antivenoms produced traditionally supports the necessity of novel alternatives that can be produced safely and with low cost in their design. The use of artificial intelligence with the information provided by venomics and antivenomics opens the possibility of creating synthetic alternatives for the neutralization of venom toxins, and their design could also be optimized to a production at large scale increasing their availabilty and reducing their cost.
Analysis of Protein Binders for Governance Goals and Actions
The World Health Organization has established a programme to evaluate the safety and effectivenes of current antivenoms intended for their use in different countries. This programme led to the recruit of several world experts, forming the Working Group on Snakebite Envenoming. Through this group, the WHO has established a goal of reducing the mortality and disabiluty of snakebite envenomings by 2030. (World Health Organization, 2019)
To accomplish this goal, the working group has developed a road map with objetives at different scales (Figure 3, Williams et al, 2019)
Figure 3: WHO snakebite envenoming road map objectives, impact goals, and timeline phases. Image gathered from Williams et al, 2019
Designing protein binders De novo fits with the objective “Safe and Effective Treatment” from the WHO roadmap. This objective proposes 5 key activities:
Make safe, effective antivenoms available, and affordable to all
Better control and regulation of antivenoms
Prequalification of antivenoms
Integrated health worker training and education
Improving clinical decision making, treatment, recovery and rehabilitation
Investing in innovative research on new therapeutics
The implementation of these protein binders as an alternative to traditionally-produced antivenoms should meet with these 5 key activites. The image below analysis how portein binders could contributo to these key activities and proposes 4 potential governance actions according to the objetive and key activites proposes by the WHO (Figure 4)
Figure 4: Analysis of protein binders and their possible relationship with government goals and actions. Figure A: Representation of key characteristics that every potential antivenom candidate must follow.(Obtained from: Thumtecho et al., 2023) Table A: Potential Governance Actions related to the use of protein binders. Table B: Possible contribution of protein binders to the key activities proposed by the WHO. Table C: Impact score of the Governance Actions proposed for each key activitiy from the WHO "Safe and Effective Treatment"
One of the most important governance actions that I would prioritize is the development of reproducible protocols for the design and use of protein binders against snake venoms. Reproducible protocols require the participation of public and private research institutions and involves the development of clear and highly reproducible strategies for de novo prediction of these protein binders, recombinant production and scalation. This action may contribute to other actions like the creation of guidelines promoted by the WHO using these protocols.
In Peru, the National Health Institute is in charge of antivenom production, the development of reproducible protocols requires the association of research laboratories with this institute. A pilot program can also be created using different species of the genus Bothrops to design and test the efficacy of protein binders.
References
Alangode, A., Rajan, K. & Nair, B. G. (2020). Snake antivenom: Challenges and alternate approaches.. Biochemical Pharmacology, 181. https://doi.org/10.1016/J.BCP.2020.114135
Lomonte, B. and Calvete, J. J. (2017). Strategies in ‘snake venomics’ aiming at an integrative view of compositional, functional, and immunological characteristics of venoms. Journal of Venomous Animals and Toxins including Tropical Diseases, 23(1). https://doi.org/10.1186/S40409-017-0117-8
Potet, J., Beran, D., Ray, N., Alcoba, G., Habib, A. G., Iliyasu, G., Waldmann, B., Ralph, R., Faiz, M. A., Monteiro, W. M., Sachett, J. d. A. G., Di Fábio, J. L., Cortés, M. d. l. Á., Brown, N. & Williams, D. (2021). Access to antivenoms in the developing world: A multidisciplinary analysis.. Toxicon: X, 12. https://doi.org/10.1016/J.TOXCX.2021.100086
Ratanabanangkoon, K. (2023). Polyvalent Snake Antivenoms: Production Strategy and Their Therapeutic Benefits. Toxins, 15. https://doi.org/10.3390/TOXINS15090517
Thumtecho, S., Burlet, N. J., Ljungars, A. & Laustsen, A. H. (2023). Towards better antivenoms: navigating the road to new types of snakebite envenoming therapies. Journal of Venomous Animals and Toxins including Tropical Diseases, 29.
Torres, S. V., Valle, M. B., Mackessy, S., Menzies, S. K., Casewell, N. R., Ahmadi, S., Muratspahić, E., Sappington, I., Overath, M., Rivera-de-Torre, E., Ledergerber, J., Laustsen, A. H., Boddum, K., Bera, A. K., Kang, A., Brackenbrough, E., Cardoso, I. A., Crittenden, E., Edge, R. & Decarreau, J. (2025). De novo designed proteins neutralize lethal snake venom toxins.. Nature, 639. https://doi.org/10.1038/S41586-024-08393-X
Williams, D., Faiz, M. A., Abela-Ridder, B., Ainsworth, S., Bulfone, T. C., Nickerson, A., Habib, A. G., Junghanss, T., Wen, F. H., Turner, M. J., Harrison, R. A. & Warrell, D. A. (2019). Strategy for a globally coordinated response to a priority neglected tropical disease: Snakebite envenoming.. PLoS Neglected Tropical Diseases, 13. https://doi.org/10.1371/JOURNAL.PNTD.0007059
OpenAI (2026). CHATGPT(GTP-5-based-model). Used for conceptual discussion and feedback on project development. https://chat.openai.com/
Week 2 Lecture Prep
Professor Jacobson Questions:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
During DNA replication our cells use DNA polymerases for DNA synthesis, these polymerases can have an error rate of 1 bp per every 100,00 nucleotides. As the human genome is composed of 6 billion bp per diploid cell, every time a cell divides DNA polymerases will make about 120,000 errors (Pray, 2008). While these errors may become mutations that could lead to new adaptations, it is important to correct these errors since they could lead to many dangerous effects on the organism’s life. To correct these errors some DNA polymerases come with an extra exonuclease 3’-5’ activity that serves as proofreading. For example, PolƐ is a DNA polymerase that is involved in the process of DNA replication of the leading strand. PolƐ is a holoenzyme compose of many subunits, when a mismatch is detected in the pol site of PolƐ the proteins arrest the pol activity and the protein moves away from the mismatched 3’end preventing additional base incorporation. Then, the proofreading region generates a change in the DNA conformation. This takes the mismatched base to the exo site of the polymerase generating the excision, after that the polymerase resumes its activity after correcting the mistake (Wang et al, 2025). Proofreading mechanisms help to reduce errors induced by the replication process, for that reason, polymerases with proofreading activity are highly important in different applications. To design complex synthetics systems, it is necessary to reduce the possibility of bp mismatches caused by the polymerase, for that reason, high fidelity polymerase with proofreading activity is available commercially
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Human genetic code is a set of three RNA bases called codon; every codon decodes a specific amino acid. The genetic code shows codon degeneracy, in other word codons that can be used to decode the same amino acid. In average most amino acids correspond to three codons, with some exceptions like Methionine and Tryptophan that only belong to a single codon. While codon degeneracy allows the use of different codons to produce an amino acid, different organisms have different preferences for the codons they use. Codon preference may occur for different reasons like metabolic pressures where some specific tRNAs are used instead of a wide variety of tRNAs for every codon available. Similarly, protein characteristics may influence the preference for some tRNAs than others (Ford, n.d).
Dr. LeProust Questions:
What’s the most used method for oligo synthesis currently?
The most used method for DNA synthesis is through Phosphoramidite chemistry. This technology consists of the use of Nucleoside Phosphoramidites, a type of modified nucleosides that allows the sequential addition of new bases in a cyclic manner. These modified nucleosides are protected in a way that chemists can control the reaction of oligonucleotide synthesis by exposing only the regions of the nucleotide they desire.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
One of the reasons why synthesis of oligos longer than 200 nt is the increase of errors caused by the natural DNA polymerases error rate or fidelity of nucleoside phosphoramidite thecnology. Another reason could be the limitations of Quality Control, since oligos require MALDI spectrometry to test their quality, this method limits the length to 10-50 nucleotides.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Production of long oligos faces the main challenge of accumulating errors in their formation making it difficult to obtain high yield of oligos with high quality (Yin et al, 2024)
George Church Questions:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
While many organisms are capable to synthesize all these 20 amino acids, some groups like ours (Metazoa) have lost the capability to synthetize nine EAAs. These amino acids are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Additionally, arginine can also be considered essential because of the incapacity of the body to synthetize it under special periods of growth (Lopez & Mohiuddin, 2024). One explanation for the loss of the synthetic capacity of these essential amino acids is because of energetic efficiency. Estimates suggest that essential amino acids have high energetic costs in their synthesis. Selective pressures towards energy efficiency may contribute to the loss of capacity to produce essential amino acids and relying on them by direct consumption (Kasalo et al., 2026).
Considering that in Jurassic Park movies the lysine contingency consists in limiting the expansion of dinosaurs by creating to them the incapacity of lysine production. This contingency now seems futile because animals have more essential amino acids than dinosaurs in that case. Animals have overcome this limitation through the ingest of these amino acids in their diets, so in consequence dinosaurs can also survive by consuming other living things that produce lysine either from animal or plant sources.
Kasalo, N., Domazet-Lošo, M., & Domazet-Lošo, T. (2026). Outsourcing of energetically costly amino acids at the origin of animals. Nature Communications. https://doi.org/10.1038/s41467-026-68724-6
Pray, L. (2008) DNA Replication and Causes of Mutation. Nature Education 1(1):214
Wang, F., He, Q., O’Donnell, M. E., & Li, H. (2025). The proofreading mechanism of the human leading-strand DNA polymerase ε holoenzyme. Proceedings of the National Academy of Sciences, 122(22), e2507232122. https://doi.org/10.1073/pnas.2507232122
Yin, Y., Arneson, R., Yuan, Y., & Fang, S. (2024). Long oligos: direct chemical synthesis of genes with up to 1728 nucleotides. Chemical Science, 16(4), 1966–1973. https://doi.org/10.1039/d4sc06958g
Week 2 HW: DNA Read, Write & Edit
Part 1: Benchling & In-silico Gel Art
Lambda Sequence: Sequence from E.coli I cl857 S7 lambda bateriophage (Daniels, et al., 1983) available at New England Biolabs (N3011)
A digest simulation was performed using the lambda sequence and 7 different restriction enzyme (EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI). The range of fragments obtained from this simulation varies depending on the enzyme used.
EcoRV, for example, has 21 restriction sites, giving 22 band in the simulation. On the other hand, KpnI, SacI, and SalI have a few restriction sites, showing only two bands in the simulation (Figure 2).
Since restriction enzymes cleave specific sequences in the genome, the difference between the number of sites for EcoRV compared to KpnI, SacI, and SalI raises the question: **Why does the lambda genome have more restriction sites for EcoRV than others?
Bacteriophages usually present fewer restriction sites as a response to this defense mechanism. This difference may change depending on the interaction between the bacteriophage and its host (Pleška & Guet, 2017)
Gel Art: Raimondi Stela
Using a simulation of the digestion of lambda genome with different restriction enzymes, I tried to portray the “god of staffs”. This is a deity found in the Raimondi Stela that belonged to the Chavin culture (Figure 3)
The gel created tried to be similar to the deity holding their staffs.
Part 3: DNA design challenge
Protein Chosen: Bothrops atrox snake venom nerve growth factor
Description: Nerve Growth Factor (NGF) is a member of the neurotrophin family that regulates the growth, differentiation, and survival of peripheral neurons during the development of the nervous system. This factor acts through two key receptors, tyrosine kinase A receptor (TrkA) and p75 neurotrophin receptor (p75NTR). The TrkA receptor activates signaling cascades that promote neuron differentiation and neurite growth.
Snake venom NGF (sNGF) is a protein that has been reported from the venom of elapid and viperid snakes. It is proposed that the presence of sNGF in the venom helps the envenomation process by causing the release of signaling chemicals that promote inflammatory reactions and increase vascular permeability, aiding the spread of other toxins and promote the apoptosis of cells (Sunagar et al., 2013).
Justification: NGFs have been proposed as promising options to treat neurodegenerative diseases and promote regenerative processes. sNGFs show high similarities to human NGFs and have been studied for many applications like chondrogenesis, neurite outgrowth, neuroprotection, tumor growth inhibition, etc. (Devi & Jayaraman, 2025).
Because sNGFs present special activities during envenomation, the study of sNGFs from other snake species may help to find new functions with a possible use in the study of regeneration and nervous development. These new functions may contribute to the design of synthetic alternatives with specific functions that can be applied for therapeutic purposes.
Protein Sequence: I have chosen the sequence of a sNGF from Bothrops atrox snake venom available at the UniProt database (ID: A0A1L8D608). The existence of this protein was proved through transcription level.
Selection of the expression system
To continue with the process of reverse translation and codon optimization, I investigated which expression system would be the most suitable to produce this protein. Schütz et al. (2023) offers a concise guide for expression system selection with a decision graph depending on the characteristics of the protein to be expressed (Figure 3).
Figure 3: Decision Scheme for gene expression system. This scheme is based on the protein characteristics. Figure taken from Schütz et al. (2023)
I gathered the following information of the protein based on four decision points proposed by Schütz:
The target is eukaryotic protein
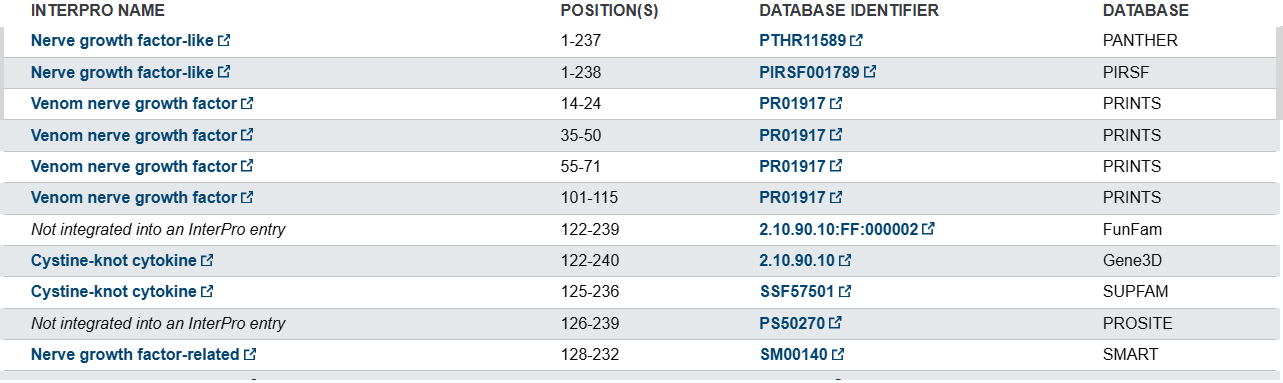
Uniprot PTM/Processing section describes that the protein contains a signal region related to its secretion between the 1-18 amino acids and three disulfide bonds (Figure 4).
