Week 4 HW: Protein Design I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average, an amino acid is ~100 Daltons)There are a variety of meats depending on the kind. Let’s take an average of about 20% of protein by mass of meat (Day, 2016). This would therefore mean that 500g of meat would contain roughly 100g of protein. The average molecular weight of an amino acid residue is 100 Daltons, which translates to about 100g per mole of the amino acid unit (100g/100g/mol = 1 mole of amino acid units) (ProPrep, 2019). 1 mole contains 6.022 x 10^23 (Avogadro’s number) amount of particles (The ChemTeam, 2026). Therefore, if you consume 500g of meat, you are also ingesting approximately 6 x 10^23 amino acid residues.
References
2.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
There are several reasons why eating another animal does not make us become that animal, and they centre on two key concepts. First, what makes us human rather than another organism is the DNA in our own cells and how our genes are regulated (Brown, 2002). Our genome encodes for specific human proteins, cells, and tissues, and this blueprint does not change just because we eat another species. Second, chemical digestion breaks food down into small building blocks before absorption. When a human consumes an organism with a different genotype, they do not absorb that organism’s genome. Instead, digestive enzymes break down animal proteins into individual amino acids and degrade the animal’s DNA into nucleotides (Patricia and Dhamoon, 2022). Because virtually all life is built from the same basic set of around 20 natural amino acids, what really matters is how these building blocks are arranged in the polypeptide chain. The human body uses the amino acids it absorbs to make human proteins according to its own genetic instructions.
References
3. Why are there only 20 natural amino acids?
Ultimately, a set of fundamental physicochemical imperatives (molecular properties such as solubility, acidity, and basicity) dictates the prebiotic selection of the canonical 20 amino acid types. The guiding principle was parsimony, where nature retained only the simplest structures with high functional value, avoiding redundancy and unnecessary complexity (Moldoveanu and David, 2022). Energy efficiency also played a key role. The study ‘Why twenty amino acid residue types suffice(d) to support all living systems (2018) used quantum chemistry and chemoinformatics to analyse a large panel of candidate chemicals, followed by statistical analysis of their complexity and property scores. The results showed that the 20 canonical amino acids were the most likely to form under prebiotic conditions and had optimal physicochemical properties (Bywater, 2018).
References
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can make other non-natural amino acids, known as non-proteinogenic or non-canonical amino acids (ncAAs), which have been routinely synthesised and engineered in labs to be added into proteins to alter certain physicochemical and biological properties for an optimal protein (e.g., reactivity), which has been a key development for enzymology and drug discovery (Adhikari et al., 2021). To design ncUAAs, you need to analyse their structure and determine what must remain the same and what may be altered. The core structure has a central alpha carbon that is bonded to an amino group, a carboxyl group, a hydrogen atom, and an R group (custom side chain). The R group can be hydrophobic, polar, or charged. The size, shape and potential for covalent interactions determine how each amino acid residue interacts with its environment (Whiteburn, 2024). It is also the area that needs to be altered/engineered to create an ncUAA.
References
5.Where did amino acids come from before enzymes that make them, and before life started?
Amino acids on prebiotic Earth formed through non-biological chemical reactions (abiogenesis), both from space delivery and surface synthesis (Cowing, 2023). Meteorites (carbonaceous chondrites) delivered large quantities of abiotically formed amino acids like glycine, alanine, α-amino-n-butyric acid, isovaline, and β-alanine, which landed in early oceans (Strasdeit, 2009). Atmospheric sparks from electric discharges (lightning) and volcanic ash-gas clouds also synthesised amino acids in water droplets (Cowing, 2025). Over time, wet-dry cycles in lagoons or tidal pools concentrated these amino acids and polymerised them into peptides, creating the building blocks for early life.
References
6.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Most amino acids come in at least two forms, whose structures are mirror images of each other and are referred to as the right-handed or left-handed optical isomers. Natural proteins use L-amino acids and form right-handed alpha helices, whereas D-amino acids are the mirror image, therefore producing the left-handed helices (enantiomers). (BOC Sciences, 2025).
References
7. Can you discover additional helices in proteins?
There may be additional helices in proteins, such as secondary-structure helices, that may be discovered. For example, the Pi-Helices, often described as rare or uncommon, are found in approximately 15% of proteins and are often overlooked despite their important functions (Cooley et al., 2010).
References
8. Why are most molecular helices right-handed?
Most protein α-helices are right-handed because L-amino acids adopt dihedral angles that minimise steric clashes between side chains and the backbone. Left-handed alpha-helices are possible on the Ramachandran plot (2D visualisation of backbone dihedral angles for amino acid residues in proteins) but suffer more side chain-backbone collisions for L-amino acids, making them energetically unfavourable and rare in nature (Robinson and Afzal, 2014).
References
9. Why do β-sheets tend to aggregate?
Beta-Sheets tend to aggregate because their exposed edges promote intermolecular hydrogen bonding that readily pairs with other beta-strands from separate proteins and misfolded chains. These associations form stable cross-beta structures, often aggregating into amyloid fibrils linked to diseases such as Alzheimer’s. (Richardson and Richardson, 2002).
References
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
For this exercise, I’m looking at Huwentoxin-IV (HwTx-IV) from Chinese bird spider venom (Cyriopagopus schmidti). It is a 35-amino-acid peptide that blocks pain signals by hitting the NaV1.7 channels (Sermadiras et al., 2013). These channels are voltage-gated sodium channels expressed in certain neurons, where they play an important role in the generation and transmission of pain-related information (Fouillet et al., 2017). This fits into one of my project propositions for the development of an analgesic cream for the treatment of arthritis pain as a non-opioid painkiller alternative. Understanding its 3D structure shows me exactly where the disulfide bonds need to form and which face binds to the NaV1.7 pain receptor, so when I clone it into E. coli, I will reduce the risks of misfolds in the protein or inactive peptide (Weiss et al., 2022). Essentially, the structure serves as the blueprint for developing a topical formulation for my design proposition.
References
2. Identify the amino acid sequence of your protein.
Full precursor sequence: MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVD DNSKGEERECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQIGK (National Library of Medicine, 2026)
The bold section of the amino acid sequence is the active section of the protein, as seen below on the RCSB PDB website, highlighted in purple:
References
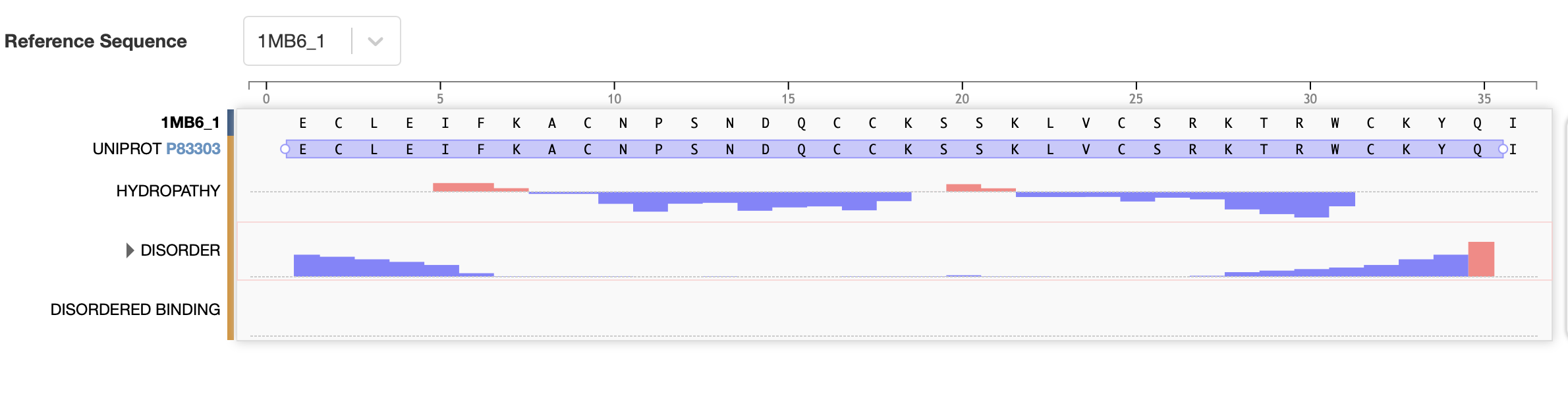
- How long is it? What is the most frequent amino acid?
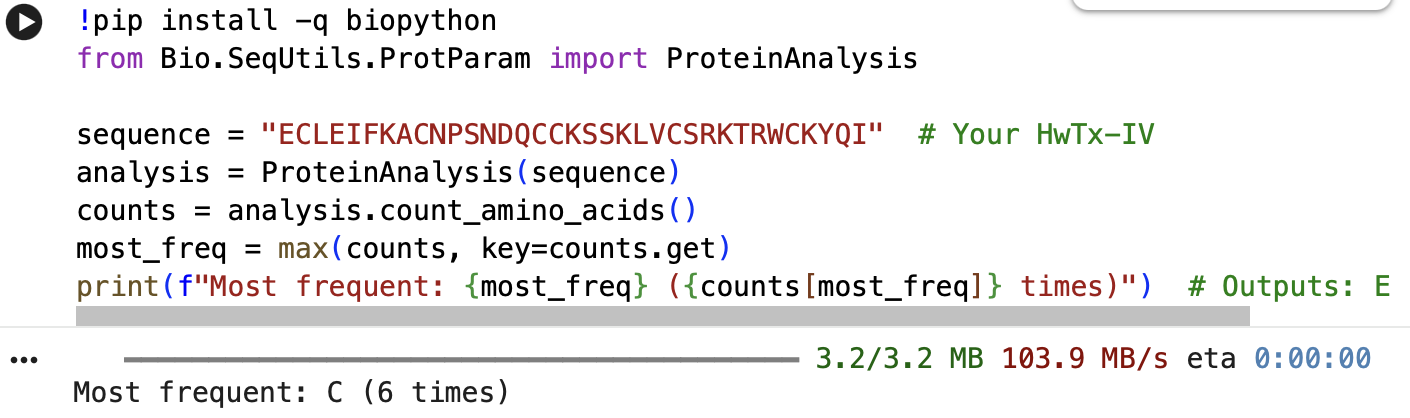
The full precursor sequence is 89aa long (UniProt, 2026) whilst the active protein is 35aa (PDB, 2017). Additionally, the most frequent amino acid is Cysteine (C) that appears 6 times in the active protein.
