Week 5: Protein Design II
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.Human SOD1 Sequence (154AA per monomer - 308AA): MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQMATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutation: The alanine to valine mutation at codon 4 (A4V) of SOD1 causes a rapid and progressive form of amyotrophic lateral sclerosis (ALS). In Uniprot, however, it appears that the sequence starts with Methionine, making the mutation from Alanine to Valine actually at codon 5.
Mutation A4V (one of the monomers): MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
References:
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
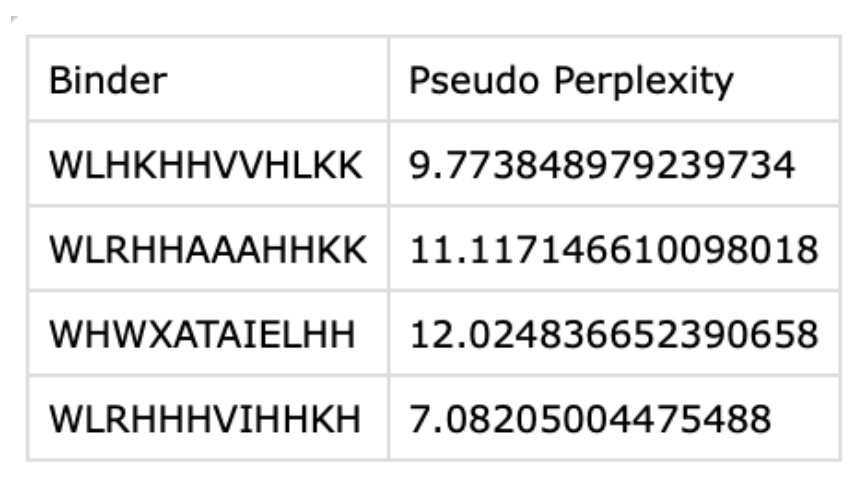 3. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
3. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
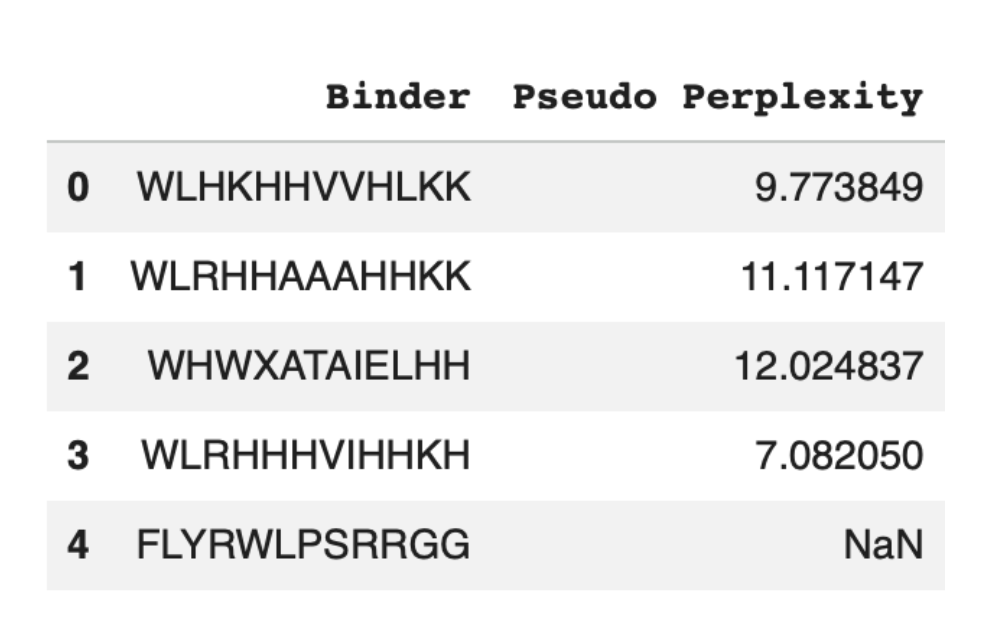
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
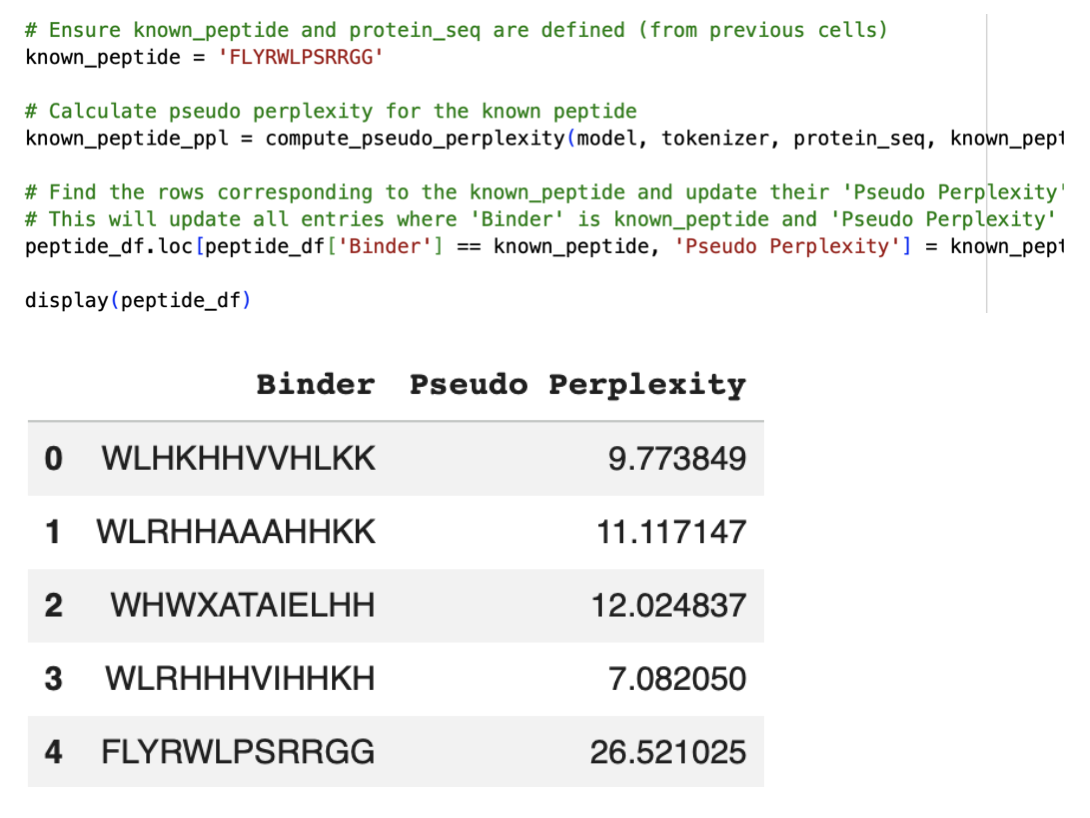
Asked for the built-in Gemini AI feature on the Colab for another code to get the pseudo perplexity of the known SOD1-binding peptide:
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
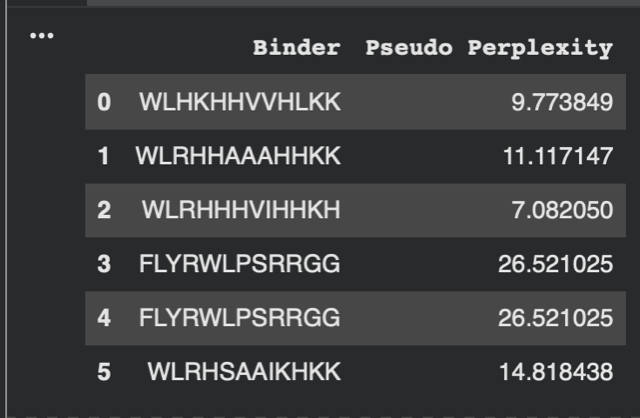
When entering the pepitdes, when adding sequence numbered 2 in the table above, the AlphaFold Server wasn’t letting me enter the ‘X’ Amino Acid as it isn’t an actual amino acid and will be used in a sequence for an unknown or undetermined AA. I therefore had to alter this sequence:
2. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localise near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
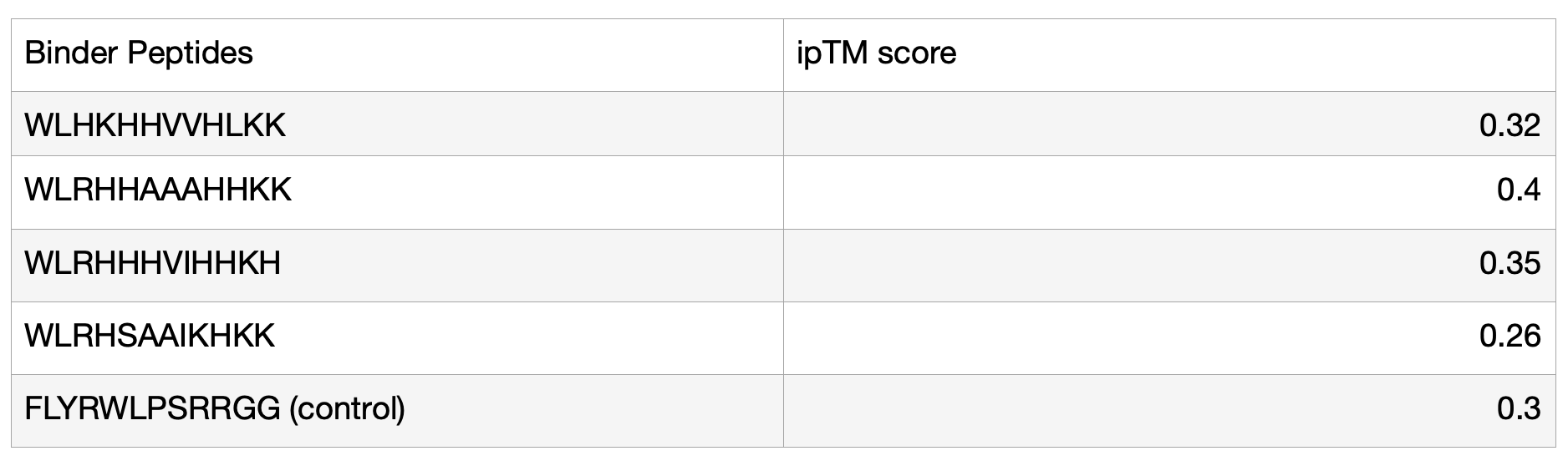
Binder 1: WLHKHHVVHLKK
The peptide doesn’t bind directly to the protein and is not located close to the N terminus (circled in red in the image above, always the orange end). It engages with the Beta barrel of the lefmonomer. It also appears to be surface-bound, as it is essentially exposed to the solvent and not in contact with the protein. I was initially confused about why I had two identical monomers, but after some research, I learned that SOD1 is a homodimer composed of two identical subunits that mirror one another.