The resulting protein would have 241 amino acids in total when expressed and 233 amino acids when secreted with a molecular mass of 27.197 KDa.
Figure 4: PTM/Processing information of B. atrox NGF available at UniProt (ID: A0A1L8D608)
Uniprot information from other NFGs does not show that requires glycosylation when expressed
In the case of this design, it wouldn’t be necessary to have an expression system with higher yield
Based on this information the decision graph suggests using an expression system using a strain of E. coli that promotes disulfide bond formation. This decision is also supported by other studies that use E. coli to express human NFGs in vitro (Tilko et al., 2016; Dicou, 1992)
Reverse Translate
Before performing the reverse translation of the protein, I decided to eliminate the amino acids 1-18 because they are part of the signal region of the protein and this won’t be used for this design. I included an initiator methionine at the N-terminus to allow translation initiation. The sequence modification was realized using the Benchling software.
Using the same software I reversed translated the protein using Escherichia coli (K12) genetic code using the method Match codon usage. The result of this process is an optimized sequence of 672 bp. This sequence was used later to perform a Blastx analysis where was found that the resulting sequence matches with other NGF from snakes (Figure 5)
Figure 5: BlastX analysis of the translated NGF using the Benchling software, the sequence showed close simmilarities with other snake NGFs and a predicted NGF group.
Codon optimization
To simulate the creation of a clonal gene using the Twist Bioscience environment, the optimized sequence was uploaded in the software. A codon optimization was performed in the application. During the configuration of the optimization, I conserved the region 321-524 since it’s predicted as the NGF region by the Blastx result.
The resulting sequence was later labeled as optimized B.atrox NGF (BatroxNGFOptimized) and finally chosed as the sequence to be used for the creation of the expression cassette.
Expression Vector Selection
To select a suitable expression vector, it is necessary to consider that the protein requires a proper environment to develop three disulfide bonds. The formation of disulfide bonds can be achieved by expressing the protein in E. coli periplasm or in the cytoplasm of engineered E coli.
A study performed by Shamriz et al. 2016 uses the pET-32a expression vector that contains the Trx-tag for increasing the solubility of the protein and its expression in E. coli Origami (DE3) to promote the correct formation of disulfide bonds in the cytoplasm of E. coli. Another strategy aims to translocate the recombinant protein into the periplasm using a signal peptide that helps the formation of disulfide bonds and increases its stability (Pouresmaeil & Azizi-Dargahlou, 2023).
Based on this information I opted for a pET-29b(+) expression vector from Twist Bioscience because it contains an N-terminal S•Tag™ sequence and may help with the protein solubility and a C-terminal His•Tag® sequence for its easy purification.
To help with the sulfide formation I selected SHuffle® strain from New England Biolabs that is engineered for the formation of disulfide bond in the cytoplasm.
Another way to express this protein is by adding signal sequence to allow the translocation of the protein to the periplasm and this could be analyzed later if possible.
Part 4: Preparation of Twist DNA Synthesis Order
A simulation of DNA Synthesis order was generated using the optimized NGF sequence obtained from the previous part and inserted into the pET-29b (+) expression vector generating the plasmid as can be observed below (Figure 6)
Figure 6: pET-29b Expression Plasmid with optimized B.atrox NGF sequence. Annotations of relevant regions where performed using the Benchling software and using information of the vector pET-29b (+) from Twist Bioscience
Part 5: DNA Read/Write/Edit
DNA ReadSequencing Idea: Genome-Wide Association Studies of Genetic Elements Related with Peanut Allergy Diversity in PeruDescription: Allergies are misdirected immune reactions against a specific molecule (Allergens) to a previously exposed patient. These reactions are associated with an immune response mediated by a particular type of antibody called IgE. Allergies are diverse in nature and involve several ambiental and congenital factors, but also genetic factors. Several genes have been investigating for their involvement in allergic reactions, showing a complex heterogeneity that varies person to person (Falcon & Caoili, 2023)
Genetic factors associated with allergies may help to elucidate the mechanisms that promote allergies predisposition. For that purpose, Genome-wide association studies (GWASs) offer a good option to study the genetic elements associated with allergies.
GWAS are used to identify the association between genotypes with phenotypes. This is performed by selecting a group of individuals to obtain their phenotypic information. Using different GWAS arrays or sequencing strategies, genotypes of these individuals are obtained. Phenotypic and genotypic information is later used to conduct association tests to obtain relevant genetic elements that may be important for the phenotype studied (Uffelmann et al., 2021)
Technologies to perform GWAS genotyping
GWAS genotyping technologies are microarrays, Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS). To study the genetic component of peanut allergy in Perú we can use previously associated genes like HLA-DQ and HLA-DR o genes located in chromosome six (Allergies and Genetics | Health and Medicine | Research Starters | EBSCO Research, n.d.). The objective of genotyping these genes is to determine Single Nucleotide Polymorphisms (SNPs) that might have strong association with peanut allergy. Whole genome sequencing technologies can be applied for SNP genotyping and involves sequencing all regions of the entire genome. On the other hand, Whole exome sequencing is a method for sequencing only the exonic region of the human genome.
Is the method first-, second- or third- generation or other?
Whole and Exome genome sequencing are part of the Next Generation Sequencing (NGS) because they are based in the massively parallel sequencing process.
What is your input? How do you prepare your input?
For this study my input is genomic DNA extracted from a representative Peruvian sample of individuals diagnosed with peanut allergy.
What are the essential steps of your chosen technology, how does it decode the bases of your DNA sample?
For WGS studies, Illumina uses a sequencing technology by synthesis, where fluorescently labeled nucleotides to sequence millions of clusters on a cell surface in parallel.
What is the output of your sequencing technology?
Illumina sequencing data is obtained through the signal intensity measurement of the labeled nucleotides that serve a terminators
DNA WriteProject Idea: Snake venom NGF from B atrox
The following idea aims to express a snake venom NGF from B. atrox. sNGFs have been applied in numerous studies to test their potential effect on regenerative processes because of their similarity with the human NGF and because of novel properties that may appear because of its evolution in the snake venom.
For its production I propose the recombinant production of this protein, for that I realized used a sequence available at UniProt (ID: A0A1L8D608) a reverse translated to then propose it cloning using a vector in E. coli and expression in the same organism.
DNA EditProject Idea: Using genetic engineered cells in hydrogels for cartilage regeneration
Hydrogels are tridimentional networks polymers that can be used as scaffold for cartilage tissue engineering. A promising approach is to modify the genome of stem cells, creating specific gene circuits to promote cartilage regeneration. Trough gene edition, we could use steam cells to modify their proliferation capacity or control it using genetic circuits, a concept that may help with this idea is the concept of BioBricks that allows to create libraries that coul be used to modify the behavior of these stem cells (Elnaggar et al., 2025).
Daniels, D.L. et al. (1983). Appendix II: Complete Annotated Lambda Sequence. R.W. Hendrix, J.W. Roberts, F.W. Stahl and R. A. Weisberg(Ed.), Lambda-II. 519-676. New York: Cold Spring Harbor Laboratory Press.
Devi, S., & Jayaraman, G. (2025). Unraveling the molecular basis of snake venom nerve growth factor: human TrkA recognition through molecular dynamics simulation and comparison with human nerve growth factor. Frontiers in Bioinformatics, 5, 1674791. https://doi.org/10.3389/fbinf.2025.1674791
Elnaggar, K. S., Gamal, O., Hesham, N., Ayman, S., Mohamed, N., Moataz, A., Elzayat, E. M., & Hassan, N. (2025). A guide in synthetic biology: Designing genetic circuits and their applications in stem cells. SynBio, 3(3), 11. https://doi.org/10.3390/synbio3030011
Falcon, R. M. G., & Caoili, S. E. C. (2023). Immunologic, genetic, and ecological interplay of factors involved in allergic diseases. Frontiers in Allergy, 4, 1215616. https://doi.org/10.3389/falgy.2023.1215616
Pleška, M., & Guet, C. C. (2017). Effects of mutations in phage restriction sites during escape from restriction–modification. Biology Letters, 13(12). https://doi.org/10.1098/rsbl.2017.0646
Pouresmaeil, M., & Azizi-Dargahlou, S. (2023). Factors involved in heterologous expression of proteins in E. coli host. Archives of Microbiology, 205(5), 212. https://doi.org/10.1007/s00203-023-03541-9
Shamriz, S., Ofoghi, H., & Amini-Bayat, Z. (2016). Soluble Expression of Recombinant Nerve Growth Factor in Cytoplasm of Escherichia coli. Iranian Journal of Biotechnology, 14(1), 16–22. https://doi.org/10.15171/ijb.1331
Sunagar, K., Fry, B. G., Jackson, T. N. W., Casewell, N. R., Undheim, E. a. B., Vidal, N., Ali, S. A., King, G. F., Vasudevan, K., Vasconcelos, V., & Antunes, A. (2013). Molecular Evolution of Vertebrate Neurotrophins: Co-Option of the Highly Conserved Nerve Growth Factor Gene into the Advanced Snake Venom Arsenalf. PLoS ONE, 8(11), e81827. https://doi.org/10.1371/journal.pone.0081827
Uffelmann, E., Huang, Q. Q., Munung, N. S., De Vries, J., Okada, Y., Martin, A. R., Martin, H. C., Lappalainen, T., & Posthuma, D. (2021). Genome-wide association studies. Nature Reviews Methods Primers, 1(1). https://doi.org/10.1038/s43586-021-00056-9
Week 3 HW: Lab Automation
Opentrons Artwork: Gel Designing
Design: Snake Trimeresurus puniceus
Inspired from a snake photo taken in the Oswaldo Meneses serpentarium, Lima, Peru. Art created Donovan’s Automation art interface
Python Script Design
Opentrons script was created following the instructions and ideas offered by the HTGAA Opentrons Colab. To create the script first I created a pseudocode with the idea of how the robot will work
Pseudocode
Get the coordinates of the art from donovan’s page in the form of a dictionary
Create a function Coordinate_per_color:
Pick up a 20 ul tip
For each coordinate
Check if the tip is empty (20 ul volume)
Aspirate an amount depending on the number of coordinates to fill (20 or less)
Get the x and y coordinates
Move to the x and y coordinates
Dispense 1 ul to the coordinate
Remove the tip
Call the function Coordinate_per_color for each color present in the dictionary
Following the idea of the pseudocode I followed the script design from Dominika Wawrzyniak, 2021 student and adapted to the coordinates from Donovan’s Automation page. For this first draft I decided to copy and paste the coordinates and give them a dictionary structure, then I changed the color names using the names from the robot deck setup constants. The resulting script is the following :
# Set the initial coordinates take from the donovan's page (Converted into a dictionary)Coordinates={"Green":[],"Red":[],"Blue":[],"Yellow":[],"Cyan":[]}#To avoid using many tips the objective is to create a function that takes up the points and add the volume per colordefCoordinate_per_color(color_string):# Pick up a 20 ul tippipette_20ul.pick_up_tip()# For every coordinate per colorforiinrange(len(Coordinates[color_string])):# i shows the number of positionsifi%20==0:# Aspirate a volume 20 if the total of remaining coord to paint is more than 20pipette_20ul.aspirate(min(20,len(Coordinates[color_string])-i),location_of_color(color_string))# Get the x and y coordinatesx_coordinate=Coordinates[color_string][i][0]y_coordinate=Coordinates[color_string][i][1]# Move to the x and y coordinatesadjusted_location=center_location.move(types.Point(x_coordinate,y_coordinate))# Dispense 1 ul to the positionpipette_20ul.dispense(1,adjusted_location)hover_location=adjusted_location.move(types.Point(z=2))pipette_20ul.move_to(hover_location)# Finishing drop the tippipette_20ul.drop_tip()#Call the function Coordinate_per_color for every color in the dictionaryfornameinCoordinates.keys():print(name)Coordinate_per_color(name)
After executing the script, I simulated the visualization and got the Image I wanted to create
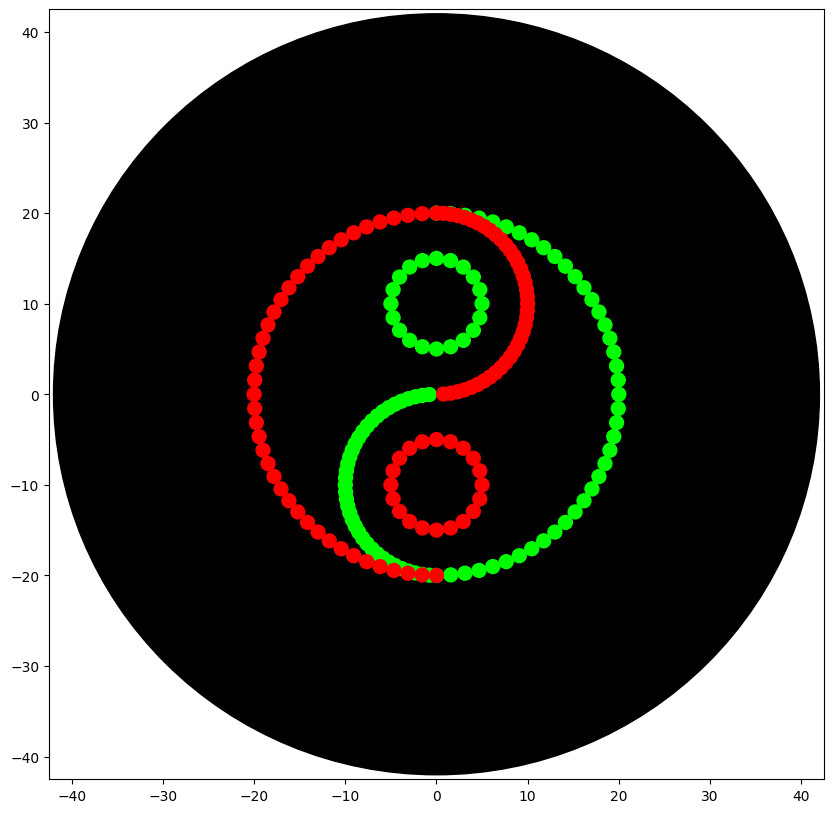
Second Design: Geometrical Green/Red Yin and Yang
After recieving the instructions from my node y change the design for a Yin-Yang inspired design. To create the design I comtemplated the idea of using mathematical formulas to desing the pattern.