Below is the Python Code as well as the initial error I got, which was fixed through Gemini Built-In Tool:
- How many protein sequence homologs are there for your protein?
When focusing specifically on the Huwentoxin-IV protein from Cyriopagopus schmidti (C. schmidti)and restricting the search by taxonomy, I identified 13 homologous sequences sharing between 42.0% and 73.8% sequence similarity with the selected protein. When all taxonomic groups were included in the analysis, the number of homologous sequences increased immensely to 193 across spider species. Among these, five sequences showed the highest similarity to Huwentoxin-IV, ranging from 89.5% to 90.7%. Notably, these highly similar sequences were derived from Cyriopagopus hainanus, another species classified as a Chinese bird spider. (Uniprot: Blast Tool, 2026)
References
- Does your protein belong to any protein family?
Yes, it belongs to the neurotoxin 10 (Hwtx-1) family, furthermore to the 22 (Htx-4) subfamily. I found this information through the Family & Domains section of my selected protein (UniProt, 2026).
3.Identify the structure page of your protein in RCSB - When was the structure solved? Is it a good quality structure? A good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)The structure was solved in August 2002 by authors Peng, K., Shu, Q., Liang, S.P. (PDB, 2017). In terms of resolution, the RCSB entry does not clearly indicate the resolution for the isolated toxin structure. However, according to the study “Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na⁺ channels in nanodisc” by Gao et al. (2020), the structure of HWTX-IV bound to human Nav1.7 was obtained at an overall resolution of 3.2 Å, with the local resolution of the toxin improving from approximately 6 Å to approximately 4 Å. In structural biology, lower resolution values indicate higher structural quality. While a resolution of 2.70 Å would generally be considered good quality, the reported resolutions of 3.2–3.5 Å are moderate rather than high. Nevertheless, these resolutions are sufficient for visualising overall toxin docking.
References
- Does your protein belong to any structure classification family?
Yes, it belongs to the inhibitor cystine knot structural family (Peng et al., 2002).
References
4. Open the structure of your protein in any 3D molecule visualization software:
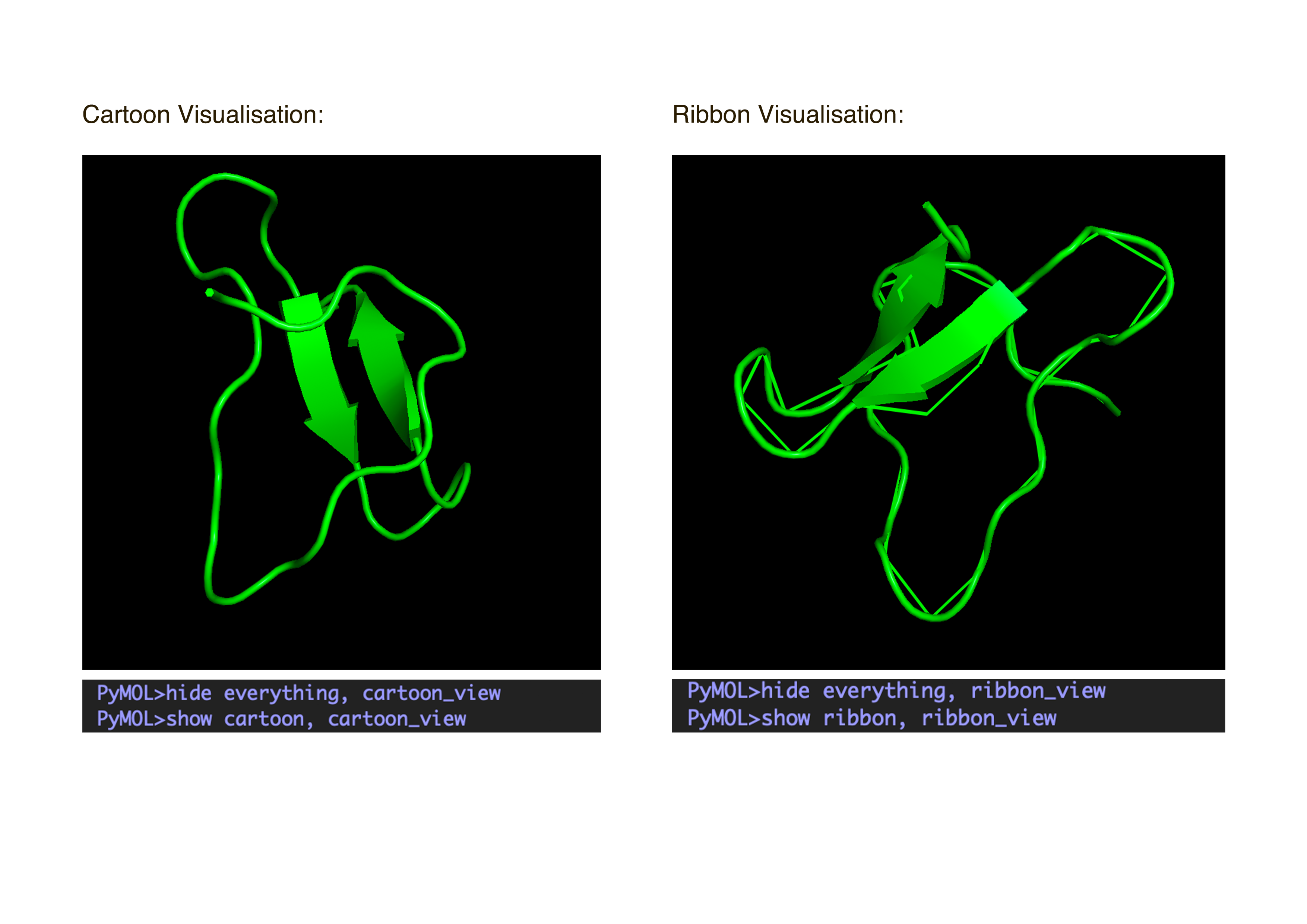
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.After downloading my protein structure from RCSB PDB as a .cif file, I was able to open it in PyMol. I then played around with the commands on the right in the ’S’ section to alter the visual structure of my selected protein.

- Color the protein by secondary structure. Does it have more helices or sheets?
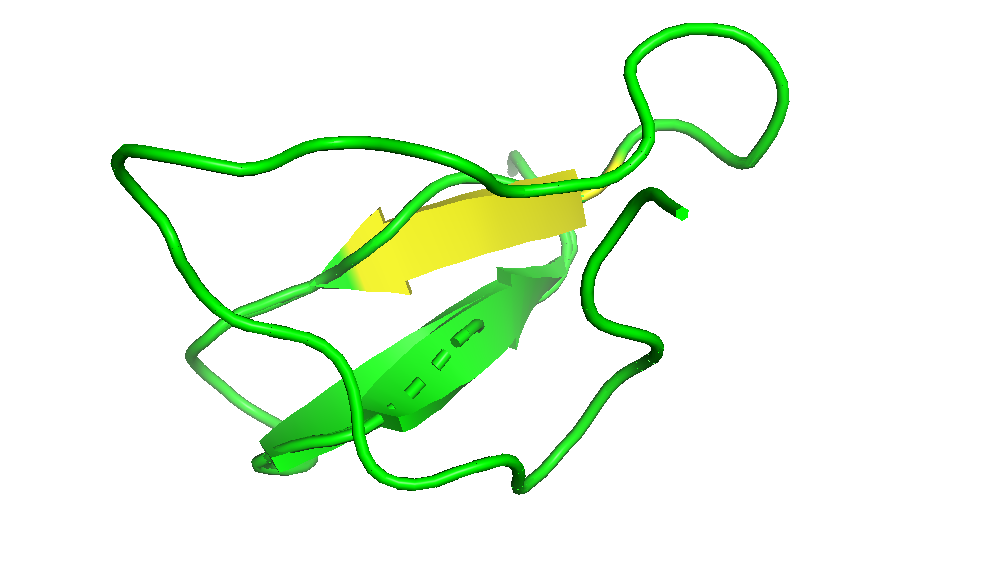
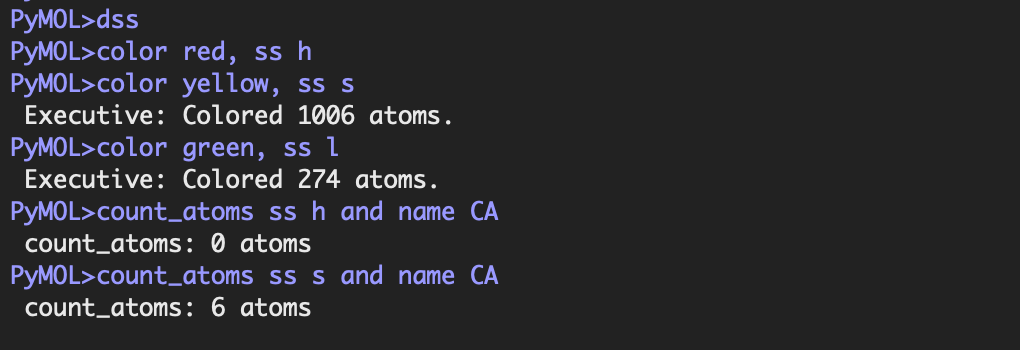
I coloured the protein by secondary structure in the cartoon representation. The green represents coils (loops), the yellow arrows represent sheets, and red represents helices. No red helices appear in the structure, while yellow sheet arrows are present. To verify this, I used the PyMOL count_items function to count residues assigned to each secondary structure type. The results showed 0 residues classified as helices and 6 residues classified as β-sheets (based on Cα atom counts). Therefore, the protein contains sheets but no helices. The remaining residues (274 atoms total) are classified as loops. Below is the following code used to colour and to verify this through PyMOL’s counting function:
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Colour Code: Yellow = hydrophobic, Green = polar (uncharged), Blue = positively charged, Red = negatively charged.