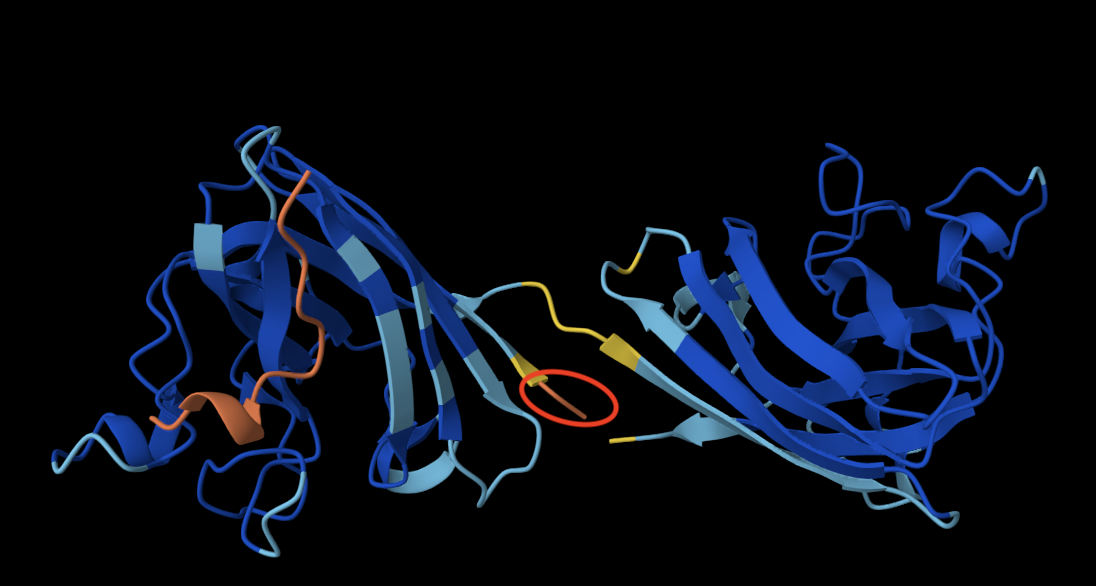
Binder 2: WLRHHAAAHHKK
Once again, the peptide doesn’t localise close to the N terminus; it is close to the Beta barrel but doesn’t interact with it and is surface-bound.
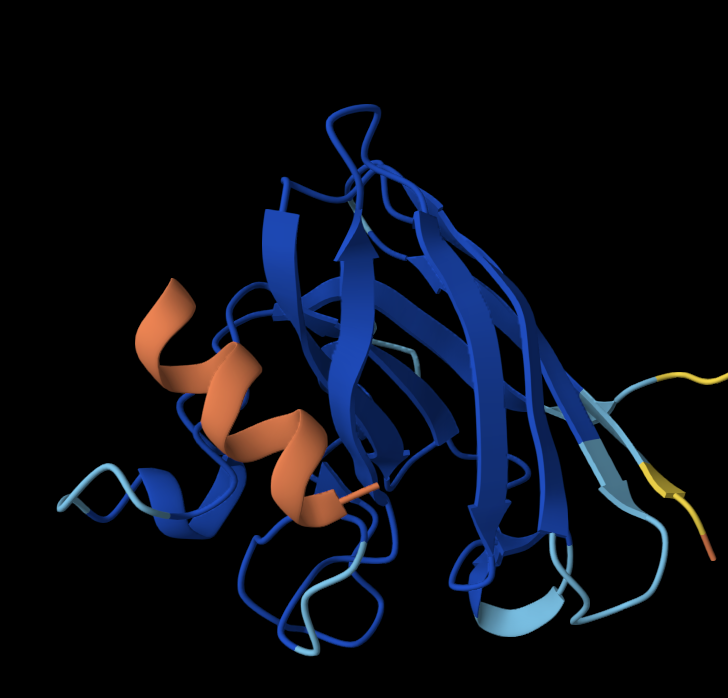
Binder 3: WLRHHHVIHHKH
The peptide doesn’t localise close to the N terminus, and doesn’t engage with the Beta barrel or the dimer interface and is surface-bound.
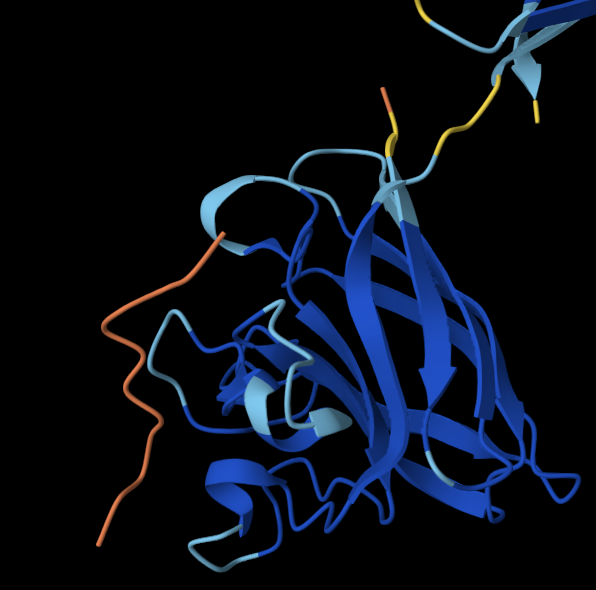
Binder 4: WLRHSAAIKHKK
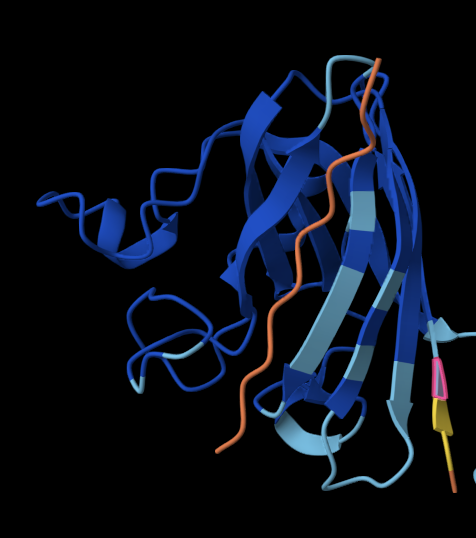
The peptide doesn’t localise close to the N terminus, and is the closest out of all the other peptides to the Beta barrel. It is also surface-bound.