AI assistance
ChatGPT was used to support conceptual understanding of the geometric contruction of the Yin-Yang symbol using circles and semicircles. All code implementation was independently developed by me, only using the artificial inteligence to offer some feedback.
First I created a code testing the mathematical approach which consisted in creating the design using circles and semicircles, first I integrated the mathematical code with the robot operation code resulting in a messy code
//FirstYin-YangCode# Start at the centercursor=center_location.move(types.Point(x=0,y=0))# Define de radius as 20 (To reduce the times to aspirate a volume)radius=20# Define the number of point (Default 40)points=40# Function to create semicirclesdefthetha(i):theta=np.pi*i/(points-1)returntheta# Fucntion aspiratedefaspirate(color):ifi%20==0:pipette_20ul.aspirate(min(20,points-i),location_of_color(color))# Function hoverdefhover():hover_location=adjusted_location.move(types.Point(x=0,y=0,z=2))pipette_20ul.move_to(hover_location)# Create a green semicirclepipette_20ul.pick_up_tip()foriinrange(points):aspirate("Green")theta=thetha(i)x=radius*np.sin(theta)y=(radius*np.cos(theta))adjusted_location=cursor.move(types.Point(x=x,y=y))pipette_20ul.dispense(1,adjusted_location)hover()# Create and S-divider in the circle (UpperSide)inner_radius=10foriinrange(points):aspirate("Green")theta=thetha(i)x=inner_radius*np.sin(theta)y=(inner_radius*np.cos(theta))+radius/2adjusted_location=cursor.move(types.Point(x=x,y=y))pipette_20ul.dispense(1,adjusted_location)hover()pipette_20ul.drop_tip()# Create a Red semicirclepipette_20ul.pick_up_tip()foriinrange(points):aspirate("Red")theta=thetha(i)x=radius*np.sin(theta)y=(radius*np.cos(theta))adjusted_location=cursor.move(types.Point(x=-x,y=y))pipette_20ul.dispense(1,adjusted_location)hover()# Create and S-divider in the circle (LowerSide)inner_radius=10foriinrange(points):aspirate("Red")theta=thetha(i)x=inner_radius*np.sin(theta)*-1y=(inner_radius*np.cos(theta))-radius/2adjusted_location=cursor.move(types.Point(x=x,y=y))pipette_20ul.dispense(1,adjusted_location)hover()
Second Attemp: To make the code more readable I created two custom functions called create_circle and create_semicircle. Also adapted the logic to get a list of coordinates so it can be used by the code example shared by the Node.
## Ying-Yang Code ## Create two functions that will give the coordinate for circles## Circle functiondefcreate_circle(x_center,y_center,radius,points):coordinates=[]foriinrange(points):angle=2*math.pi*i/pointsx=x_center+radius*math.cos(angle)y=y_center+radius*math.sin(angle)coordinates.append((x,y))returncoordinates## Semicircle functiondefcreate_semicircle(x_center,y_center,radius,points,direction="left"):"""
Four semicircle orientations:
- right
- left
- up
- down
"""coordinates=[]foriinrange(points):angle=math.pi*i/(points)# Change directionifdirection=="right":# Base x and y coordinates (Default = right)x=x_center+radius*math.sin(angle)y=y_center+radius*math.cos(angle)coordinates.append((x,y))elifdirection=="left":x=x_center+radius*math.sin(angle)y=y_center+radius*math.cos(angle)coordinates.append((-x,-y))elifdirection=="up":x=x_center+radius*math.cos(angle)y=y_center+radius*math.sin(angle)coordinates.append((x,y))elifdirection=="down":x=x_center+radius*math.cos(angle)y=y_center+radius*math.sin(angle)coordinates.append((-x,-y))else:raiseValueError("direction must be: right, left, top or bottom")returncoordinates# Ying-Yang Design: Using the robot script offered by the node# Green parts# Green middle circle boundary pipette_20ul.pick_up_tip()green_big_circle=create_semicircle(0,0,20,40,"right")forx,yingreen_big_circle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Green"))dispense_and_detach(pipette_20ul,1,adjusted_location)# Semicircle which center is the middle inferior part of the circlegreen_S_semicircle=create_semicircle(0,10,10,40,"left")forx,yingreen_S_semicircle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Green"))dispense_and_detach(pipette_20ul,1,adjusted_location)# Small circle whose center is at the center of the s_semicirclegreen_small_circle=create_circle(0,10,5,20)forx,yingreen_small_circle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Green"))dispense_and_detach(pipette_20ul,1,adjusted_location)pipette_20ul.drop_tip()# Red parts# Red middle circle boundary pipette_20ul.pick_up_tip()red_big_circle=create_semicircle(0,0,20,40,"left")forx,yinred_big_circle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Red"))dispense_and_detach(pipette_20ul,1,adjusted_location)# Semicircle which center is the middle inferior part of the circlered_S_semicircle=create_semicircle(0,10,10,40,"right")forx,yinred_S_semicircle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Red"))dispense_and_detach(pipette_20ul,1,adjusted_location)# Small circle whose center is at the center of the s_semicirclered_small_circle=create_circle(0,-10,5,20)forx,yinred_small_circle:adjusted_location=center_location.move(types.Point(x=x,y=y))ifpipette_20ul.current_volume==0:pipette_20ul.aspirate(1,location_of_color("Red"))dispense_and_detach(pipette_20ul,1,adjusted_location)pipette_20ul.drop_tip()
The final code allowed me to obtain the desired Yin-Yang Design
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications
The article I found interesting is developed by Kverneland et al (2024). In this article an automated workflow is designed with the objective of preparing protein samples for LC-MS/MS Analysis. Using Opentons OT-2 robot Hela cells samples and plasma serum from patients were prepared with a shotgun approach to prepare the sample for the proteomic analysis. From this approach they analyzed 192 HeLa samples and consistently identified approximately 8000 protein groups and 130,000 peptide precursors.
The importance of this study relies on the necessity of identifying and analyzing protein profiles of many samples. Proteomics approaches offer valuable information that can be used to discover novel biomarkers and contribute to the development of personalized treatments. Also, this article provides a potential approach to creating databases containing proteomic information that could be used for novel synthetic technologies like, for example, de novo design of proteins.
Write a description about what you intend to do with outomation tools
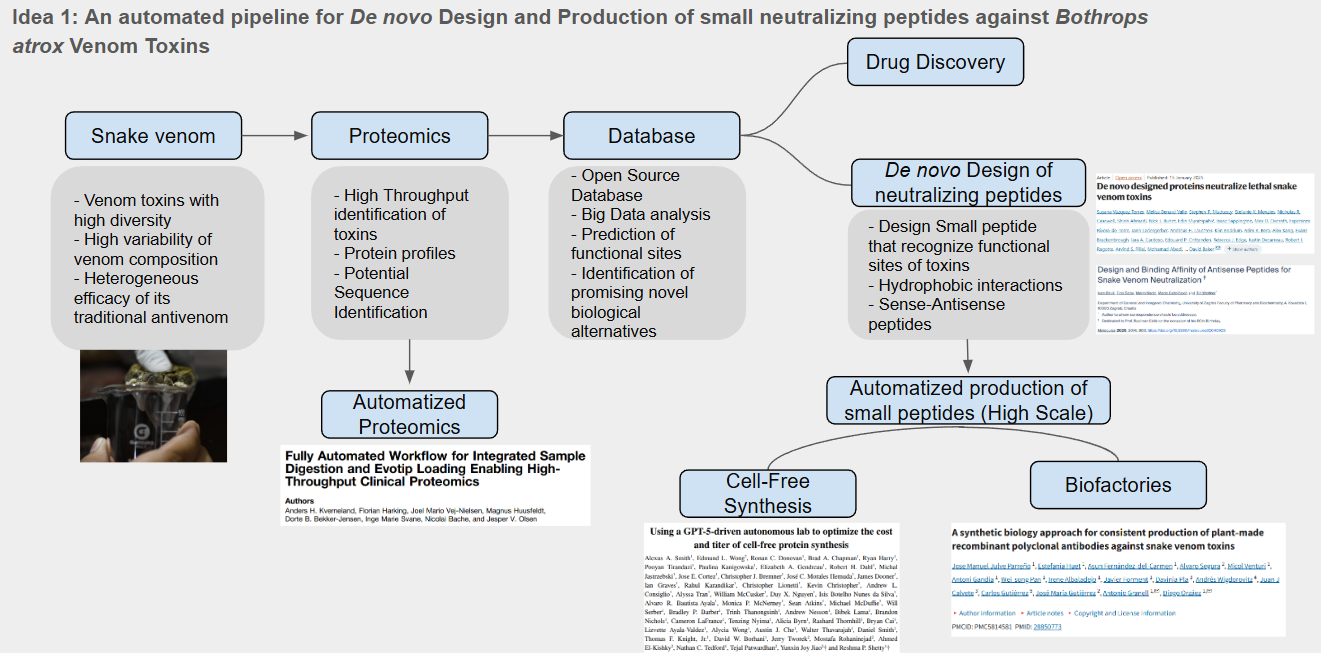
Idea 1: An automated pipeline for De novo Design and Production of small neutralizing peptides against Bothrops atrox Venom Toxins
For this idea I designed a pipeline and identified the use of automation approaches in two key activities as shown below
The use of automated proteomic can help us to identify novel proteins with potential therapeutical applications and also identify protein families and relevant sequences, this sequences can be used for the desing of small neutralizing peptides that may be used against snake venoms. Since the design would produce a high amount of potential candidates it is necessary an automated process of protein synthesis. For this I identify the use of AI driven cell-free protein synthesis as promising aproach to produce and test possible candidates to neutralize snake venom toxins.
Final Project Ideas
I created three slides containing my three final project ideas using the lessons learned until now:
How many molecules of amino acids do you take with a 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming whole composition of meat is protein, the number of amino acids molecules in 500 grams is 3.011 x 1024 molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This is because our digestion breaks down macromolecules into their monomers. Proteins are broken down into amino acids that later are used for the biosynthesis of proteins. The phenotypic characteristic of an organism is defined in its principally by its genome, and it’s not affected by the food they consume.
Why are there only 20 natural amino acids?
Amino acids origin is a complex field of study of many theories indicating why living organisms have conserved 20 amino acids. Crick for example mention the “Frozen accident theory” which states that amino acids have been kept the same as living beings’ complexity cross a limit where other amino acids cannot compete with the gold standard. On the other hand, there is a close relationship between these 20 amino acids and living being’s metabolism, suggesting that these amino acids are the result of metabolism optimization (Kirschning, 2022).
Can you make other non-natural amino acids? Design some new amino acids.
Non-natural amino acids are modified amino acids that don’t belong to the traditional 20 amino acids, these amino acids can be produced by several organisms. Non-natural amino acids can be designed by modifying natural amino acids using techniques grouped in the field of Peptidomimetics.
Where did amino acids come from before enzymes that make them, and before life started?
Several hypotheses have been proposed for the origin of amino acids, for example the “ARN World” hypothesis proposes the existence of a precursor to RNA whit the capability of processing information and catalyze chemical reactions. Evidence of this hypothesis is the presence of Ribozymes, RNA molecules with catalytic activity (Higgs & Lehman, 2014). Other theories propose other molecules like coenzymes or the association of metabolic pathways and amino acids for their production and the possible apparition of amino acids from meteorites (Kirschning, 2022)
If you make a -helix using D-amino acids, what handedness (right or left) would you expect?
L- and D-Amino Acids are chiral molecules, meaning that L-Amino Acids are the reflection of D-Amino acids. Alpha-helix using L-amino Acids results in right handedness helix. D-Amino Acids Could result in left handedness in their alpha helix (Novotny & Kleywegt, 2005).
Why are most molecular helices right-handed?
Most helices are right-handed because they are formed by L-amino Acids which are more stable at that orientation.
Why do β-sheets tend to aggregate?
Aggregation may be promoted by the presence of Aggregation Prone Regions (APRs) that are 5-15 residue long stretches in proteins. These APRs regions are usually buried inside the hydrophobic core of the protein. A protein with more APRs may aggregate because this rearing can link together through hydrogen bones which is also promoted by their hydrophobicity forming a “steric zipper” (Aggregation Prone Regions (APRs), n.d.). a. What is the driving force for β-sheet aggregation?
Β-sheet aggregation is promoted by intermolecular forces like hydrogen bonds and hydrophobic interaction between the Aggregation Prone Regions
Can you use amyloid β-sheets as materials?
A study made by Cheng et al. (2012) proposes the use of amyloid β-sheets mimics (ABSMs) to antagonize the aggregation of amyloid proteins and reduce their toxic activity.
Cheng, P., Liu, C., Zhao, M., Eisenberg, D., & Nowick, J. S. (2012). Amyloid β-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nature Chemistry, 4(11), 927–933. https://doi.org/10.1038/nchem.1433
Higgs, P. G., & Lehman, N. (2014). The RNA World: molecular cooperation at the origins of life. Nature Reviews Genetics, 16(1), 7–17. https://doi.org/10.1038/nrg3841
Kirschning, A. (2022). On the Evolutionary History of the Twenty Encoded Amino Acids. Chemistry - a European Journal, 28(55), e202201419. https://doi.org/10.1002/chem.202201419
Novotny, M., & Kleywegt, G. J. (2005). A survey of left-handed helices in protein structures. Journal of Molecular Biology, 347(2), 231–241. https://doi.org/10.1016/j.jmb.2005.01.037
Protein Analysis and Visualization
Protein Description: Bothrops atrox snake venom nerve growth factor (NGF)
Nerve Growth Factor (NGF) is a protein involved in the process of neurite differentiation and growth through the activation of a receptor that promotes signaling cascades. Snake venoms possess NGFs that might contribute to the toxicity of the venom by activating pro-inflammatory signals that increase vascular permeability. Snake NGF has been proposed as an alternative for therapeutic strategies aimed at regenerating tissues.