ANALYSIS: The positively charged residues (blue and red) are strongly hydrophilic and seem to be concentrated more on the outer loops than the centre. The polar residues are usually also hydrophilic and appear spread throughout the protein structure. The hydrophobic residues (yellow) appear to be seen in the core of the sheet, but are also spread out around the full structure.
- Visualise the surface of the protein. Does it have any “holes” (aka binding pockets)?

ANALYSIS: Across the whole surface of the protein, only a single small cavity is apparent and could be identified as a binding pocket. Overall, the protein surface is relatively smooth and compact, which would explain why the protein doesn’t have any other molecules, as it may be limiting interactions by providing an unstable environment for binding.
Part C: Using ML-Based Protein Design Tools
1. Choose your favorite protein from the PDB
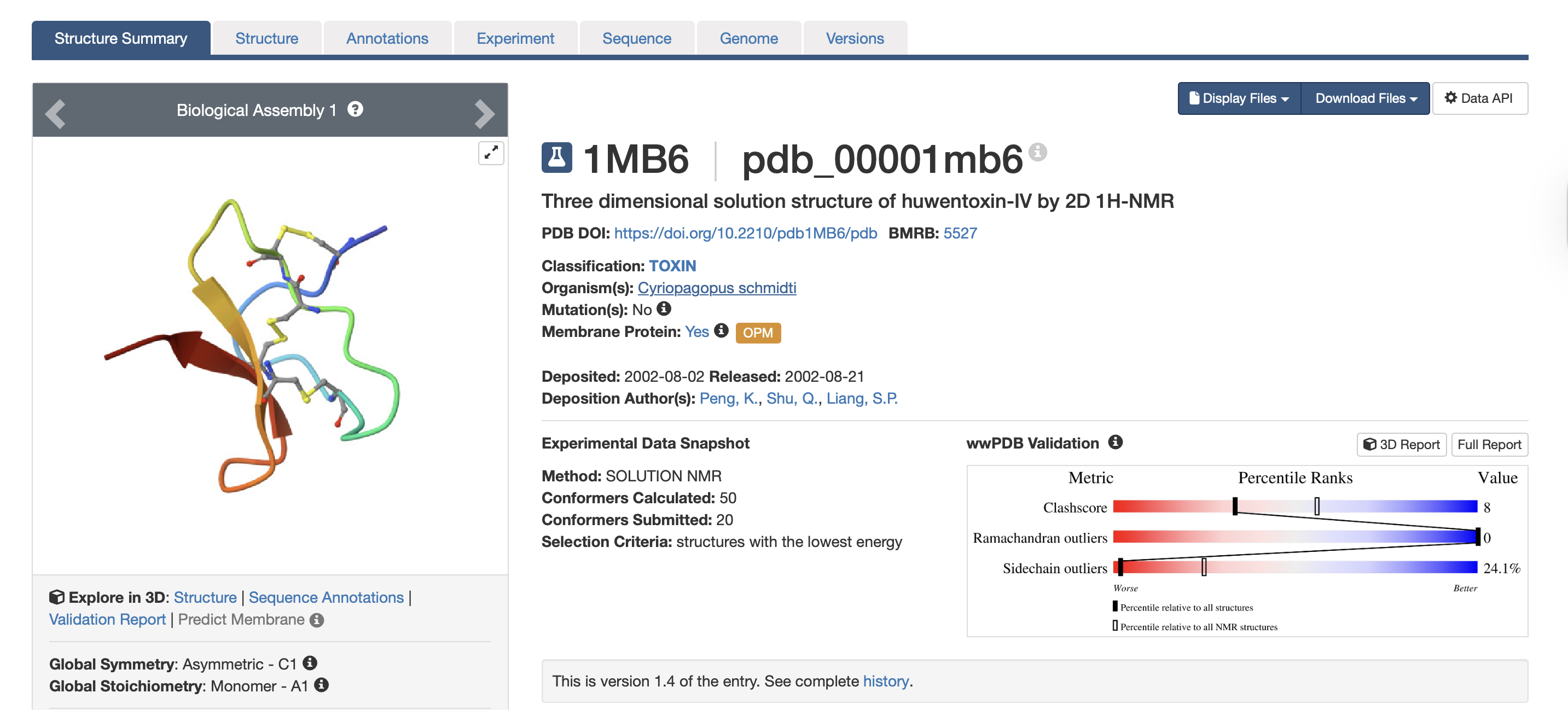
C.1 Protein Language Modeling
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
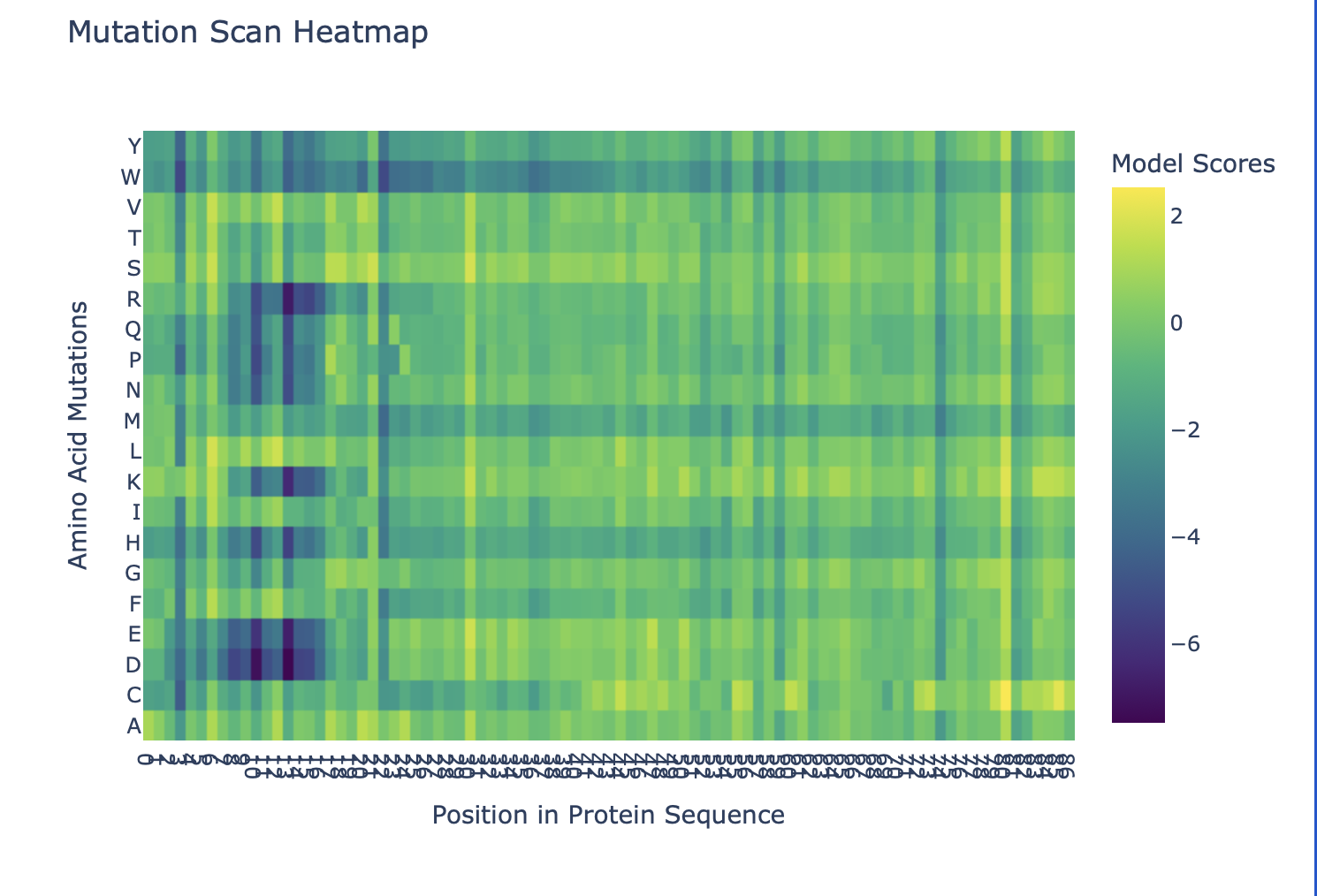
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Overall, the heatmap shows many positions with predominantly high scores (yellow/green), indicating mutation-tolerant sites, which contradicts the idea that smaller proteins would be more susceptible to mutations, disrupting protein function. Positions 8–16 stand out with a high concentration of blue (-4/-6), suggesting high mutation intolerance here. W mutations seem to be specifically prone to mutations wherever they are located in the sequence implyging implying that mutations in these amino acids could critically disrupt core stability or binding despite the region's flexibility.