Controlled Binder: FLYRWLPSRRGG
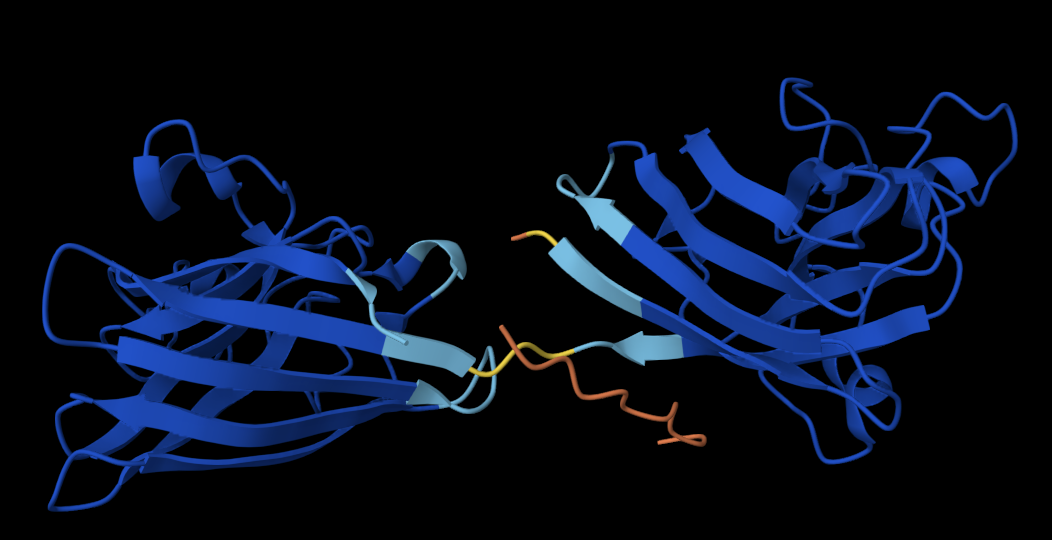
The peptide doesn’t localise close to the N terminus, is slightly partially buried and approaches the dimer surface, where the two monomers join.
3. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The ipTM values of my generated peptides are in a range of 0.26 to 0.4, with an average of 0.33. In comparison, the known (control) binder has a score of 0.3. Ultimately, they are relatively similar, but 3/4 of my generated peptides have a higher score than the known binder. Overall, all the binders have a relatively low confidence in the predicted interface, translating to weak interactions between the protein and the binder.
Evaluate Properties of Generated Peptides in the PeptiVerse
1. Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:Binder 1:
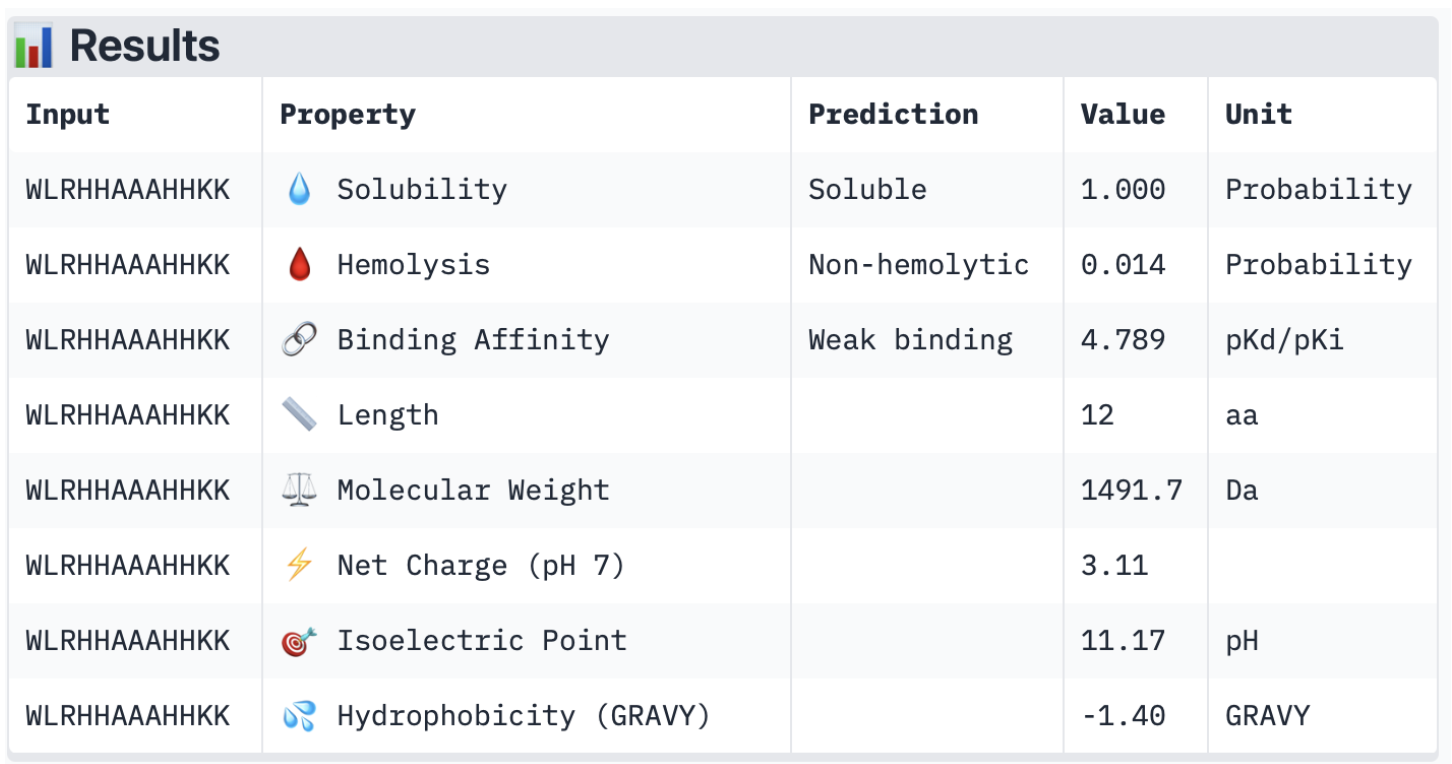 Binder 2:
Binder 2:
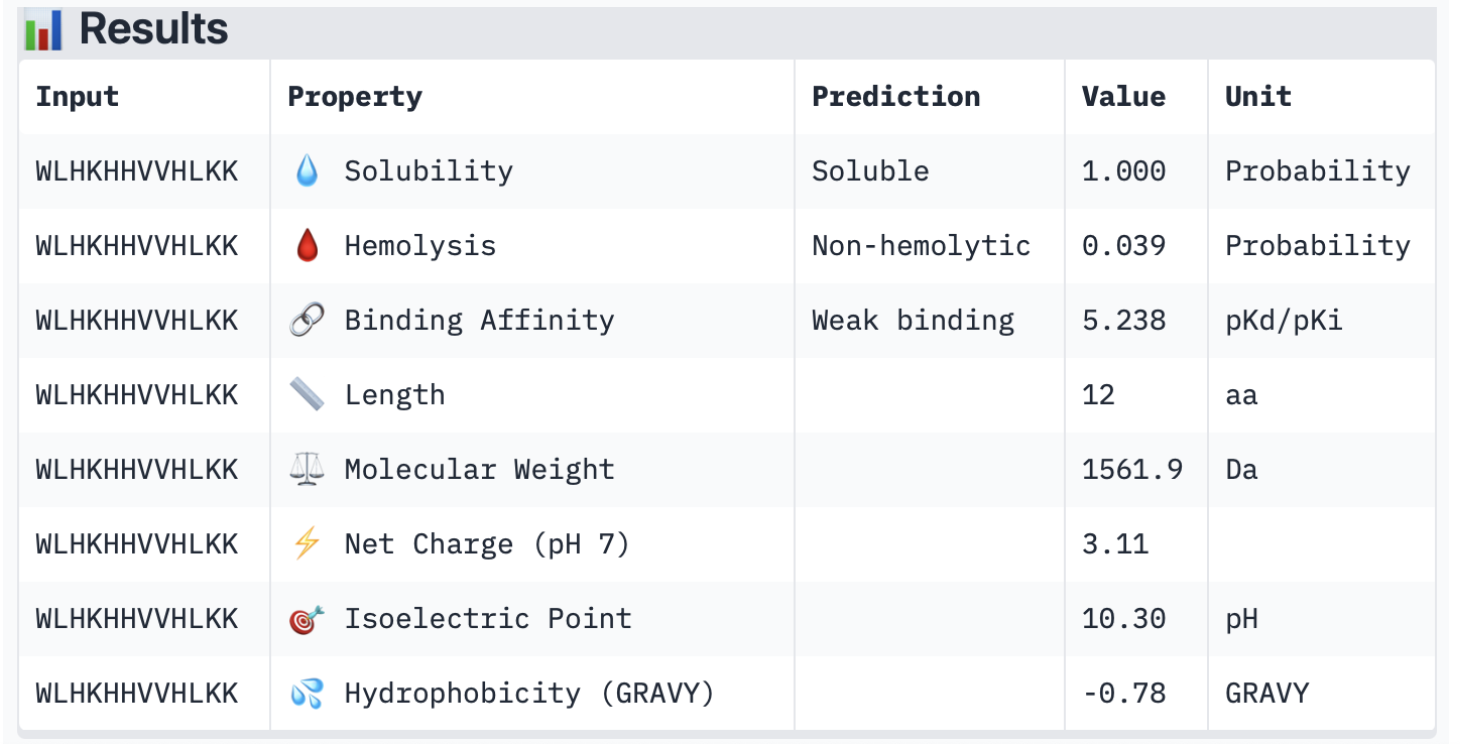 Binder 3:
Binder 3:
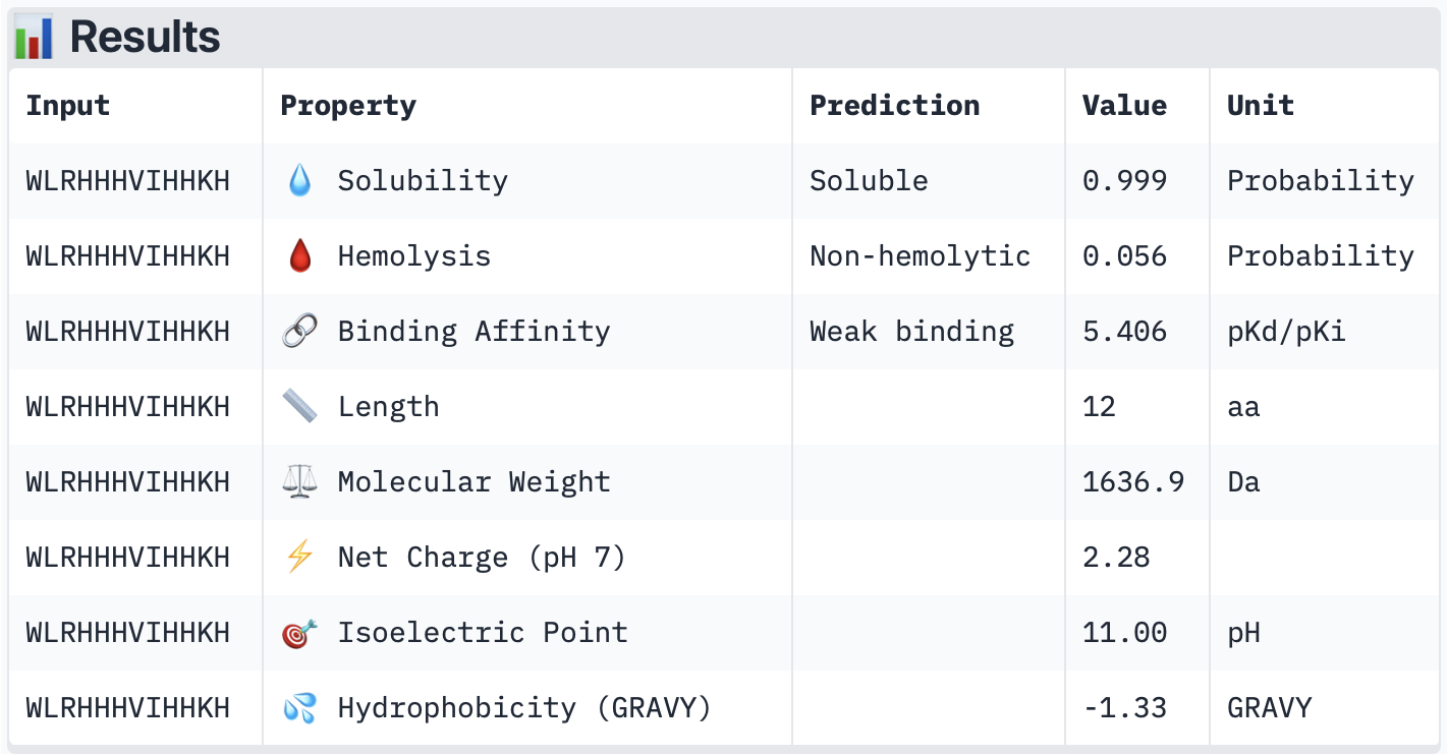 Binder 4:
Binder 4:
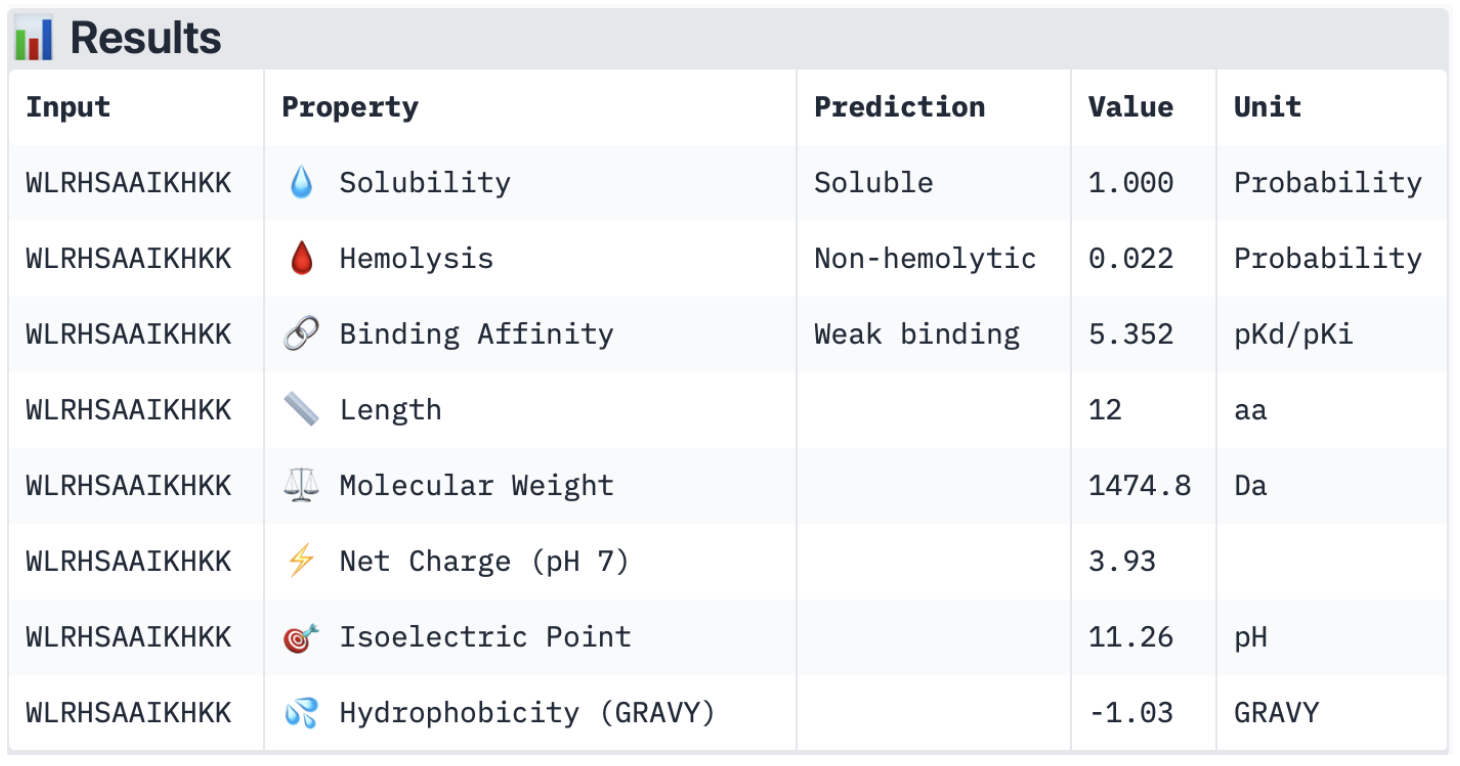
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Overall, all the binders have a weak binding affinity, which matches the ipTM scores described earlier. This also backs up the 3D structures where none of the peptides was closely interacting with the protein. In terms of solubility, all of them are soluble, which is therapeutically beneficial as it improves dissolution rates and increases absorption (Saeed et al., 2009). Furthermore, they are all non-hemolytic (low hemolysis probability), which is also therapeutically beneficial, as it ensures minimum red blood cell destruction (Cleveland Clinic, 2022). Looking at the net charge, SOD1 is negatively charged (-6 monomers) with the physiological pH of 7.4 (Shi et al., 2014), and all the binders are positively charged with a pH ranging from 2.28 to 3.93. This is generally advantageous as the positively charged binders will be attracted to the negatively charged SOD1 surface; however, when they are too positively charged, it may also cause the binders to bind to other negatively charged molecules. Lastly, in terms of molecular weight, all binders have a relatively low value, ranging from 1474.8 to 1636.9 Da, which minimises aggregation risks.