Amino acid sequence description
The amino acid sequence was obtained from the Uniprot database (ID: A0A1L8D608) predicted at a transcriptomic level
Using Uniprot’s Blast tool I found that the protein has high homology with a broad number of NGFs from other species of snakes and also from other reptiles and fishes
Uniprot’s database informs that this protein belongs to NGF-beta family and other related families according to different databases
NFG structure page
The B. atrox NGF doesn’t have a crystallyzed protein so its structure haven’t been resolved. To answer the protein structure questions another NGF from Mus musculus (ID: pdb_0001btg)
This protein has been resolve with 2.50 A structure resolution. This makes the structure a good model to study
Mus musculus Domains are part of the Neurotrophin family according to the Scop Database, each chain is linked with the 1BET A: 10-116 Domain (SCOP ID: 8026411)
Protein Visualization
For the visualization of the B. atrox NGF a model was created using the SWISS-MODEL tool to generate an predicted structure base on homology (A0A1L8D608)
Protein Visualization was performed using Pymol Software
NGF model was visualized as ribbons, cartoon and sticks. This model is a homo-2-mer
Coloring the protein by secondary structure shows that the protein lacks of alpha helix structures and is mainly formed by b-sheets structures and loops.
Hydrophobic (orange) and Hydrophilic (skyblue) residues were colored in the model. The model contains a homogeneous distribution of these residues along the beta sheets structure while the loops are mainly hydrophobic.
Part C1: ML-Based Protein Design Tools
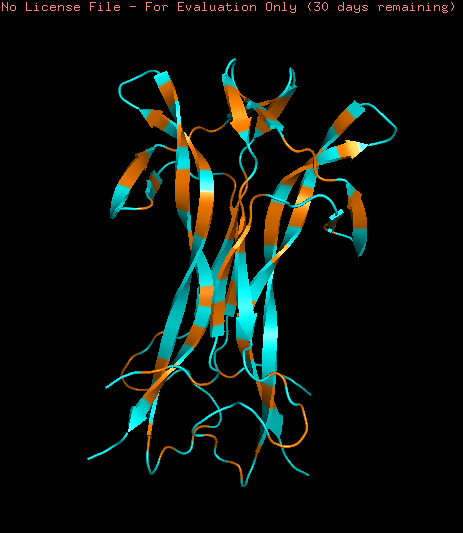
The protein selected is the crystal structure of beta nerve growth factor at 2.5 A resolution from Mus musculus (ID: pdb_00001btg).
Deep Mutational Scans:
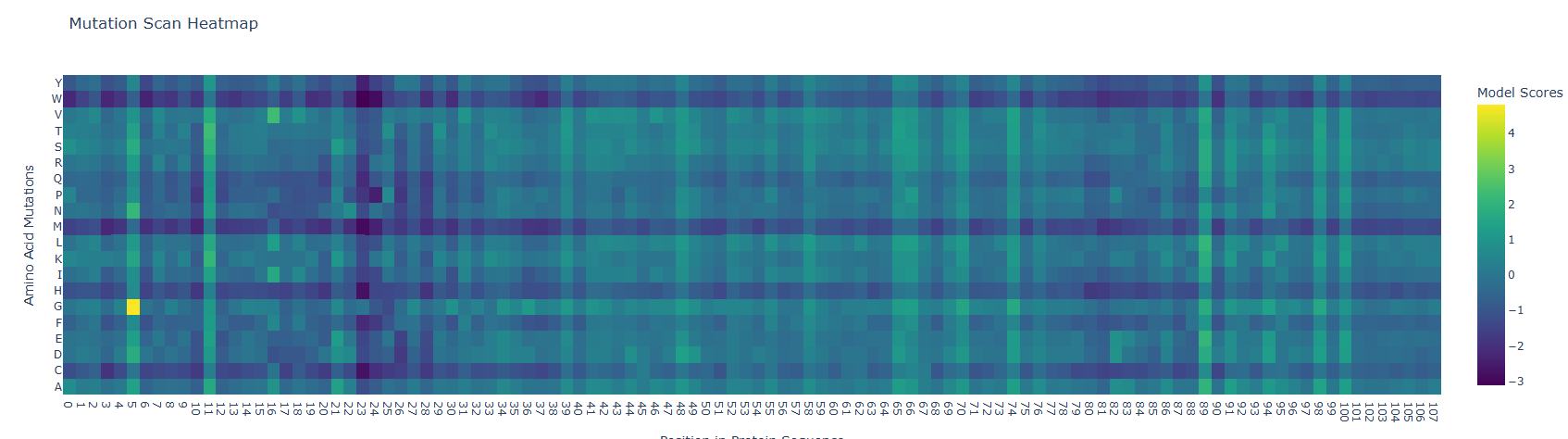
Mutation Scan Heatmap results show low scores along the protein sequence as can be observed below. However the fifth residue (S) showed a high mutational score with the glycine amino acid. Low scores in the mutation heatmap may be related with their protein conservation. A study from Barker et al. (2020) states that Mammalian nerve growth factors (NGF) are conserved, this could be related with a low mutational capacity. To corroborate this hypothesis I suggest comparing mutational scores of NGF of mammals and other animal groups.
Latent Space Analysis
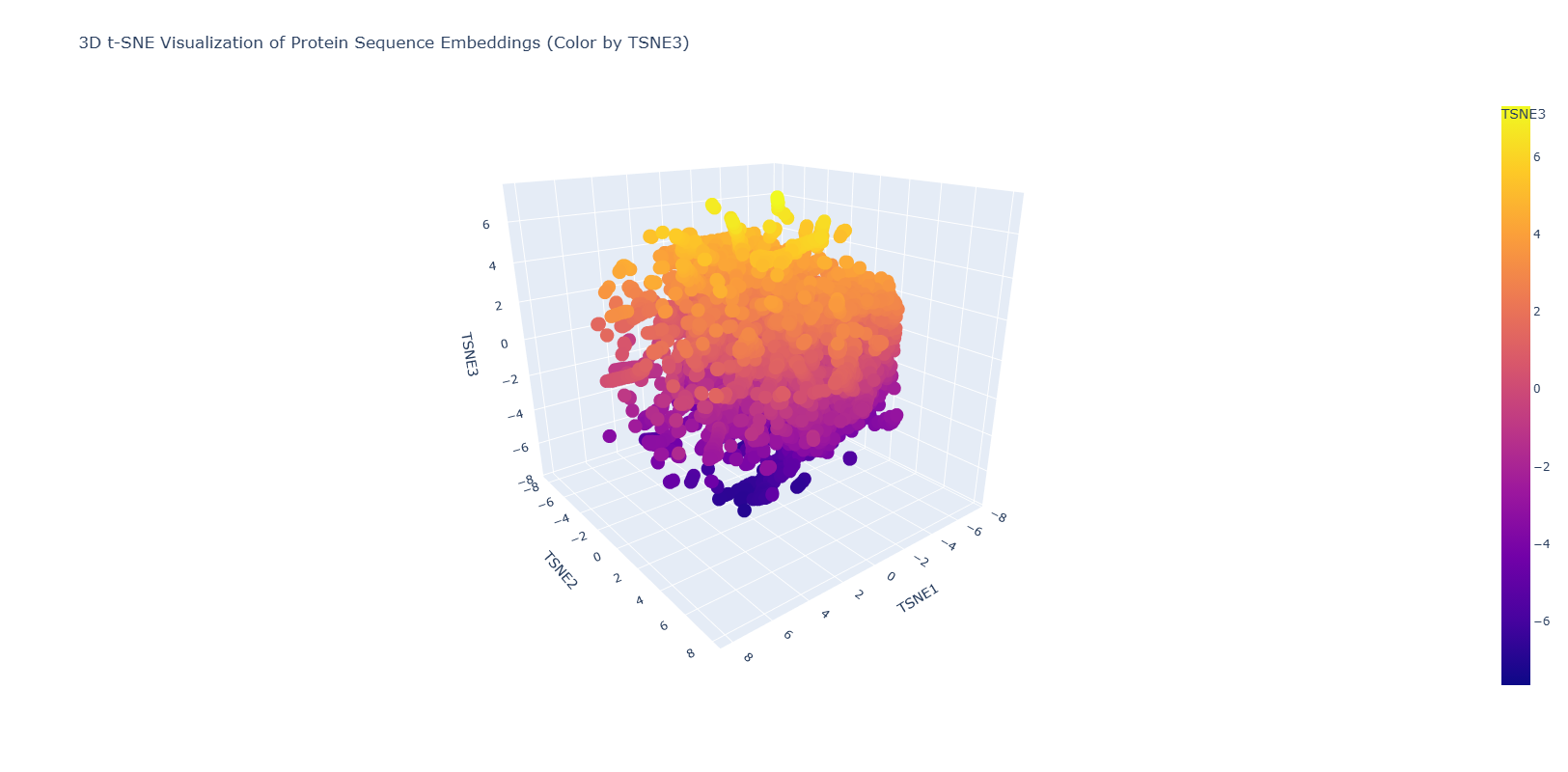
The figure below shows the latent space result after introducing the NGF sequence.
The resulting figure shows a homogeneous distribution of the proteins in the database with small dispersed regions. This could suggest a gradual evolution of the proteins used in this database. Dispersed regions are diverse in origin and also comprises automated matches.
Part C2: Protein Folding
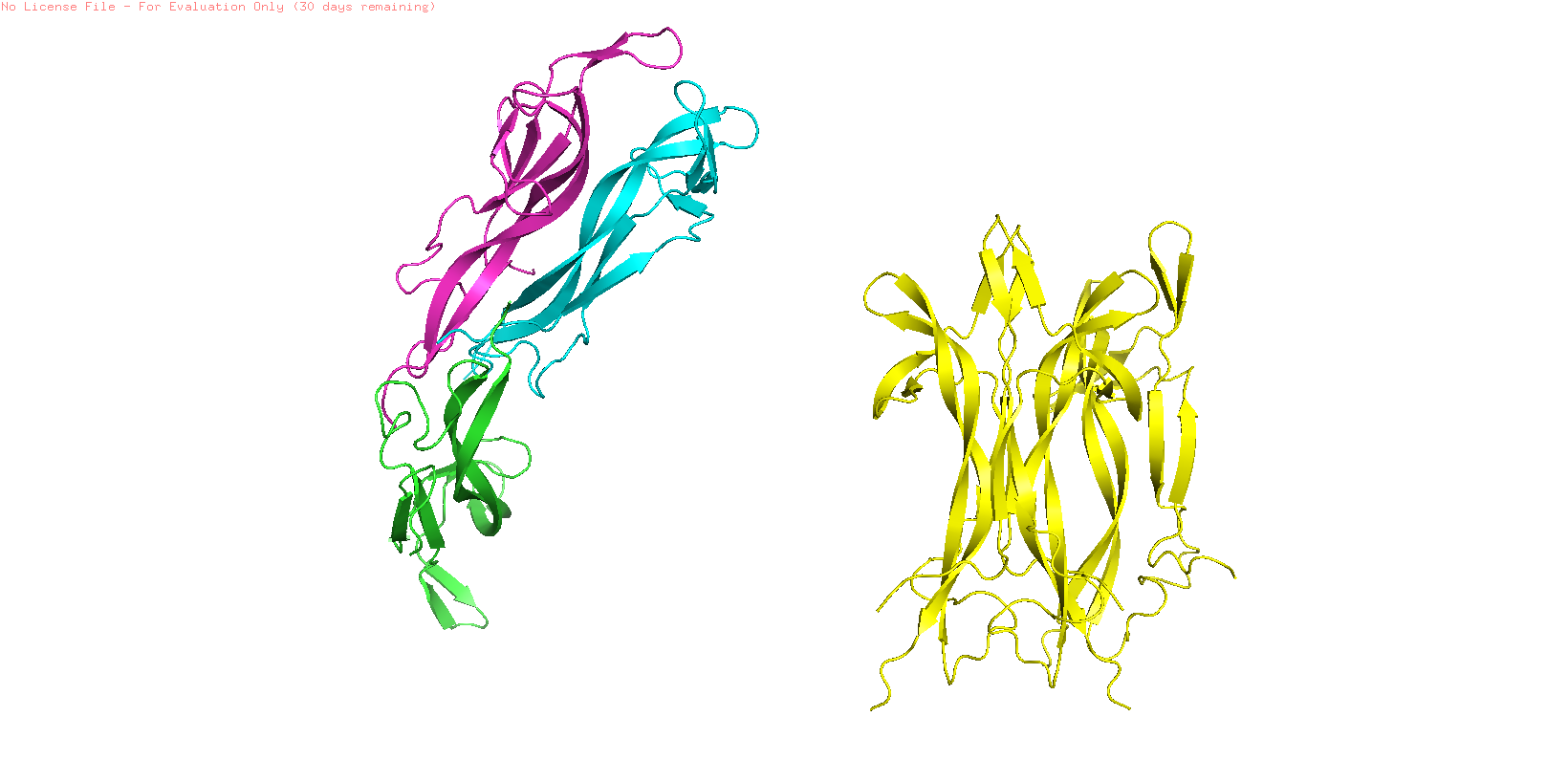
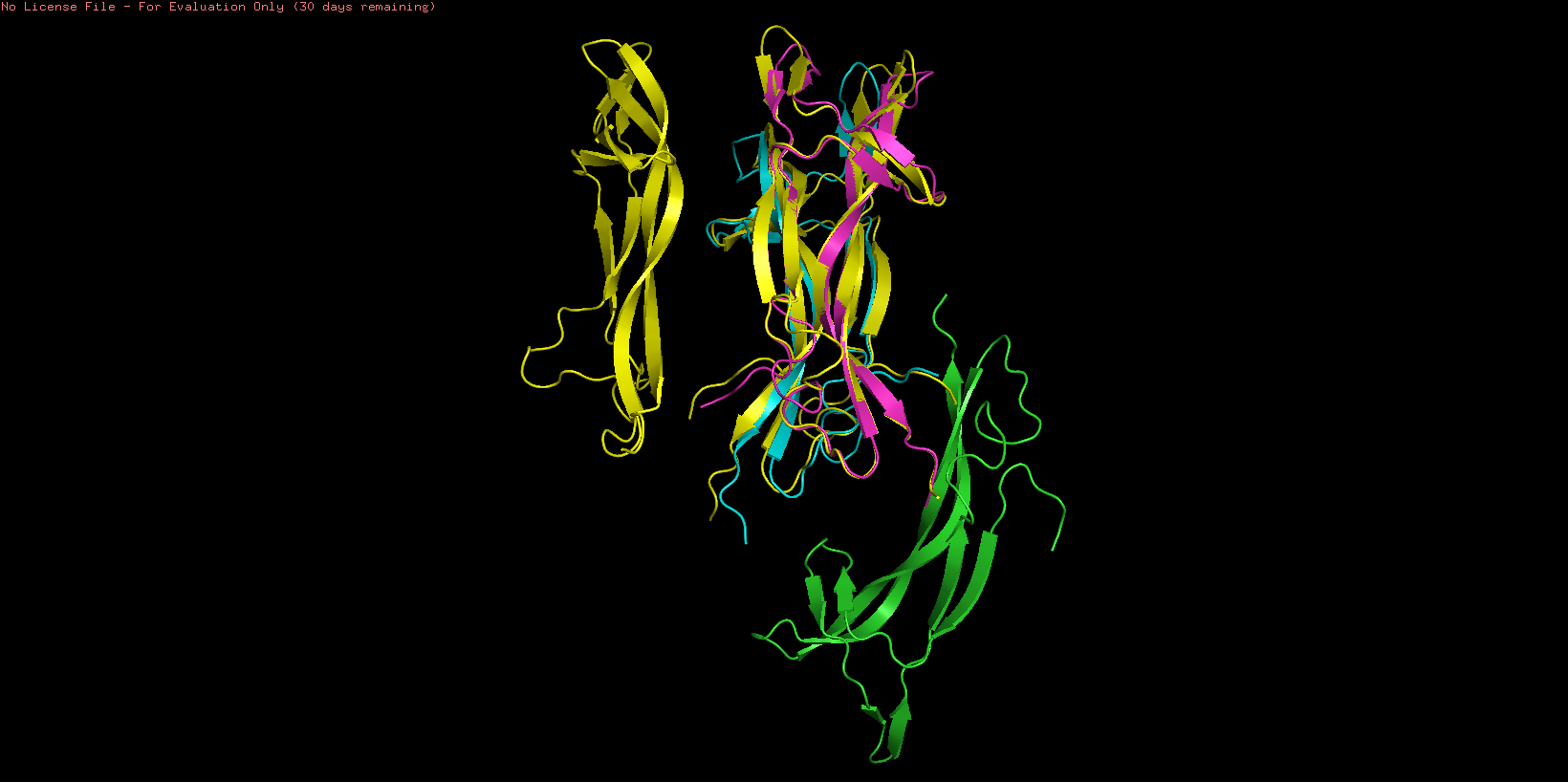
NGF folding obtained using the ESMFold program was created using the sequence from the Mus musculus NGF (ID: pdb_00001btg). The sequence was used creating 3 copies since the original PDB sequence presents a homotrimeric structure. The resulting prediction (Yellow) was compared with the original structure (Multicolor) using the Pymol software
To study the quality of the protein folding the predicted region was aligned with the original structure
As showed in the image above, an aligment was performed using only two chains of both structures (B and C) resulting an RMSD = 0.487 indicating a close alignment between the predicted structure and it’s original model. Similar result can be observed comparing the resulting structures, observing similarities en their secondary structure. This result suggests the correct application of the ESMFold language and indicates that the predicted protein can be used for other structural studies.