C.2 Latent Space
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
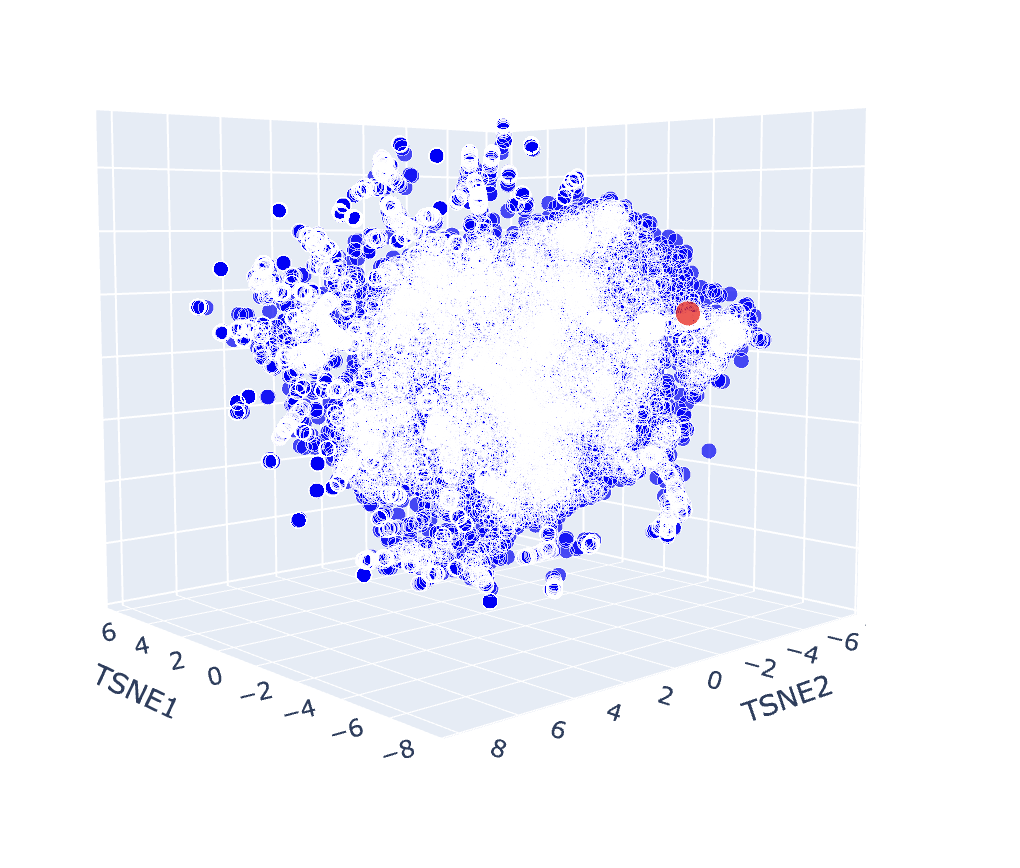
b. Analyse the different formed neighbourhoods: do they approximate similar proteins?
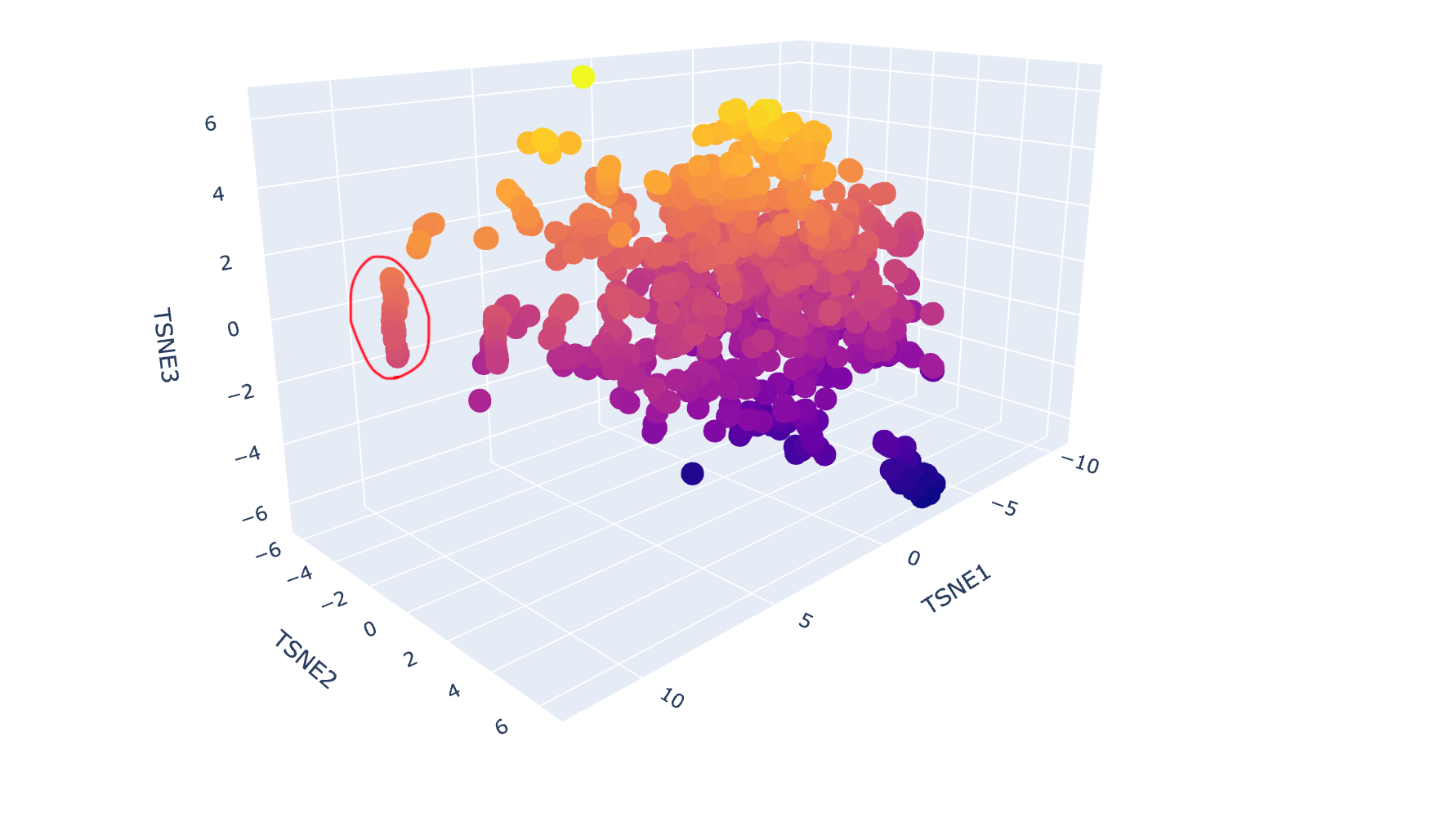
The latent space map groups proteins into neighbourhoods where closer points share similar sequences, structures, or functions. For example, the circled group in the image below includes proteins from soil bacteria like Bacillus subtilis and Enterococcus faecalis, which have overlapping membrane and stress-response features, showing that the map captures biological similarity.
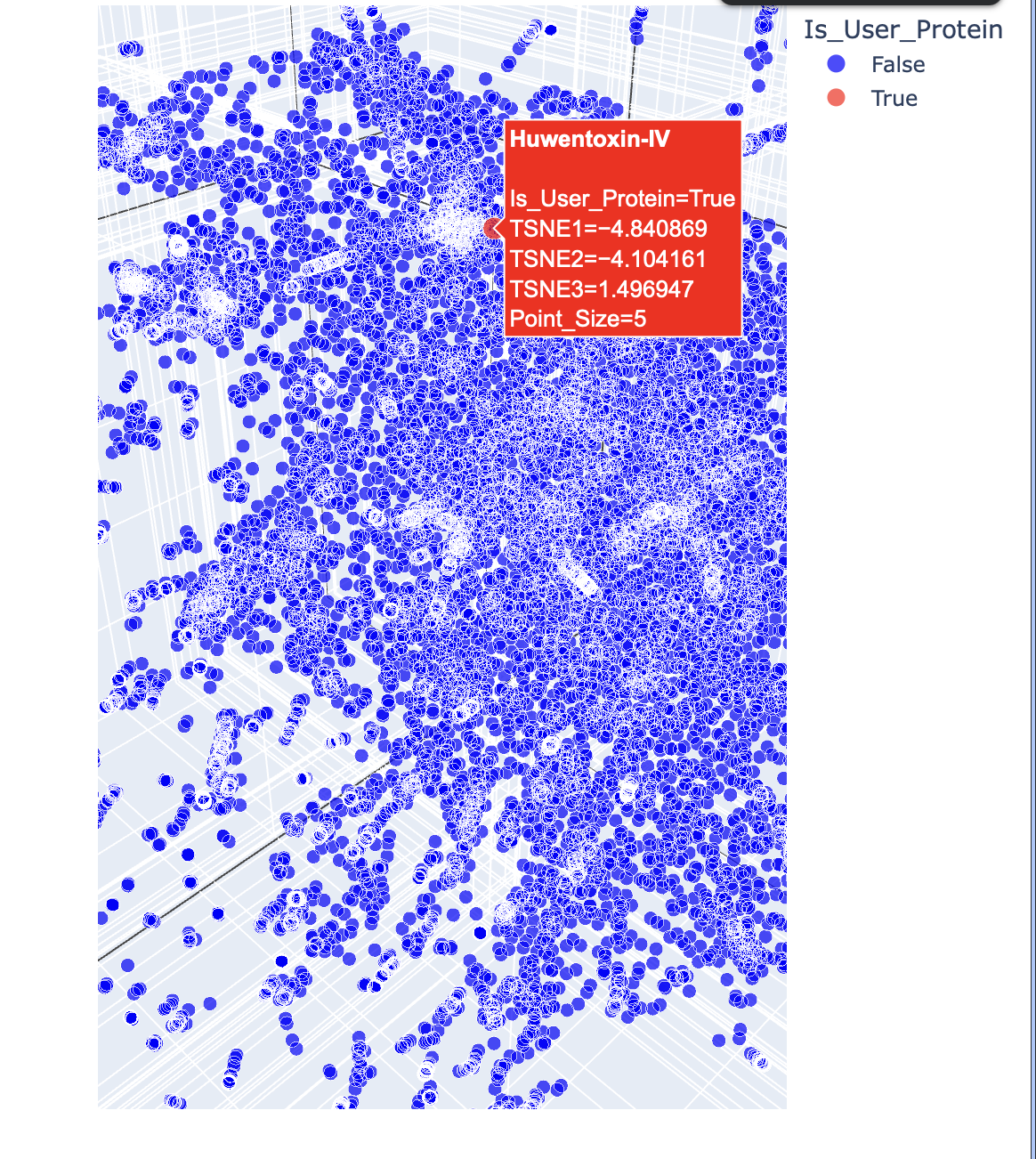
c. Place your protein in the resulting map and explain its position and similarity to its neighbours.