I believe the peptide which best balances predicted binding and therapeutic properties would be peptide 3 (WLRHHHVIHHKH), firstly due to having the highest ipTM score. Secondly, in terms of its therapeutic properties, it has the highest binding affinity out of all the peptides, as well as having high solubility and being non-hemolytic. Although all peptides had similar properties and scores, it appears to be the most appropriate option for further development.
References:
Generate Optimised Peptides with moPPIt
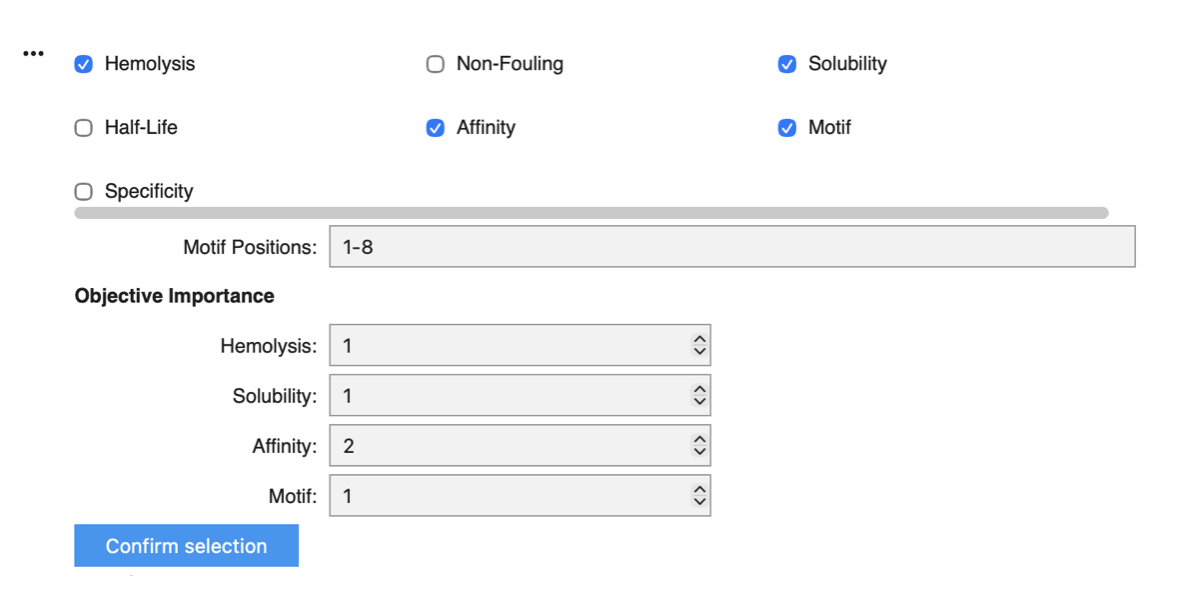
I focused on Motif positions 1–8 to keep the design space close to the mutation site at position 4. I increased the weight of binding affinity to 2, as this is ultimately the most critical objective, while assigning similar weights to hemolysis, solubility, and motif to preserve overall therapeutic properties.