Part C3: Reverse Folding
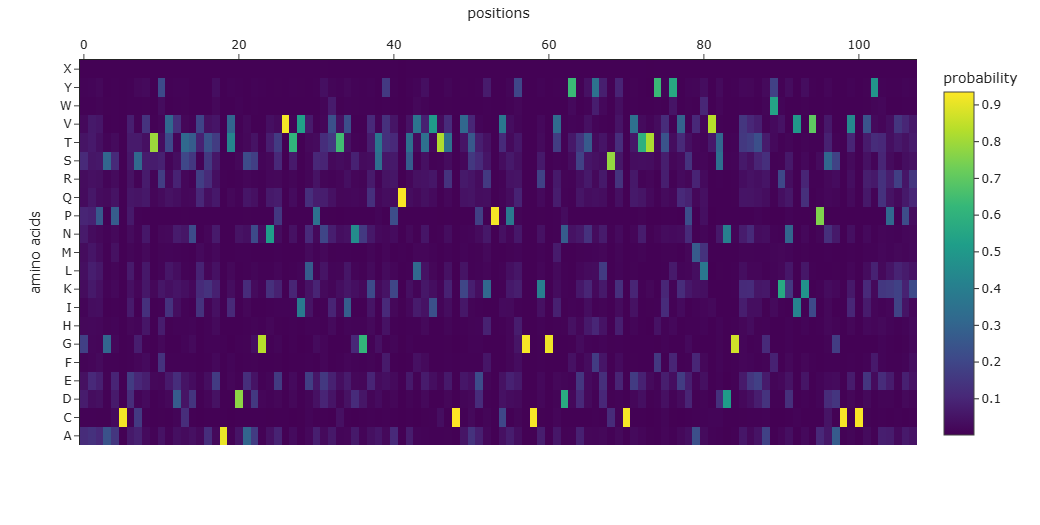
The Mus musculus NGF was used with the Protein MPNN tool to predic the protein sequence based on the structural information. Using the default parameters of the tool a new sequence was obtained:
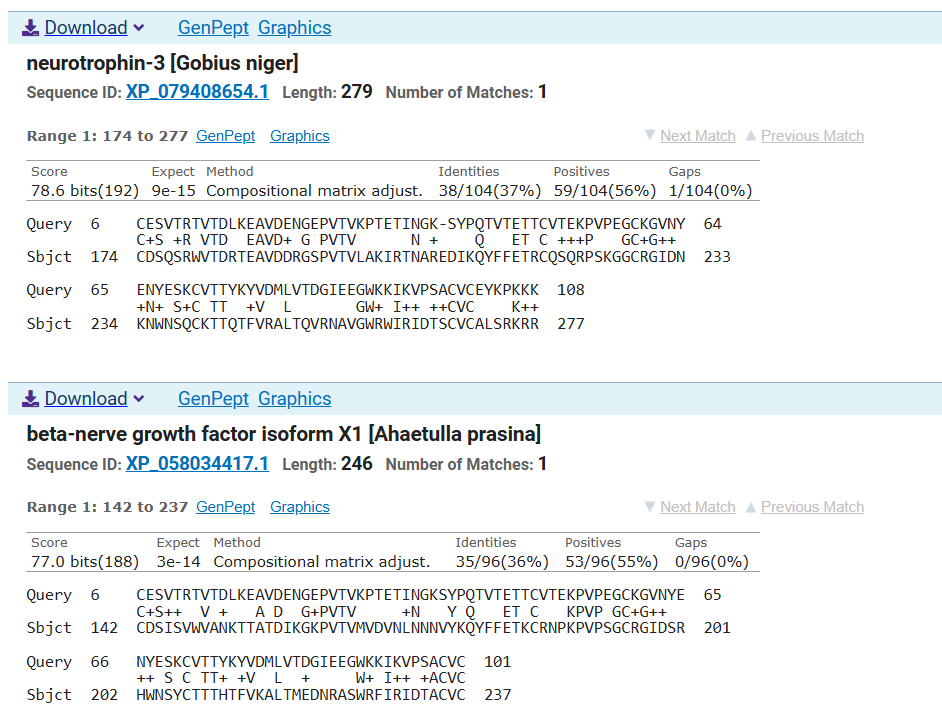
The resulting sequence presents a recovery score of 0.3519 wich suggest a low recovery percentage of the original sequence. Similar result can be observed when aligning both original and predicted sequence using Clustal Omega and through the probability graph of every aminoacid:
However Blast analysis predicted a close relationship of the predicted sequence with other NGFs of different species and other neurotrophins which could be supported with their evolutionary conservation of this family of proteins
Part D. Group Brainstorm on Bacteriophage Engineering
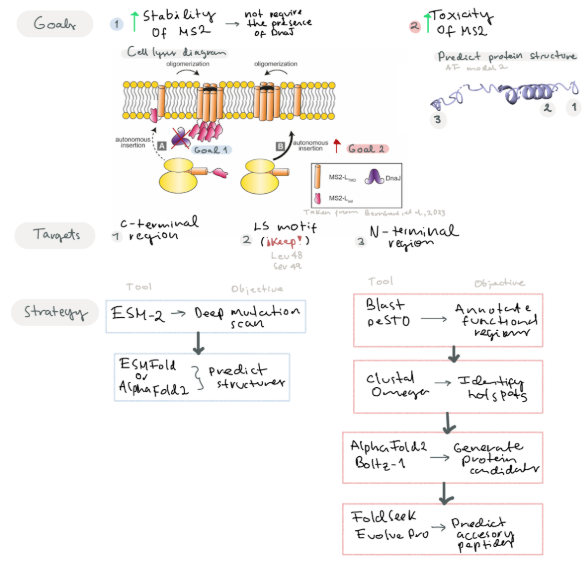
Main Goals:
Goal 1: Increase the stability of the MS2 lysis protein by predicting mutations of residues near the C-terminal region and surrounding the LS motif
Goal 2: Improve the N-terminal region by modifying residues to contribute to its toxic activity or add new functional regions that may increase its toxicity.
Improving MS2 lysis protein by modifying regions not related with the Leu48 and Ser49 (LS motif) and surrounding to improve protein toxicity (Chamakura et al, 2017). Predict mutations that may improve its stability. We suggest that by increasing protein stability, the protein would not require the presence of the DnaJ for its action.
Another goal is to design new accessories to the N-terminal region to improve lysis toxicity. Berkhout et al, 1985 suggest C-terminal region is key for protein activity, so taking this in consideration we can try to modify the N-terminal region to improve protein stability or add a new characteristics that may improve the toxicity of the protein
Strategy
Given our two main goals, we propose different strategies to address each objective
For the first goal, we propose using a protein language model such as ESM-2 to perform in silico deep mutational scan that evaluates the plausibility of all possible single-point mutations in the MS2 L protein. Subsequently, we will employ ESMFold or AlphaFold2 to predict the resulting 3D structural variations.
For the second goal:
Step 1: Identify and Annotate key functional regions near the C-terminal motif and LS motif
Software: Blast (For conserved domains), PeSTO (Functional motifs)
Predict mutations near the N-terminal and C-terminal site that may improve protein stability
Software: Clustal Omega (To identify hotspots for mutations)
Generate different protein candidates with mutations and evaluate their stability
Software: Alpha-Fold Multimer, Boltz-1
We propose using Alpha-Fold with a specific training set for bacteriophages
Predict accessory peptide sequences to insert in their N-terminal region and improve its toxicity
Software: FoldSeek (To find remote sequences with similar folding), EvolvePro (To suggest optimized N-terminar sequences)
Test suitability of these protein candidates by performing docking essays with a bacterial membrane model, etc.
The model can predict structures that look stable and coherent, but it does not measure real folding energy, membrane insertion, or toxicity. “Looks good” in silico ≠ “works better” in vivo.
Selection of variants that appear structurally improved but do not increase stability or toxicity — or even reduce lytic activity.
Phage-specific training / limited viral datasets
If the model is trained or fine-tuned on a small, biased set of phage proteins, it may learn dataset-specific patterns instead of general biological rules (overfitting).
Mutations may seem optimized in the model but fail outside that narrow dataset. Reduced generalizability and misleading predictions.
References:
Chamakura, K. R., Edwards, G. B., & Young, R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology, 163(7), 961–969. https://doi.org/10.1099/mic.0.000485
Berkhout, B., De Smit, M., Spanjaard, R., Blom, T., & Van Duin, J. (1985). The amino terminal half of the MS2-coded lysis protein is dispensable for function: implications for our understanding of coding region overlaps. The EMBO Journal, 4(12), 3315–3320. https://doi.org/10.1002/j.1460-2075.1985.tb04082.x
Week 5 HW: Protein Design Part II
Part 1: SOD 1 Binder Peptide Design
Superoxide dismutase 1 sequence was retrieved from Uniprot database (P00441), this protein has a length of 154 amino acids.
The mutated version of the human SOD1 caused by an A4V mutation was retrieved from the PDB database that contains a structure obtained from an X-Ray Diffraction study with a resolution of 1.90 Å (Hough et al., 2004)
1UXM_1 Superoxide Dismutase Mutated from Homo sapiens:
An alignment between normal SOD 1 and mutated SOD 1 was performed using Clustal Omega to corroborate the mutation at position four, an initial methionine was included into the mutated SOD 1 to have a protein of the same length. (Figure 1)
Figure 1: Multiple sequence alignment Clustal between SOD1 sequence (POO441) retrieved by Uniprot database and mutated SOD1 sequence available at PDB database (1UXM_1). Alignment shows a single point mutation in the residue 4 A/V that has been reported in several studies a the cause of the amyotrophic lateral sclerosis (ALS) disease
Small protein binders were generated using the PepMLM model made by Chen et al (2025). Four peptides were generated with a length of 12 amino acids and a Top K value of 3.
index
Binder
Pseudo Perplexity
0
WRYYAVVVAHKX
12.802906286585648
1
WHYGVVALAHKX
7.909934706159041
2
WLSYPAALRHKX
11.125327842529979
3
WRSPAAAVRWKE
11.952399811426888
The four candidates have low pseudo perplexity values (< 20) indicating confidence from the model to the peptides designed (Chen et al. 2025). A fasta document was created including the four candidates with the mutated SOD sequence and SOD-1 binding peptide FLYRWLPSRRGG as a control. However Generated candidates contained an X amino acid coded that means an unknown residue.
These candidates were aligned with the original protein using Clustal Omega (Figure 2)
Figure 2: Multiple sequence alignment Clustal between three small binders candidates and the mutated SOD1 sequence, another peptide was used as control to compare the suitability of the generated binders. Results shows close similarities between the three candidates and the region 32-44 of the mutated SOD protein, while the control didn't show the same similarity with the candidates
Part 2: AlphaFold 3 Binders
Peptide candidates were modeled using the AlphaFold Server together with the mutated SOD 1 sequence. The control peptide was also modeled and showed a close integration into the SOD 1 structure. Candidates 1, 2, and 3 haven’t shown an integration into the internal structure of SOD 1 (Figure 3)
Figure 3: AlphaFold Generation of the interaction between the candidates and mutate SOD1 sequence. Candidates 1,2 or 3 don't show a possible insertion to a pocket region in the target sequence while the control seem to interact and insert well into the protein
Confidence metrics are presented in the table below where pTM and ipTM scores are shown for each Candidate and the control. These scores measure the accuracy of the structures generated. For all candidates and the control, the pTM scores are more than 0.5, suggesting some confidence that the structure is like its true structure. On the other hand, ipTM value suggests poor confidence in the relative position of the subunits within the complex
Peptide
ipTM
pTM
Control
0.26
0.78
Candidate 1
0.36
0.76
Candidate 2
0.45
0.83
Candidate 3
0.36
0.87
Part 3: PeptiVerse Evaluation
PeptiVerse was used to predict several characteristics that are required for proposing a binding peptide with therapeutical application.