Below are the names and structures of three proteins in the same neighbourhood as my selected protein:
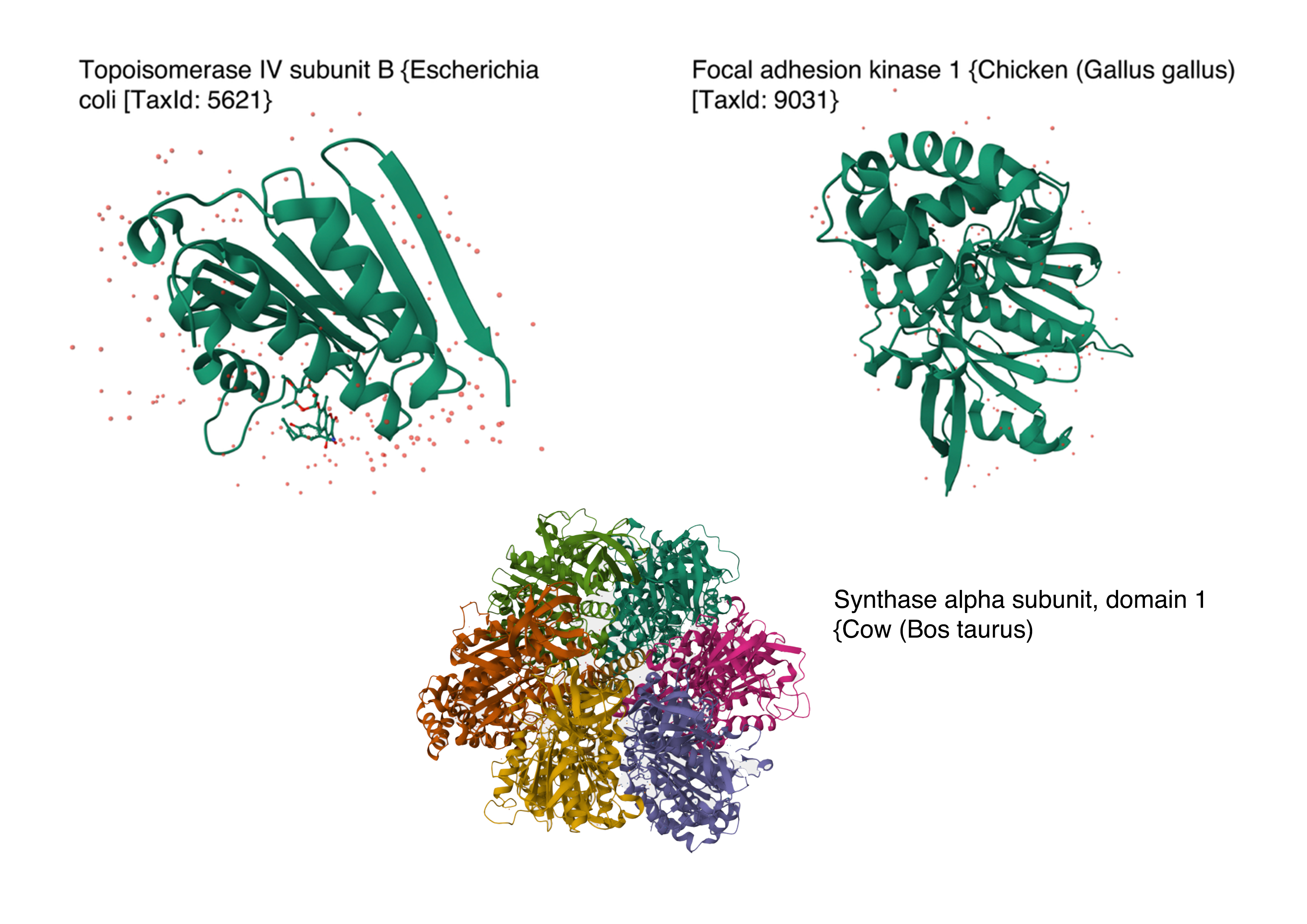 Image references:
Image references:
- Topoisomerase IV subunit B and Focal adhesion kinase 1 from RCSB PDB Website
- Synthase alpha sunbunit, domain 1 from Uniprot Website
In the protein latent space, Huwentoxin-IV from Cyriopagopus schmidti sits in a region populated more by regulatory and interaction-focused proteins than by purely structural ones. Its closest neighbours include an ATPase domain from topoisomerase IV, along with fragments of chicken focal adhesion kinase and a synthase alpha subunit. Despite their differences in size, fold, and cellular context, these proteins all seem to rely on chemically specialised surfaces to influence the behaviour of other macromolecules. This suggests that the model is picking up on less global structural similarity and more on shared biophysical features, such as molecular interactions and regulation.
C.2 Protein Folding
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The 35aa active part of the 89aa Huwentoxin-IV mini-protein, which I looked at in my Part B homework: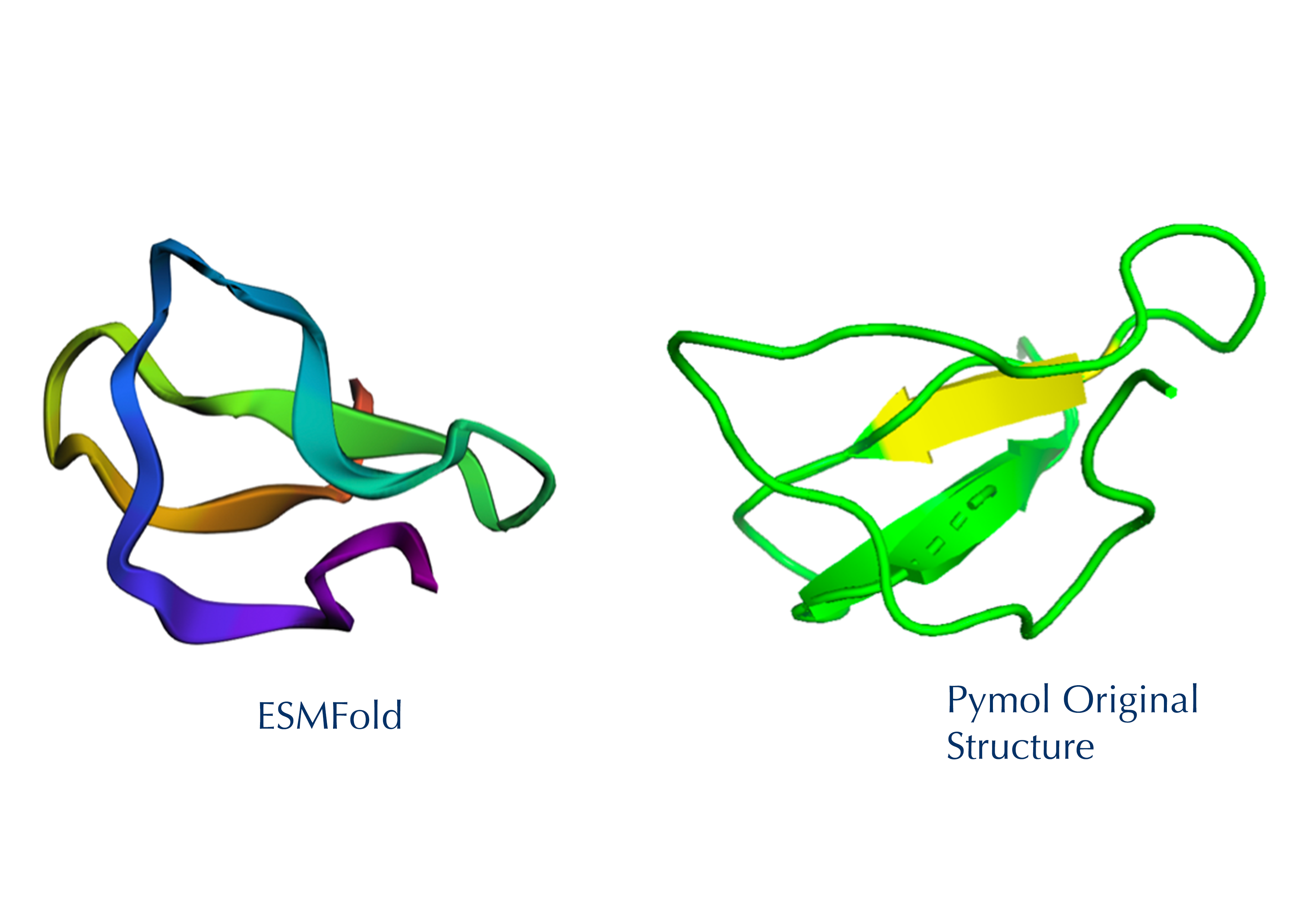
Yes, the predicted coordinates from ESMFold appear to match the original structure quite well. The overall beta sheet-rich fold is preserved, and the main secondary structure elements align closely with the original 35-aa active region of Huwentoxin-IV. While there may be minor differences in loop orientation or flexible regions, the core remains essentially the same.