Candidate
Solubility
Hemolysis
Binding Activity
pH
Length
Molecular Weight
Candidate 1
Soluble
Non-Hemolytic
Weak
9.70
12
1373.7 Da
Candidate 2
Soluble
Non-Hemolytic
Weak
9.99
12
1323.8 Da
Candidate 3
Soluble
Non-Hemolytic
Weak
10.84
12
1456.7 Da
Control
Soluble
Non-Hemolytic
Weak
11.71
12
1507.7 Da
Candidates 1, 2, and 3 showed high solubility and low hemolytic probability, indicating their possible expression and use. However, pHs obtained a highly basic making it difficult to keep their structure in blood. Predicted Binding activities suggest that the candidates would have a weak interaction with their target. This result is also supported by the ipTM values gotten indicating that these candidates could not be able of binding to the target.
Part 4: Optimized Peptides Generation with moPPIt
Peptide binders were produced using the moPPIt using the mutated SOD1 N-terminal as target region. I propose that these candidates would bind to the mutated region and prevent the aggregation by stabilization of the structure. Peptides were generated considering as objectives and weights their Hemolysis probability, Solubility, Affinity and Specificity.
A total of 4 candidates who were generated have low pseudo-perplexity values indicating low uncertainty for the model to the predicted sequence (OFS Pseudo-perplexity for Protein Fitness, n.d.)
Candidates
Sequence
Pseudo-Perplexity
Candidate 1
WRYYAVVVAHKX
12.80
Candidate 2
WHYGVVALAHKX
7.90
Candidate 3
WLSYPAALRHKX
11.12
Candidate 4
WRSPAAAVRWKE
11.95
A Clustal Omega alignment was performed for all the candidates generated by moPPIt and PEPMLM showing close similarities in their sequences (Figure 4)
Figure 4: Multiple alignment between PepmLM and moPPit generated peptides. Alignment shows close similarities with the peptides generated by both language models
moPPIt candidates were evaluated using the PeptiVerse programs to evaluate their main characteristics and therapeutical applicability.
Candidate
Solubility
Hemolysis
Binding Activity
pH
Length
Molecular Weight
Candidate 1
Soluble
Non-Hemolytic
Weak
9.70
12
1373.7 Da
Candidate 2
Soluble
Non-Hemolytic
Weak
8.61
12
1262.7 Da
Candidate 3
Soluble
Non-Hemolytic
Weak
9.99
12
1323.8 Da
Candidate 4
Soluble
Non-Hemolytic
Weak
10.84
12
1456.7 Da
All candidates were predicted with weak affinity and presented a pH superior to 7 making them difficult to use directly in a human.
References
Hough, M. A., Grossmann, J. G., Antonyuk, S. V., Strange, R. W., Doucette, P. A., Rodriguez, J. A., … & Hasnain, S. S. (2004). Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proceedings of the National Academy of Sciences, 101(16), 5976-5981.
Chen, L. T., Quinn, Z., Dumas, M., Peng, C., Hong, L., Lopez-Gonzalez, M., … & Chatterjee, P. (2025). Target sequence-conditioned design of peptide binders using masked language modeling. Nature Biotechnology, 1-9.
Zhang, Y., Tang, S., Chen, T., Mahood, E., Vincoff, S., & Chatterjee, P. (2026). PeptiVerse: A Unified Platform for Therapeutic Peptide Property Prediction. bioRxiv, 2025-12.
The MS2 lysis proteins is a small protein resposible for host cell lysis during bacteriphage infection and can be used as antimicrobial candidates. The aim of this project evaluate mutants of the MS2 lysis protein to improve the stability of the lysis protein and its interaction of de DnaJ protein. The following protein sequences were used:
MS2 lysis protein sequence:
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
MS2 lysis sequence was analyzed using Blatp tool most related sequences were extracted. Clustal Omega tool was used to align the lysis protein with their most related sequences to identify possible conserved sites (Figure 1).
Figure 1: Clustal Omega Multiple Alignment of the MS2 lysis protein and other lysis sequences extracted from Blastp
Clustal alignment showed a strong conserved N-terminal region with a conserved hydrophobin core, however some variable residues were found in the region 23-29 and 44-49.
Using the google collab offered in the activity a mutation heatmap was produced to see the mutational scores of the MS2 protein (Figure 2)
Figure 2: Mutational Heatmap of the MS2 lysis protein
Heatmap results show lower mutation scores in predicted conserved sites in Clustal Omega suggesting that these conserved regions could have important functions in the protein. Comparing multiple alignment with the heatmap suggest that residues 5, 24-29, 38, 44-49 may be mutated.
These regions were analyzed by comparing their function with Chamakura et al (2017) that suggests that regions close to the C-terminal region like the LS motif can affect the integration of the protein to the membrane, for that reason these region were avoided. Additionally, residues 44-49 have different effect in the activity of the protein and these residues were also considered as potential mutation sites.
By comparing Clustal aligment, mutational heatmap and experimental values I selected the following residues to generate mutants
Residues 23 - 29 based on conserved and mutational sites
Variable residues 44, 47, 49 based on experimental studies
To determine the type of mutation, experimental results found in the cvs document were use to determine the types of subtitutions that doesn’t affect the activity of the protein lysis. Mutated sequence is shown below:
Mutated MS2 Lysis Protein (K23E, C29R, L44P, F47Y, S49L)
Figure 3: Clustal Omega Multiple Alignment of the Mutated MS2 Lysis protein and Native protein
Stage 2: Interaction Analysis using Boltz
Boltz interactions were produced between the native and mutated lysis proteins and DnaJ proteins as a target. Streptococcus pneumonia DnaJ pdb was used based on similarity search in the PDB database (ID: 6JZB)
Figure 4 shows the interaction of the mutated protein (green) and native protein (red). Mutated lysis protein showed a loop around the DnaJ protein, but this wasn’t observed in the native protein, however binding confidence values in both structures were low indicating that these results aren’t conclusive.
Figure 4: Boltz interaction analysis of the mutated MS2 lysis protein and native protein
Modification of residue 49 seems to be related with the change of the folding, to corroborate this the mutation of the residue 49 was reverted (Figure 5) showing that the protein have lost the loop that generates it folding around the DnaJ protein, suggesting that residue 49 may have an important structural role in the protein, however further studies are required.
Figure 5: Boltz interaction analysis of the mutated MS2 lysis protein with the residue 49 reverted and native protein
Week 6 HW: Genetic Circuits Part 1
Part 1: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fifelity PCR Master Mix offered by New England Biolabs is a product that contains a DNA polymerase with high fidelity useful for cloning and amplification of difficult amplicons. Phusion DNA polymerase contains proofreading activity (3’ -> 5’ exonuclease) and a higher fidelity 50X greater that Taq polymerase. Thermo Fisher Scientific Phusion High-Fidelity PCR Marter Mix is composed of a HF Buffer or GC Buffer; both buffers are used to reduce the error rate of the DNA polymerase. HF Buffer contains a lower error rate (4.4 x 107) than GC Buffer (9.5 x 107), however GC buffer can improve the performance of the polymerase on some difficult or long templates with high GC-rich templates or with secondary structures. The master mix is also provided with a optimized concentration of MgCl2 which is an essential cofactor that stabilizes the DNA double helix and facilitates primer annealing. High Fidelity PCR reaction can also include DMSO that is used to reduce the melting temperature through its association with Cytosine residues that changes the conformation of the DNA template.
What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature is affected by the Primer melting temperature, annealing temperature is usually 3-5 °C below the primer Tm this temperatura depends on the GC content of the primers or primer length and the prescense of secondary structures. Another factor that can modify the Ta is the MgCl2 concentration indicating the higher concentrations can increase the Ta.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR is a laboratory method often used to increase the amount of a desired DNA sequences, it uses primer to determine the boundaries of the fragment and uses a DNA polymerase that can be genetically engineered to get different results. PCR is often used to amplify sequences for other analysis like sequencing or cloning. Resctriction enzyme digest used restriction endonucleases that recognize specific palindromic sequences and cuts these sites, it doesn’t not amplify the target sequence and is often used for its integration in a cloning vector for recombinant technologies.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences will be appropiate for Gibson cloning the regions must have overlaping regions that can be used to connect correctly with the vector. The digested vector mus be linear and it must be purified to prevent re-ligation with the vector.
How does the plasmid DNA enter the E. coli cells during transformation?
The era multiple transfection methods use to insert a target DNA sequence inside the E. coli cells different methods can be used, one method called electroporation uses an electric shock to disturb the membrane and insert the DNA vector, another method uses a heat shock to disturb and insert the desired vector.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golde Gate Assembly uses Type IIS restriction enzyme and T4 DNA ligase that cuts outside their recognition sequence, this allows to desing overhangs that can be ligated later, later this recognition sites are removed during assembly. Golden gate method allows assembling up to nine fragments at a time in a recipient plasmid. This can be done in a single tube that contains the donors, the recipient vector, a type IIS restriction enzyme and ligase (Engler & Marillonnet,. 2013)
Some advantages of the Golden Gate Assembly are:
The overhang sequence creates is not dictated by the REase, so no scar sequence is introduced
The fragment-specific sequence of the overhangs allows orderly assembly of multiple fragments simultaneously
The restriction site is eliminated from the ligated product, so digestion and ligation can be carried out simultaneously
Figure 1: NEB Golden Gate Assembly Workflow
References:
Engler, C., & Marillonnet, S. (2013). Golden gate cloning. In DNA cloning and assembly methods (pp. 119-131). Totowa, NJ: Humana Press.
Part 2: Asimov Kernel
Represilator Recreation
To test the different parts of the Kernel options a recreation of the Demo Repressilator was made (Figure 2). The repressilator consists in three genes LacI, LambdaCI and TetR and their following repressor sequence pTetR that supress LacI expression in the presence of TetR, pLacI that supress LambdaCI expression in the presence of LacI and pLambdaCI that supress the expression of TetR. The logic inside this construct is that at the beginning LacI, LambdaCI and TetR are expressed, the expression of these three proteing will supress the expression o the other proteins. The presence of a protein can modify the expression of the the other protein following a domino effect that could be used to control de expression of a protein depending the input introduced to the system
Figure 2: Repressilator construct made in Asimov Kernel
Simulation of the genetic circuit can be observed in figure 3. RNA and Protein concentrations varies over time having peak when it relative repressor expression is lower. For example LambdaCI concentrations increases when LacI increases and TetR concentrations are higher, but increasing the concentration of LambdaCI decreases the concentration of TetR over time and Increases LacI concentration that ends reducing LambdaCI, this result shows the ciclic nature of this genetic circuit making them useful to control de expression of these proteins over time
Figure 3: Simulation of Repressilator Results
Own Constructions
LacI progressive repressor
The objective of this construct was to creater a circuit that proggressively reduced the expression of a protein. For that purpose a LacI promoter was selected and build with and test BBa test protein, then a weaker promoter was added with the LacI sequence. The results obtained shows a weak repression of the test protein however I coudn’t see if the repression could increase progressively until the expression of the test protein decreases completely, this is because of the absence of RBS regions making it difficult to express the desire proteins (Figure 4, 5)
Double Repressor
A lac activator was built as genetic circuit, this circuit consists in three proteins TetR, LamdaCI y BBa_K2150013 as the targe protein. The expression of the target protein is suppressed by the pLamdaCI sequence in the presence of the LabmdaCI protein. The expression of LambdaCI is suppressed by TetR that is suppressed by a LacI protein. The logic of this circuit is that at the beginning when no LacI is addedd to the system the expression of the target protein is off caused by the presence of LambdaCI but the addition of LacI or TetR to the system can supress the expression of the target protein, with this construct I aim to suprress the expression of the target protein depending the presence of to supressors or only one and investigate if the can affect the yield of the expression.
Figure 4: Double Repressor simulation
Gradual expression of a targe protein
The following genetic circuit consist in three parts, a TetR protein that is regulated by a pLacI repressor, a LambdaCI protein controlled by a TetR suppressor and a BBa_K125013 that acts as the target protein followed by another LacI protein. The logic of this circuit is to produce a circuit that starts with the expression of the target protein, TetR and LacI. The expression of LacI along with the target protein slowly reduces the expression of the TetR protein, increasing the expression of LamdaCI that switch off the expression of the target protein and LacI, the reduced espression of LacI activaes the expression of TetR regenerating the genetic circuit. This circuit aims to produce the target protein at fixed amount by controlling is expression using the LacI protein.
Figure 5: Gradual Expression construct
Week 7 HW: Genetic Circuits Part II
Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
One characteristic of IANNs is that they can process continuous inputs rather than only discrete boolean data, this resembles with the natural process of a living system that is composed of a complex network of molecular interactions. Boolean login can simplify complex living activities making it difficult to process and engineer systems with this logic but IANNs uses complex analysis that can be used to reproduce complex process in living beings that uses millions of parameters. By training an IANN is possible to simulate complex genetic activities that resemble more to the living processes than only using boolean functions (Nilsson et al, 2022)
Because IANNs works with continous data they can work better where a system have more molecular noise and context effect thant Boolean switches making them useful in therapetic context where there is a high amount of molecular noise that must be processed to produce a suitable answer (Müller et al., 2025)
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Proposal: Intracellular articial neural networks for close monitoring and pain modulation in rheumatoid arthritis
The goal of this proposal is to evaluate the implementation of a cell-based system that implements IANNs logic to detect inflammatory signals in tissues affected by rheumatoid arthritis, computed a graded pain/inflammation state, and regulate de release of neuromodulatory or anti-inflammatory effectos to reduce pain and tissue damage.
Input molecular: Molecules related with rheumatoid arthritis like IL-1, TNF-alpha, IL-6. Also some other conditions related with proinflammatory conditions like local pH and specific neurotransmissors
Sensors: Promoter or riboswith that can detect each input and transform it into signals that can be processed by the neural network. For example Choi et al (2021) used carlaginous implants that contain a series of genetic circuits that detect inflammatory factor of the K/BxN model of inflammatory arthritis in the figure below
Figure 1: Experimental design a of a gene circuit that expresses a inhibitor of IL-1 in response of a proinflammatory ligan and is application in a implant to modulate inflammatory responses in K/BxN model of rheumatoid arthritis (Extracted from Choi et al., 2021)
Output: Controlled expression an secretion of anti-inflammatory factor or neuromodulatory peptides for local pain modulation. The evaluation of the model can be performed through fluorescent reporters that can be measured experimentally
Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials can have the following applications:
Mycelium-based composites (MBCs): This type of materials is composed of bio-composite waste bonded with dense mycelium to generate a material that can be used in constructions, insulating material or packaging. The importance of these type of materials is that can replace materials that used fossil fuels for their manufacturing offering a ecological alternative. However these materials require optimization studies that for the correct growth of the material (Camilleri et al., 2025). Additionally MBCs can have biomedical application as biocompatible drug delivery systems of scafolds.