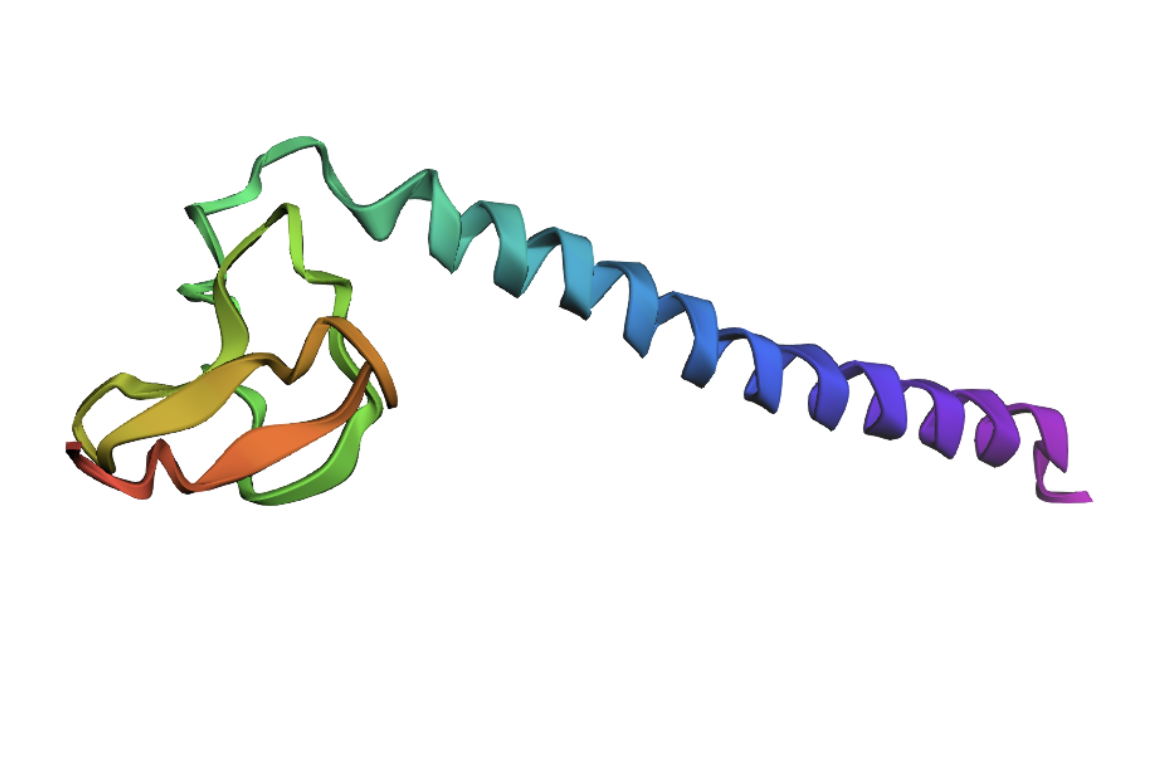
I decided to also look at the full precursor protein, since I had not done so in Section B because of some initial confusion. Most papers mainly mention and highlight the 35-amino-acid mature toxin, and although the 89-aa precursor is listed on the PDB mini-protein page, opening the .cif file in PyMOL only shows the structure of the active toxin region. Since this is also the main focus of my final project idea (Option 3), it makes sense that this is where most researchers have concentrated their attention. Looking at the full precursor, we can see that what was previously a structure dominated by beta sheets now also includes a long alpha helix, which is therefore not part of the active toxin site.
2.Try changing the sequence: first try some mutations, then large segments. Is your protein structure resilient to mutations?
Processor Sequence:
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQIGK (Bold: active protein)
MUTATION 1
The active part of the protein, located at 50-85 in the full precursor sequence, Amino acids (Tryptophan) W, Histidine (H) and Methionine (M) seem to have lower tolerance to mutations throughout that section of the sequence. I want to see if altering one of these specific amino acids in the active protein will produce a visible change in the structure.
Mutated Sequence 1: Changed Lysine (K) to Tryptophan (W) on 57aa
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWACNPSNDQCCKSSKLVCSRKTRWCKYQIGK
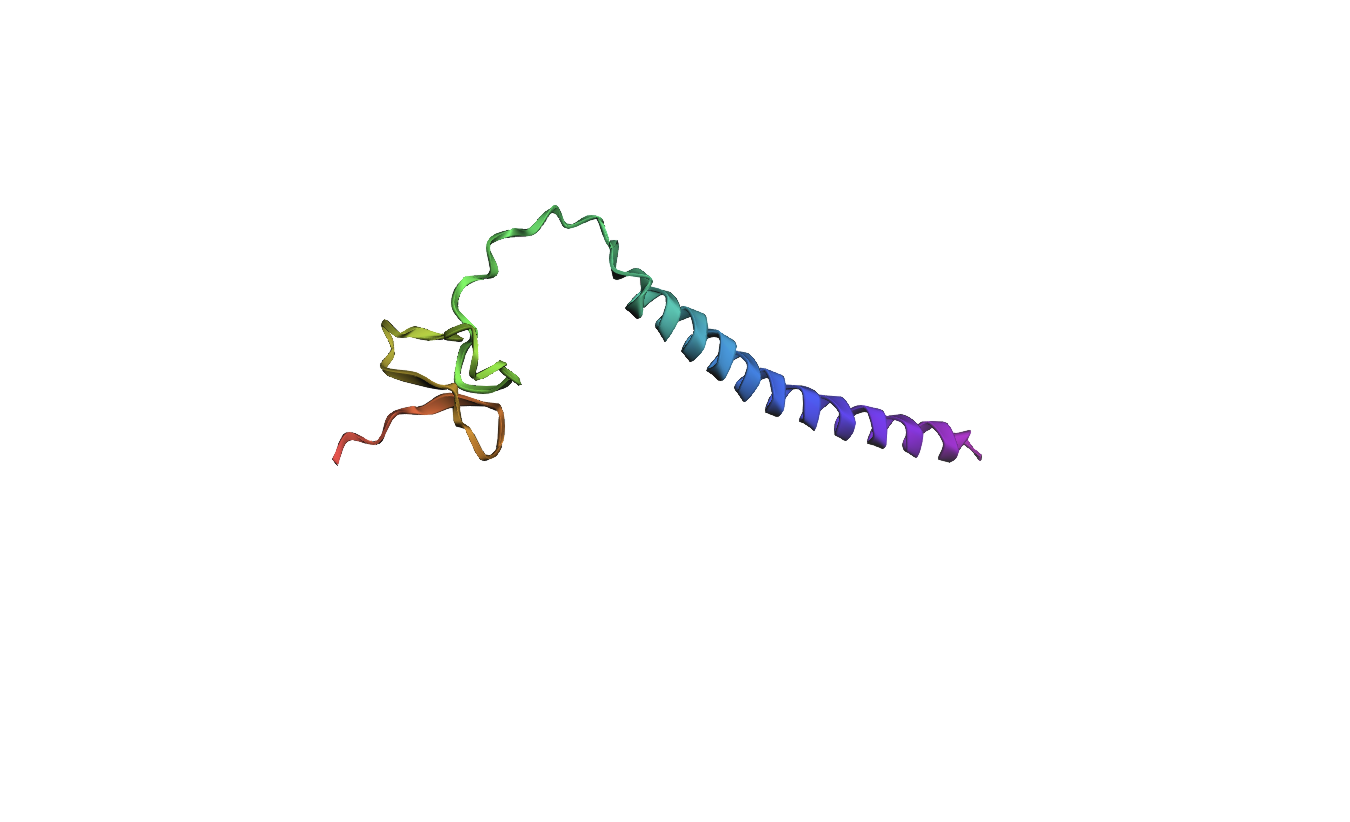
Analysis: It appears that even changing a singular amino acid in the active site of the protein has already significantly altered the local structure.
MUTATION 2
Secondly, I changed 3 amino acids in the protein’s active region, once again focusing on those at higher risk of mutation.