Mycellium foams: Mycellium foams uses the properties of chitin and chitosan, polysaccharide found in fungus. This mycellium foams can resist strenghts of up to 25 MPa and up to 200 MPa and have high performance as natural insulators. Becuase of these characteristics Mycellium foams can be used replacing thermoplastics in insulation foams, furniture, decking, etc (Majib et al 2025).
Mycellium Leather: Mycellium can be used to produce a fungal-based leather-like material that can be used in the production of chlotes and other leather articles. However this alternative faces the lack of internationally recognized standards such as ISO making them difficult to implement commercially yet (Elsacker et al., 2023).
Fungal materials face some challenges like the limited documentation available which indicates the need of further studies for these alternatives. Fungal materials also have high water absorption making them unsuitble for huming environments and the have a slower production cycle since Fungi growth takes many days.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For Mycellium-based composites (MBCs) would require the genetic modification of the fungi species to increase improve their growth and reduce the time it take to proudce the materials, aditionally more resistant fungi would be require for materials because of the constant interaction of these materials with harsh environmental conditions.
One advantege of fungi over bacteria is their capacity of expressing proteins with post-translational modifications and proper folding making them more suitable for the production of animal-based molecules. Also their capacity to produce large amounts of enzyme makes them more useful for biomass degradation.
Part 3: Final Project Expression Cassette Design
A DNA linear expression cassette was produced for the final project. This expression cassette was designed in Benchling using sequences from several scientific papers. The expression cassette contains some protective 5’ and 3’ sequences to prevent the degradation of the cassette in the cell-free system, a T7 promoter and terminator with, a Ribosome Binding Site, and a His-Gst taq for its purification and improve the folding of the protein binders. Figure 2 presents the DNA expression cassette made in Benchling.
Figure 2: Linear DNA expression cassette made for the final project
References:
Nilsson, A., Peters, J. M., Meimetis, N., Bryson, B., & Lauffenburger, D. A. (2022). Artificial neural networks enable genome-scale simulations of intracellular signaling. Nature Communications, 13(1), 3069.
Müller, M. M., Arndt, K. M., & Hoffmann, S. A. (2025). Genetic circuits in synthetic biology: broadening the toolbox of regulatory devices. Frontiers in Synthetic Biology, 3, 1548572.
Choi, Y. R., Collins, K. H., Springer, L. E., Pferdehirt, L., Ross, A. K., Wu, C. L., … & Guilak, F. (2021). A genome-engineered bioartificial implant for autoregulated anticytokine drug delivery. Science Advances, 7(36), eabj1414.
Camilleri, E., Narayan, S., Lingam, D., & Blundell, R. (2025). Mycelium-based composites: An updated comprehensive overview. Biotechnology Advances, 79, 108517.
Majib, N. M., Yaacob, N. D., Ting, S. S., Rohaizad, N. M., & Azizul Rashidi, A. M. (2025). Fungal mycelium-based biofoam composite: A review in growth, properties and application. Progress in Rubber, Plastics and Recycling Technology, 41(1), 91-123.
Elsacker, E., Vandelook, S., & Peeters, E. (2023). Recent technological innovations in mycelium materials as leather substitutes: a patent review. Frontiers in Bioengineering and Biotechnology, 11, 1204861.
Week 9 HW: Cell Free Systems
Part A: General and Lecturer-Specific Questions
General Homework Questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production:
Cell-free protein synthesis (CFPS) has greater flexibiltiy and experimental control that recombinar expression systems because the entire reaction happens outside a living organism. Without the use of a cell system to express the protein we can control and optimize every component of the system, making them scalable a can be applied in low amount making them suitable to analyze multiple protein candidates in parallel.
Cell-free systems don’t depend on a host reducing the neccesity of chosen an appropiate host depending on the characteristics of the target protein, for example the image below (Figure 1) shows a decision scheme for gene expression system based on the characteristics of the target protein (Schütz et al,. 2023). Cell-free systems can avoid this decision scheme because component for protein expression can be controlled.
Figure 1: Decision Scheme for gene expression system. This scheme is based on the protein characteristics. Figure taken from Schütz et al. (2023)
CFPS systems can lower the expression times by avoiding the growth of the expression host for the production of the protein.
A case where CFPS is more benefitial than recombinant technologies can be found in an article made by Pandi et al,. (2023) that uses a deep learning system to desing several antimicrobial peptides. The production of multiple candidates makes important to create expression system that can be use to express multiple candidates and evaluate them in parallel allowing the screening of multiple antimicrobial peptides.
A second case where CFPS is more benefitial than recombinant systems could be found in testing genetic circuits. Since genetic circuits require the manipulation of multiple factors a cell free system can be used to test the efficiency of a genetic circuit before its implementation in a living system. This could allow the optimization of the genetic circuit (Li et al., 2023)
2. Describe the main components of a cell-free expression system and explain the role of each component
A CFPS is composed of several components that can be grouped as:
Cell extract (lysate)
Contains the molecular component required for protein transcription and translation, the components found in a cell extract are:
Ribosomes: Translate mRNA intro protein
tRNAs: deliver amino acids
Transcription Factors
Chaperones
Endogenous enzymes
Energy regeneration system: Contains the molecules require to regenerate the ATP required for the expression of the protein, these systems are commonly composed of:
ATP or GTP
Phosphoenlopyruvate, creatine phosphate or 3-PGA
Regeneration enzyme like for example creatine kinase
Amino acids: A complete set of the 20 amino acids requires for the production of the protein
Nucleotides: Required for transcription and providing energy for some enzymatic reactions
Salts and cofactors and Buffers
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is essential in cell‑free systems because ATP is consumed extremely rapidly during transcription and translation and CFPS reactions cannot produce ATP on their own. Without an external regeneration system, protein synthesis stops almost immediately.
A common method to maintain ATP levels is to include phosphoenolpyruvate (PEP) in the reaction. PEP donates phosphate groups that pyruvate kinase converts into ATP, sustaining the reaction for hours. By preparing a PEP stock, adding it to the reaction mix, and maintaining optimal ionic and temperature conditions, you can ensure continuous ATP supply and maximize protein yield (Cui et al., 2022).
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic CFPS
Eukaryotic CFPS
Fast protein syntheis
Support post-transcriptional modifications
Simple transcription-translation machinery
Accurate folding of complex proteins
Low cost and easier to scale
Better handling of large, multidomain secreted proteins
Prokaryotic CFPS system candidate: Green Fluorescent Protein (GFP)
GFP is a commnon protein that has been expressed in prokaryotic systems because they don’t require complext post-traductional modifications, and has been optimized in E. coli extracts
Eukaryotic CFPS system candidate: Venom proteins
Venoms are a complex cocktail of multiple proteins that present multiple modifications and complex subunits, Cell-free protein expression systems could be used for produce selected venom components at high speed. The expression of these venom toxins can have therapeutical potential.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To express a cell free membrane a common problem is that the cannot be solubilized for it integration in the membrane, to solve that problem CFPS can express the membrane protein in special mediums like detergents or lipids to keep the protein soluble in hydrophobic conditions (Schneider et al., 2009). It would be required the use of chaperones to avoid misfolding and aggregation a and also use specific tags to avour protease degradation.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield can be related with a poor desing of the template expression DNA cassette caused by wrong promoter or the presence of secondary structures that can affect the transcription efficiency. To solve this problem it would be require to analyze the expression cassette and optimize before the expression process.
Low yield can also be cause by suboptimal concentration of key components like Mg/K, aminoacids or NTPs. to address this problem is necessary to screen different concentration and compare the resulting yields (Batista et al., 2021)
Translation can be reduce if the protein have toxic activities or is difficult to fold, to test this effect it is require to add chaperons or molecules to promote correct folding and stability
Kate Adamala Homework Questions
Minimal quorum-sensing biosensor cell
Function: Create a minimal biosensor cell that detects extracellular acyl-homoserin lactone and converts it into a fluorescent output. Acyl-homoserin lactone (AHL) was chosen because is a common signal molecule uses by gram neagtive bacteria depending on the concentration and poulation density and can be used in circuits based in quorum sensing (Fuqua et al., 2001)
Output: Strong green fluorescent protein (sfGFP) which expression is propotironal to the AHL concentration
For the desing I propose using a genetically modified E.coli strain that expresses luxR and a Plux-sfGFP reporter plasmis that acts as AHL biosensor.
Cell-free Tx/Tl could work but because the purpose of this desing is recreate a quorum sensing system it wouldn’t provide the requirements for that system
Desired outcome: When exposed to AHL the E. coli activetes a Plux promoter that initiate the expression of sfGFP. This will result in the apparition of green fluorescen in the colonies and the degree of fluorescence could be controlled with the concentration of AHL given.
Components of the synthetic minimal cell
Membrane: Phospholipid membrane gotten from the E coli strain
Inside the membrane will be a Plasmid for the expression of the LuxR with a weak promoter and a second plasmis containing the AHL-responsive reporter.
Form the membrane comminication native E. coli envelope will be used and since AHLs are membrane-permeable they wont require and special chanell
Experimental details and measurements:
Cell growth with appropiate antibiotics, growth can be measure by OD600 and GFP fluorescence
Cell distributions can be analyzed using flow cytometry or Fluorescence microscopy
Peter Nguyen
Proposal: Create a freeze-dried cell free system embedded in a building panel that detect indoor air pollutants and changes color to warn the occupants.
Description: The building panel contains micro-domains of freeze-dried cell-free systems programmed with a transcriptional biosensor for air pollutants. When pollutans diffuses into de material and moisture activae the cell-free spost triggering the expression of a visibles pigment. The color change can accumulates over hours, which can provide a clear indicator of poor air quality. Because this system doens’t use a living organism as biosensor there is low chance to spread making it dangerous.
The societal challenge that this system aims to solve is the air pollution that can affect many places. Existing sensor are often expensive, require power and often fail to provide a intuitive visualization.
Addressing limitations:
Activation with water: The scafold of the material could be engineered to absorb ambient humidity slowly
Stability: Freeze-dried systems ramin stable for month at room temperature
One-time use: Would be require a reversible system, this could be done using a pigment that changes depending the pH or oxidation state of the system
Ally Huang
Idea: Radiation-responsive DNA repair biosensor
Concept: Use BioBits to report how space‑relevant radiation doses affect DNA repair pathway activity using fluorescent reporters.
Background angle: Space radiation damages astronaut DNA; understanding repair capacity is key for long‑duration missions.
Target: Promoter regions or transcription factors of DNA damage response genes (e.g., p53 pathway motif, or bacterial SOS promoter like recA/lexA).
bHypothesis:“Radiation dose and quality will produce distinct fluorescence signatures from DNA damage–responsive promoters in BioBits reactions.”
Plan sketch:
miniPCR: amplify promoter–GFP constructs and damage‑responsive elements.
BioBits: expose DNA or lyophilized reactions to simulated radiation, rehydrate, measure GFP with P51.
Schütz, A., Bernhard, F., Berrow, N., Buyel, J. F., Ferreira-da-Silva, F., Haustraete, J., … & Remans, K. (2023). A concise guide to choosing suitable gene expression systems for recombinant protein production. STAR protocols, 4(4).
Pandi, A., Adam, D., Zare, A., Trinh, V. T., Schaefer, S. L., Burt, M., … & Erb, T. J. (2023). Cell-free biosynthesis combined with deep learning accelerates de novo-development of antimicrobial peptides. Nature Communications, 14(1), 7197.
Li, W., Xu, Y., Zhang, Y., Li, P., Zhu, X., & Feng, C. (2023). Cell-free biosensing genetic circuit coupled with ribozyme cleavage reaction for rapid and sensitive detection of small molecules. ACS Synthetic Biology, 12(6), 1657-1666.
Cui, Y., Chen, X., Wang, Z., & Lu, Y. (2022). Cell-free PURE system: evolution and achievements. BioDesign Research, 2022, 9847014.
Schneider, B., Junge, F., Shirokov, V. A., Durst, F., Schwarz, D., Dötsch, V., & Bernhard, F. (2009). Membrane protein expression in cell-free systems. In Heterologous expression of membrane proteins: Methods and protocols (pp. 165-186). Totowa, NJ: Humana Press.
Batista, A. C., Soudier, P., Kushwaha, M., & Faulon, J. L. (2021). Optimising protein synthesis in cell‐free systems, a review. Engineering Biology, 5(1), 10-19.
Fuqua, C., Parsek, M. R., & Greenberg, E. P. (2001). Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annual review of genetics, 35(1), 439-468.
Week 10 HW: Imaging and Measurement
Homework: Final Project
Figure 1 below presents some key aspects of my final project that require experimental testing and quantitative evaluation. These aspects refer to the expression of the protein binders generated using a Deep Learning Model and selected after in silico prediction of their therapeutic characteristics.
Figure 1: Experimental aspects of the final project: AI-driven Antivenom: A Generative Pipeline for De novo Neutralizing Peptides Against Snake Toxins. Image generated using: Copilot AI
Aspect 1: Cell-Free Expression System (CFS)
Cell-Free Expression System (CFS) enables rapid expression of multiple protein variants in parallel. Since this project aims to predict and express different protein binders, CFS provides a scalable and automatable platform that avoids cell culture and allows preliminary functional testing without full purification (Cui et al., 2022)
To ensure reproducibility, the CFS workflow must be standarized and quantitatively monitored. Expression efficiency will be measures by using detection tags like biotinylated lysine or His-tag into the peptide of interest, enabling detection through SDS-PAGE followed by Western Blotting. A colorimetric readout using biotin-binding secondary antibody and chromogenic substrate will aloow quantification of expression levels (Hunt et al., 2024)
Quantitative outputs for this aspect
Relative Expression Intensity (Densitometry of Wester Blot bands)
Expression yield per reaction
Reproducibility across replicates (CV%)
Figure 2 illustrates the Promega FluoroTect and Trascend detection systems that portrays the methodology to evaluate the expression of protein from a CFS.