Mutated Sequence 2: Changed KAC to WHM on 57-59aa
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
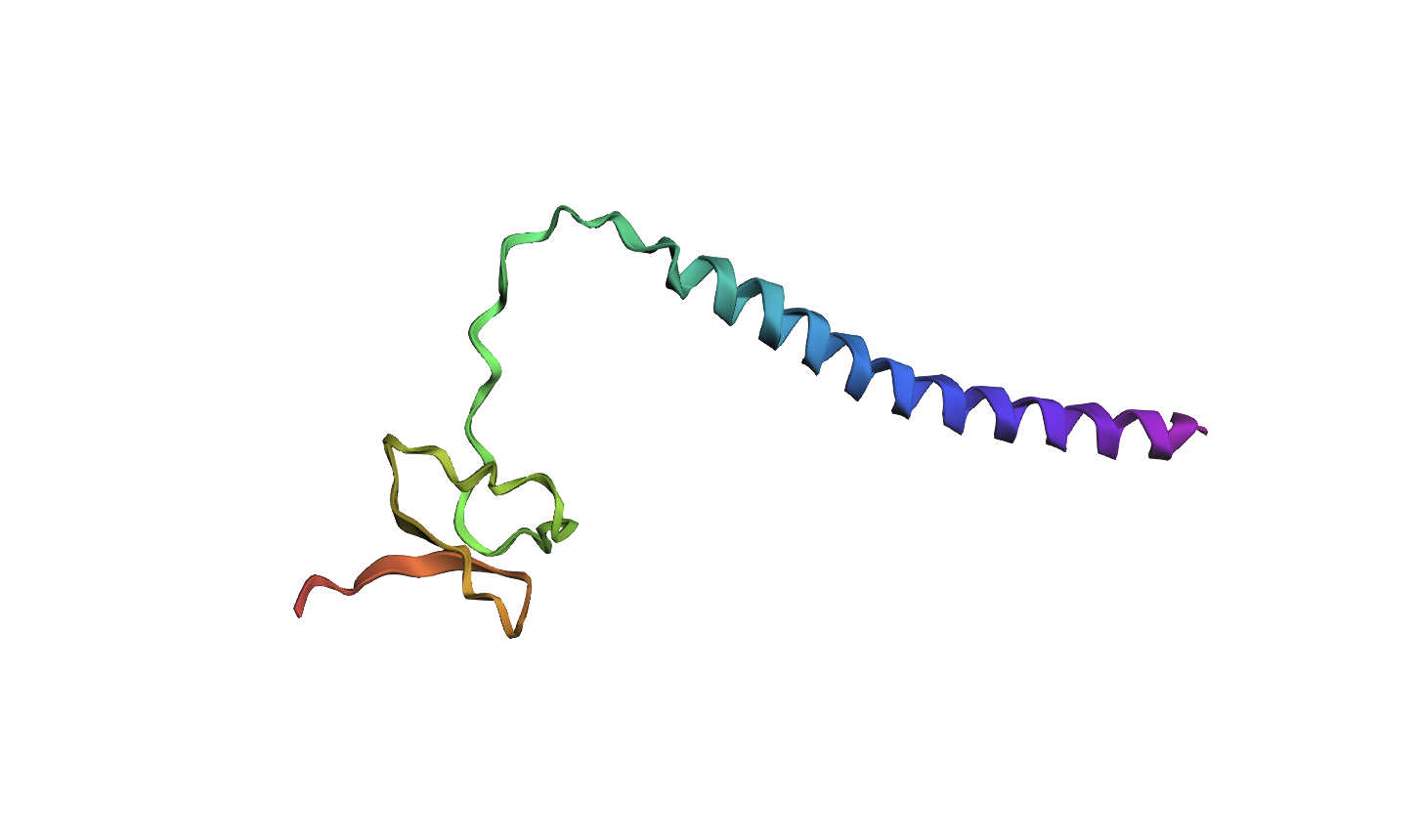
Analysis: Similar to the previous mutation, altering amino acids in the active site continues to modify the local structure. However, the active-site fold remains broadly similar across both mutations. Relative to the original structure, the beta strands seem to become progressively more separated, suggesting that increasing the number of mutations may gradually reduce the compactness of the native fold.
MUTATION 3
Additionally, I wanted to focus on the active protein as it also showed interesting results on the mutation heat map. Specifically, in amino acids E and D towards the start of the sequence (positions 11 and 13), which are located in the alpha helix section of the protein, show significantly low numbers on the scale (-6), suggesting high mutational intolerance. I therefore predict this would alter the structure/position of the helix.
Mutated sequence 3: Changed MFLALAGL to EDEDDEED on 8-16aa
MVNMKASEDEDDEEDVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
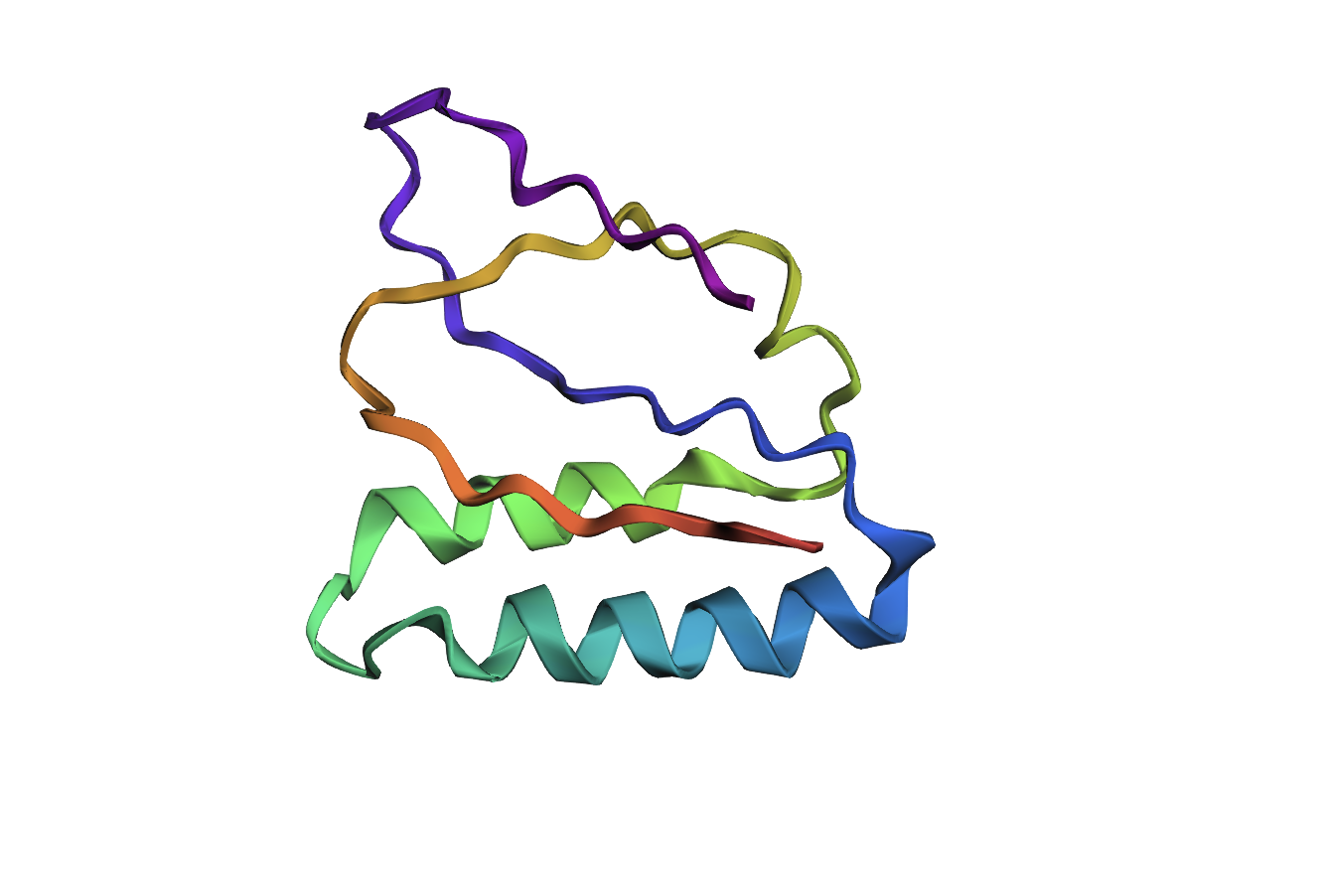
Analysis: The mutation caused a major alteration in the protein’s overall structure, leading to significant distortion of the alpha helices and beta sheets and a noticeable change in the overall folding pattern.
MUTATION 4
Lastly, I also wanted to look at changing a larger section of my sequence and opted for the first 20 amino acids, as overall, the mutation heat map suggests a high tolerance to mutations. Therefore, I predict that altering this part of the sequence shouldn’t alter the overall structure too much.
Mutated Sequence 4: Changed MVNMKASMFLALAGLVLLFV to EDWWHMELFALALSQKICVD on 1-20aa
EDWWHMELFALALSQKICVDVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
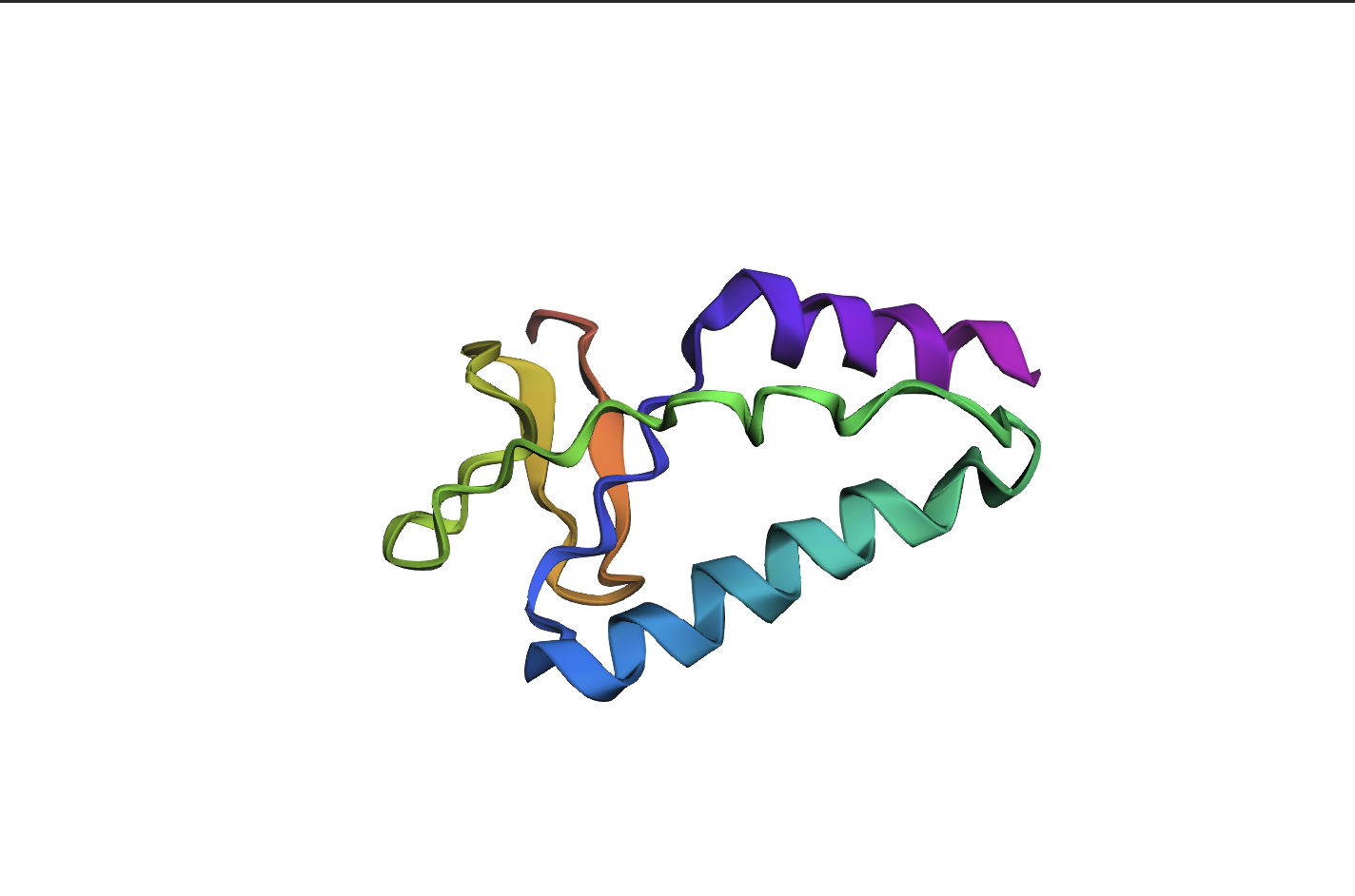
Analysis: The mutation in the first 20 amino acids resulted in a significant alteration of the overall protein structure, with major disruptions to the alpha helices and beta sheets. This indicates that, despite predictions from the mutation heat map, this region is less tolerant to sequence changes than expected.