Figure 2: Promega detection system for their cell-free expression systems
Aspect 2: Peptide Purification
To evaluate neutralization capacity, the expressed peptides must be purified from the CFS reaction. PURExpress In vitro Protein Synthesis Kit (New England Biolabs) for example uses reverse purification, a method that uses magnetic affinity resing based on metals that can selective capture His-tagged peptides (Figure 3)
Figure 3: Reverse purification of His-tagged proteins, this method could be use to extract peptides and later eliminate the tag by the use of a Tag Removal by Enzymatic Cleavage system
This approach could enable rapid purification suitable for small peptides or mini-proteins and is compatible with downstream biochemical assays.
Quantitative outputs for this aspect
Purification yield
Purity (Assessed by SPS-PAGE)
Recovery efficiency to crude expression levels
The metrics allow can be use later to generated an automated system for protein binders expression and purification
Aspect 3: Reactivity and Neutralization
The functional evaluation of the binders focuses on their ability to interact and neutralize the target toxin.
In vitro Neutralization: Neutralization can be test in vitro through the determination of the enzymatic activity of many protein targets (e.g. PLA2 or SVMP activity) in the presence of each binder. This provides a measureable neutralization parameter such as % inhibition or IC50.
Another strategy is the use of proteomics technique that involve the formation of the toxin-binder complex and and analyzed the interaction using a liquid chromatography-ms thechnique, which enables:
Confirmation of the complex formation
Identification of bound toxin fragments
Comparizon of binding efficiency acroos binder variants
Proteomics thecnique are most suitable to analyze different binder-protein interaction in an automated process
Quantitative outputs for this aspect
% Reduction in toxin enzymatic activity
IC50 values for each binder
LC-MS peak intensities corresponding to toxin-binder complexes
References:
Cui, Y., Chen, X., Wang, Z., & Lu, Y. (2022). Cell-Free PURE System: evolution and achievements. BioDesign Research, 2022, 9847014. https://doi.org/10.34133/2022/9847014
Hunt, A. C., Rasor, B. J., Seki, K., Ekas, H. M., Warfel, K. F., Karim, A. S., & Jewett, M. C. (2024). Cell-Free gene Expression: methods and applications. Chemical Reviews, 125(1), 91–149. https://doi.org/10.1021/acs.chemrev.4c00116
Waters Part I: Molecular Weight
Theoretical Molecular Weight and Isoelectric Point
The molecular weight (MW) of the eGFP protein was calcultad using the Expasy tool. Prior the calcultion, the His-tag and linker sequences were removed to obtain the MW corresponding to the native eGFP amino-acid sequence:
Native eGFP MW: 26.941 kDa
Isoelectric point: 5.58
The isoelectric point, being below than 7, indicated that the protein behaves as a weak acid, carrying a negative charge at a physiological pH
Expasy calculates MW using the average isotopic masses of amino acid and stimates the isoelectric point using the Bjellqvist pK set.
Adjacent Charge State Apprach for Experimental MW Determination
The molecular weight of eGFP was determined using LC-MS data provide in the homework figure. Two of the most intense adjacent charge-state peaks were selected for analysis (Figure 4)
Figure 4: Determination of the experimental MW of the eGFP protein through LC-MS analysis
Using this approach a charge stat of 31 was stimated and the experimental MW was estimated near to 28.05 kDa
Comparison with the theoretical MW of native eGFP revealed a difference of 1.108 kDa. This deviation cannot be explained by common post‑translational modifications (PTMs), which typically contribute mass shifts in the range of +14 to +80 Da. Furthermore, eGFP expressed in E. coli does not undergo glycosylation, eliminating the possibility of large PTM‑related mass additions.
The calculated accuracy of the experimental result relative to the native theoretical MW was 3.95%, which is significantly higher than the accuracy typically reported for intact‑protein mass determination using this methodology (Durbing et al., 2025). This discrepancy strongly suggested that the analyzed protein did not correspond to the native eGFP sequence.
Molecular Weight including His-Tag and Linker
It was propose that the theoretical MW include the his-tag and linker sequences so a new theoretical MW was determine including this sequences:
Molecular Weight (MW): 28.006 kDa
Isoelectric point (pI): 5.90
When compared to the experimental MW (28.05 kDa), the difference was only 0.044 kDa, corresponding to an accuracy of 0.09%. This level of agreement is consistent with the expected performance of intact‑mass LC‑MS analysis and suggests that the protein analyzed retains its purification tag and linker.
Part 3: Waters Part 3
The eGFP peptide was analyzed using the Benchling software that states the sequence contains 6 arginines and 20 lysines that compose 2.4% and 8.1% of the whole protein respectively (Figure 5)
Figure 5
Using the Expasy Peptide mass tool it is predicted that the eGFP would have 19 fragments after the digestion with trypsin.
Figure 6: Trypsin digestion simulated using the Expasy Peptide mass tool
Peptide peaks were identified determining the count of the highest peak as 100% of abundance (1.2e7) and drawing a line the represent the 10% percents abundance and selecting those peaks that significatively surpasse the 10% line (Figure 7)
Figure 7: Determination of peptide peaks with a relative abundance superior to 10%
From figure 7 a total of 19 peaks were detected, however other peaks were discharged because they were close to the 10% line determined, this could suggest that there are more thant 19 peaks experimentally.
Calculation were performed for the isotope peaks of the fragment 11 as can be observe in figure 8. A z value of 2 was obtained and the molecular weight of the predicted fragment was about 1049 Da. The accuracy of this calculation was measure by comparing the experimental value and the thoeretical value obtained from the Expasy tool, getting an accuracy of 9.54 x 10^4.
Figure 8
According to the peptide mapping the percentage of the sequence that was confirmed is about 88%
According to the results obtained the peptide molecular weight matches with the fragment 11 obtained in the expasy tool that has the following sequence: FEGDTLVNR
Peptide map shows close simmularities with the eGFP however because the coverage is about 88% we cannot conclude the protein in mention is the standar eGFP based only in the experimental results.
Waters Part IV: Oligomers
Based on the subunit masses provide in the Table 1 we can determine the oligomeric species by multiplying the number of subunits in each oligomer
Name
Number of Subunits
Subunit Mass
Total Mass
7FU Decamer
10 subunits
340 kDa
3400 kDa = 3.4 MDa
8FU di-decamer
20 subunits
400 kDa
8000 kDa = 8.0 MDa
8FU 3-decamer
30 subunits
400 kDa
12000 kDa = 12.0 MDa
8FU 4-decamer
40 subunits
400 kDa
16000 kDa = 16.0 MDa
Using the figure 7 in the homework we can predict that the peak 3.4 is the 7FU Decamer, peak 8.33 is close to the 8FU di-decamer, 12.67 is related with 8FU 3-decamer and a peak close to the 8FU 4-decamer is not present
Week 11 HW: Bioproduction and Cloud Labs
Part A: Artwork Canvas
Contibution: I contribute with 52 dots making the “Love” Desing at the bottom left plate. I liked the collaborative working of the canvas and the constant organic evolution of the design that resembles very well with the development of synthetic biology. For a next collaborative art experiment I could propose an experiment that serves as a competence of different groups, or a collaborative project that shows thet location of the participant in the world.
Part B: Cell-Free Protein Synthesis
Components of the cell-free reaction:
E. coli lysate
BL21 (DE3) Star Lysate: Cell extract that contains all the components neccesary for transcription, translation, folding a nd energy regeneration of the cell-free system. E. coli BL21 strains is typically chosen for cell-free expresion because of its enhance mRNA stability and decrease protease activity (Hunt et al. 2024).
Salts/Buffer
Potassium and Magnesium Glutamate: These salts are used to have different uses. Potassium and Magnesium ions are use to balance the charge of negatively charge biomolecules like DNA (Hunt et al., 2024). The ions are also involved in mantaining the integrity of stability of ribosomes, for example lack of magnesium is related with the loss of ribosome integrity and lack of potassium is related with the loss of ribosome activity (Nierhaus, 2014)
Glutamate acts a anionic counterion but also is used as secondary energy source. Optimizing glutamate amount is commonly done to regulate the amount of NAD and CoA cofactors because these tend to increase the cost of the expression system (Cai et al., 2015)
HEPES-KOH and Potassium phosphate monobasic and dibasic: HEPES is traditionally used a buffer to control the pH level under acidic subproducts created from glucose metabolism (Hunt et al., 2024). Potassium phosphate can be also use as buffer and also to control ATP concentrations by supporting glycolisis and pyruvate metabolism.
Energy / Nucleotide system
Energy supplements are used for ATP regeneration through metabolic pathways. ATP is required as energy molecule for transcription and translation. Glucose is used a primary energy source for ATP generation through glycolisis but requires phosphate as source (Hunt et al., 2024)
NMPs (AMP, CMP, GMP, UMP): Are used a cost-effective supplement of nucleotides replacing NTPs. This monophosphates. Incorporation of NMPs is well stablished as the cytomin/glutamate-phosphate/NMP batch system because of its high yield and batch production (Jewett et al., 2008)
Translation Mix
17 amino acid mix: Aminoacids are neccesary for the translation process. Traditionally the 20 amino acid can be supplemented in excess to the reaction, however optimization studies have shown that several aminoacids like aspartate, lysine, and tyrosine can be produced in the cell-free reaction using the extract source materials (Jewett et al., 2008). Simmilarly other study has shown thet effect of halving every single aminoacid in the reaction showing that some aminoacids can be obtained through native metabolic process (Pedersen et al., 2010).
Cystein also plays a key role in protein synthesis, for example increasing the concentration of cystein can be related with and increase of glutathione S-transferase (Shingaki & Nimura, 2011), also cystein can cause inespecific disulfude bonds so its optimization is important.
Additives
Nicotinamide: Nicotinamide is a regulator of NAD metabolism and synthesis being an important regulate of the energy production.
Backfill
Nuclease-Free Water: Nuclease free water acts as the medium for all metabolic reactions and transcription-translation process. It is required to provide water free of nucleases and protease to avoid degradation of the desired products
Differences between 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix
Figure below shows master mix compositions for the cell free system
The principal difference that can be found is the source of energy and nucleotides used. For the 1 hour optimized system it can be observed that is base on Phosphoenol pyruvate (PEP), which is often used as energy soruce for the cell-free reaction. PEP is an expensive additive to the CFS reaction but initially showed better performance than glucose (Calhoun & Swartz, 2005). For the second master mix Ribose and Glucose are used a main source of energy, this could decrease the pH of the reaction and for that the addition of other buffers like Potassium phosphate are included.
Another main difference between both systems is the used of tri-phosphate nucleotides for the 1-hour master mix and monophosphate nucleotides for the 20 hours master mix, According to Jewett et al (2008), NMPs are cost-effective replacements of NTPs and can be used in batch systems with high yields.
Some additives are used in the 1 hour master mix to improve the reaction, for example Spermidine is usually used to increase transcription fidelity (Igarashi et al., 1982) and DMSO is commonly used to dissolve water-insoluble substances. In the case of the 20-hour master mix this additives can be avoided to decrease the cost of the reaction.
Part C: Planning the Global Experiment
Fluorescent Proteins
sfGFP: Superfolded GFP is a variant of GFP from Aequorea victoria that is superfolded to avoid the effect of GFP folding in oxidizing environments (Aronson et al., 2011). One characteristic that can affect the role of GFP is the formation of inespecific agrupation because the protein tends to form weak dimers that can produce inclusion bodies.
mRFP1: mRFP1 main disadvantages are the slow maturation and tendecy to form oligomers
mKO2: mKO2 shows moderated acid sensitivity with a pKa of (5.5) indicating the necessity to control the pH of the medium
mTurquoise2: This protein is sensitive to Mg and ionic concentration that can alter the brighness of the protein.
mScarlet-I: mScarlet-I is prone to misfolding at high expression rates, it would be necessary a chaperone or low expression rate to avoif misfolding
Electra2: Electra2 pH-sensitive is pH sensitive causing the fluorescence to drop as cell-free reactions acidify over time
Hypothesis
Electra2: Increasing buffer concentration can help stabilize Electra2’s structure and mitigate its pH sensitivity mantaining its color for 36 hours.
References
Aronson, D. E., Costantini, L. M., & Snapp, E. L. (2011). Superfolder GFP is fluorescent in oxidizing environments when targeted via the SEC Translocon. Traffic, 12(5), 543–548. https://doi.org/10.1111/j.1600-0854.2011.01168.x
Calhoun, K. A., & Swartz, J. R. (2005). Energizing cell‐free protein synthesis with glucose metabolism. Biotechnology and Bioengineering, 90(5), 606–613. https://doi.org/10.1002/bit.20449
Cai, Q., Hanson, J. A., Steiner, A. R., Tran, C., Masikat, M. R., Chen, R., … & Yin, G. (2015). A simplified and robust protocol for immunoglobulin expression in E scherichia coli cell‐free protein synthesis systems. Biotechnology progress, 31(3), 823-831.
Hunt, A. C., Rasor, B. J., Seki, K., Ekas, H. M., Warfel, K. F., Karim, A. S., & Jewett, M. C. (2024). Cell-Free gene Expression: methods and applications. Chemical Reviews, 125(1), 91–149. https://doi.org/10.1021/acs.chemrev.4c00116
Igarashi, K., Hashimoto, S., Miyake, A., Kashiwagi, K., & Hirose, S. (1982). Increase of fidelity of polypeptide synthesis by spermidine in eukaryotic Cell‐Free systems. European Journal of Biochemistry, 128(2–3), 597–604. https://doi.org/10.1111/j.1432-1033.1982.tb07006.x
Jewett, M. C., Calhoun, K. A., Voloshin, A., Wuu, J. J., & Swartz, J. R. (2008). An integrated cell‐free metabolic platform for protein production and synthetic biology. Molecular Systems Biology, 4(1), 220. https://doi.org/10.1038/msb.2008.57
Pedersen, A., Hellberg, K., Enberg, J., & Karlsson, B. G. (2010). Rational improvement of cell-free protein synthesis. New Biotechnology, 28(3), 218–224. https://doi.org/10.1016/j.nbt.2010.06.015
Shingaki, T., & Nimura, N. (2011). Improvement of translation efficiency in an Escherichia coli cell-free protein system using cysteine. Protein Expression and Purification, 77(2), 193–197. https://doi.org/10.1016/j.pep.2011.01.017