Conclusion: Overall, the protein shows very low resistance to mutations, likely due to its small size and peptide nature, which makes structural stability more sensitive to sequence changes.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.

Using the backbone from Huwatoxin (PDB: 1MB6, active 35aa region) in ProteinMPNN, I generated a new sequence candidate:
EPKGENTPCTEENQNCDKEKNVECSPEKGACAPP
It scored 1.1293, lower than the original backbone’s 2.3287, showing it’s a plausible but less optimal fit to the structure. The sequence recovery of 0.2286 (23%) indicates low similarity to the wild-type (~9% identity), meaning a substantial redesign. Compared to the original’s mutation heat map, this designed sequence appears much more mutation-tolerant, suggesting improved structural robustness for engineering.
Heat map of the generated sequence candidate:
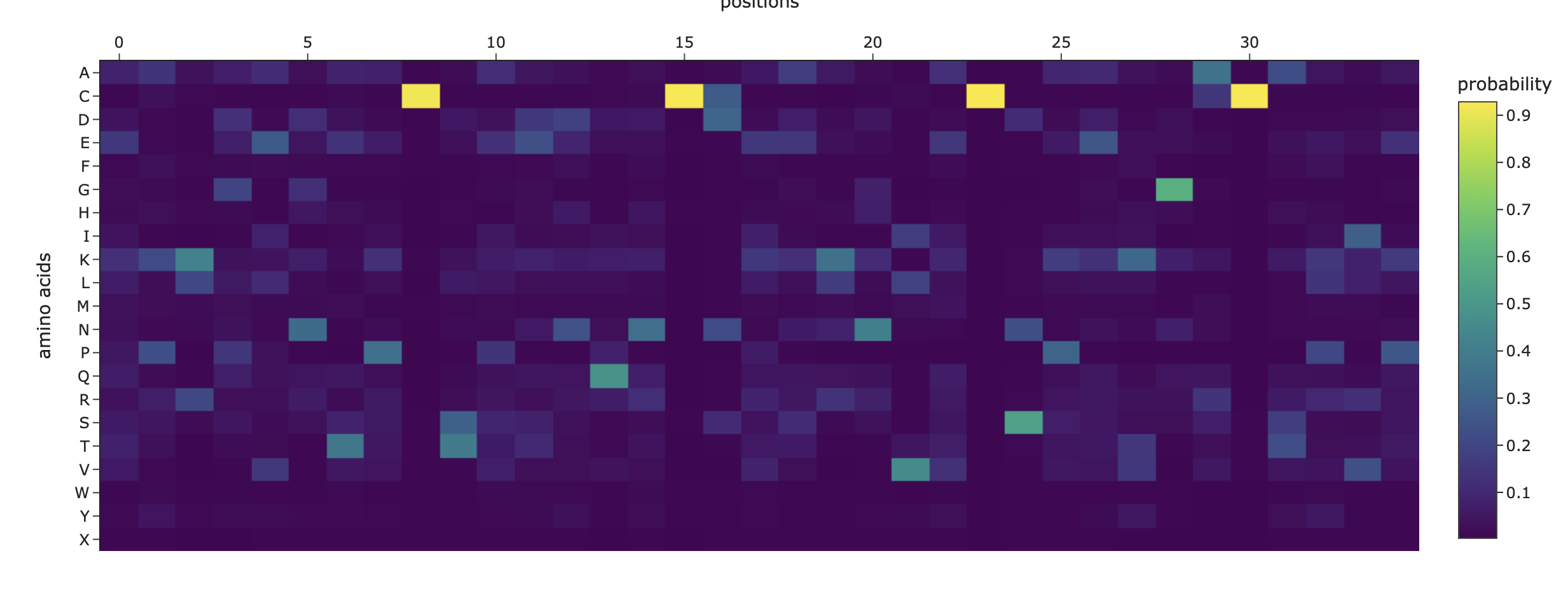
2. Input this sequence into ESMFold and compare the predicted structure to your original.
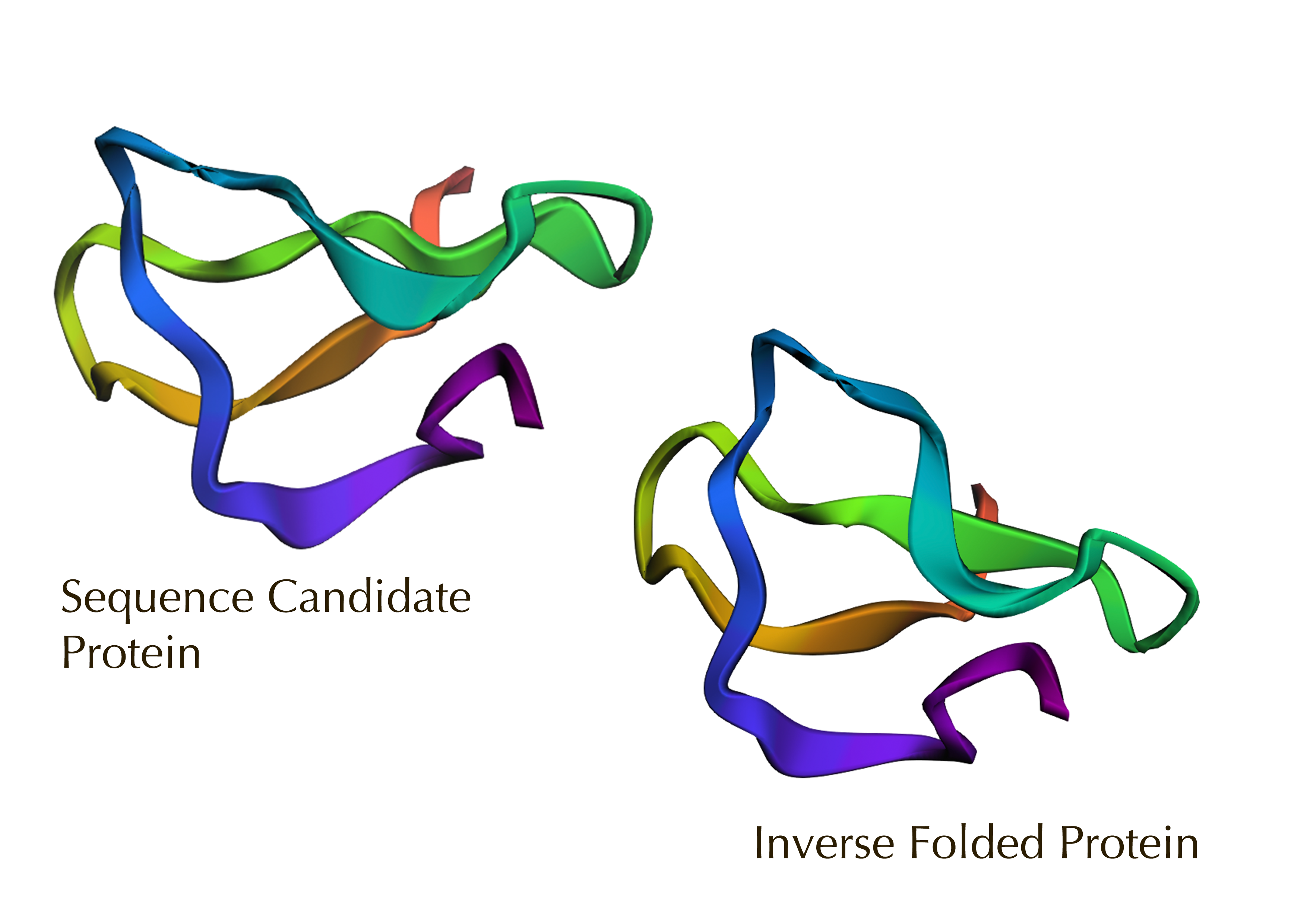
The sequence candidate model appears more looped and flexible, while the inverse-folded model on a fixed backbone is more compact and helical. Overall, this confirms a high fidelity visual overlap despite sequence changes, validating the design’s structural stability.
Part D. Group Brainstorm on Bacteriophage Engineering
Group of Commited Listeners LifeFabs: Sara Gaviria Escobar, Ruben Janssen, Justine de RiedmattenProposal: Engineering the MS2 Lysis Protein L to Enhance Stability Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis; this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been widely used in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.References