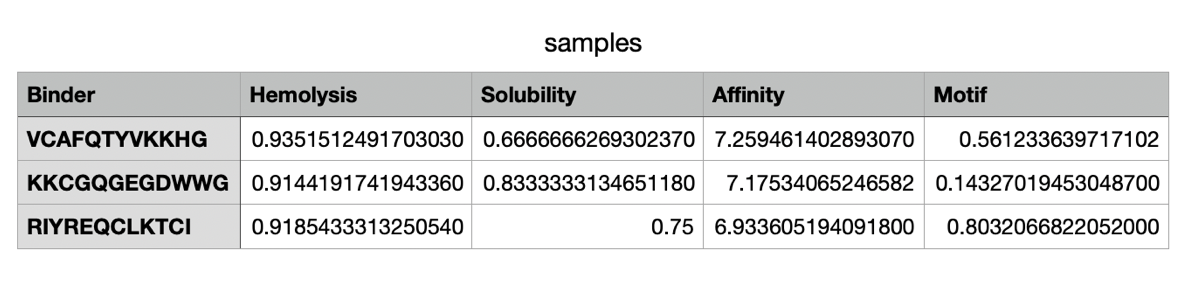
The moPPit peptides generated differ from the ones I have generated on PepMLM in their binding affinity, solubility and hemolysis risks. moPPit peptides have a significantly higher affinity score (6.93-7.26) in comparison to the PepMLM ones. This could essentially be due to the motif position, as well as increasing the weight of affinity in the settings. In terms of solubility, the moPPit peptides have a lower solubility value than the PepMLM peptides, which makes them slightly less advantageous in this specific therapeutic property. Additionally, they have a significantly higher hemolysis value, resulting in potentially higher risks of red blood cell destruction. Ultimately, the moPPit peptides appear to bind more strongly but appear less favourable for safety and developability. To evaluate these peptides before advancing them to clinical studies, I would begin by testing their predictions in an in-silico model through in vitro experiments before any animal or human trial. Furthermore, I would conduct a safety screening (Cyprotex in vitro hemolysis testing) to evaluate the level of toxic hemolysis (Bauch, 2026).
References:
Final Project: L-Protein Mutants Option 1: Mutagenesis
Step 1: Gathering information about Lysis proteins
DNA sequence of Lysis Protein Phage MS2 (E. coli) 225bp: atggaaacccgctttccgcagcagagccagcagaccccggcgagcaccaaccgccgccgcccgtttaaacatgaagattatccgtgccgccgccagcagcgcagcagcaccctgtatgtgctgatttttctggcgatttttctgagcaaatttaccaaccagctgctgctgagcctgctggaagcggtgattcgcaccgtgaccaccctgcagcagctgctgacc
Reference: EMBL-EBI (2026). ENA Browser. [online] Ebi.ac.uk. Available at: https://www.ebi.ac.uk/ena/browser/view/V00642 [Accessed 26 Mar. 2026].
Chaperone DNAj sequence (E. coli, strain K12) 1358bp: TCGACGCTGAATTTGAAGAAGTCAAAGACAAAAAATAATCGCCCTATAAACGGGTAATTA TACTGACACGGGCGAAGGGGAATTTCCTCTCCGCCCGTGCATTCATCTAGGGGCAATTTA AAAAAGATGGCTAAGCAAGATTATTACGAGATTTTAGGCGTTTCCAAAACAGCGGAAGAG CGTGAAATCAGAAAGGCCTACAAACGCCTGGCCATGAAATACCACCCGGACCGTAACCAG GGTGACAAAGAGGCCGAGGCGAAATTTAAAGAGATCAAGGAAGCTTATGAAGTTCTGACC GACTCGCAAAAACGTGCGGCATACGATCAGTATGGTCATGCTGCGTTTGAGCAAGGTGGC ATGGGCGGCGGCGGTTTTGGCGGCGGCGCAGACTTCAGCGATATTTTTGGTGACGTTTTC GGCGATATTTTTGGCGGCGGACGTGGTCGTCAACGTGCGGCGCGCGGTGCTGATTTACGC TATAACATGGAGCTCACCCTCGAAGAAGCTGTACGTGGCGTGACCAAAGAGATCCGCATT CCGACTCTGGAAGAGTGTGACGTTTGCCACGGTAGCGGTGCAAAACCAGGTACACAGCCG CAGACTTGTCCGACCTGTCATGGTTCTGGTCAGGTGCAGATGCGCCAGGGATTCTTCGCT GTACAGCAGACCTGTCCACACTGTCAGGGCCGCGGTACGCTGATCAAAGATCCGTGCAAC AAATGTCATGGTCATGGTCGTGTTGAGCGCAGCAAAACGCTGTCCGTTAAAATCCCGGCA GGGGTGGACACTGGAGACCGCATCCGTCTTGCGGGCGAAGGTGAAGCGGGCGAGCATGGC GCACCGGCAGGCGATCTGTACGTTCAGGTTCAGGTTAAACAGCACCCGATTTTCGAGCGT GAAGGCAACAACCTGTATTGCGAAGTCCCGATCAACTTCGCTATGGCGGCGCTGGGTGGC GAAATCGAAGTACCGACCCTTGATGGTCGCGTCAAACTGAAAGTGCCTGGCGAAACCCAG ACCGGTAAGCTATTCCGTATGCGCGGTAAAGGCGTCAAGTCTGTCCGCGGTGGCGCACAG GGTGATTTGCTGTGCCGCGTTGTCGTCGAAACACCGGTAGGCCTGAACGAAAGGCAGAAA CAGCTGCTGCAAGAGCTGCAAGAAAGCTTCGGTGGCCCAACCGGCGAGCACAACAGCCCG CGCTCAAAGAGCTTCTTTGATGGTGTGAAGAAGTTTTTTGACGACCTGACCCGCTAACCT CCCCAAAAGCCTGCCCGTGGGCAGGCCTGGGTAAAAATAGGGTGCGTTGAAGATATGCGA GCACCTGTAAAGTGGCGGGGATCACTCCCATAAGCGCT
Reference: EMBL-EBI (2026). ENA Browser. [online] Ebi.ac.uk. Available at: https://www.ebi.ac.uk/ena/browser/view/M12565
Conserved sites and known mutational effects:
Lysis proteins, and in particular single-gene lysis (Sgls) proteins, often feature conserved sequence sites despite their diversity. Analysis of L sequences from related F-specific MS2-like phages reveals four key elements in the L protein: a basic N-terminus, a hydrophobic region, a conserved Leu(48)-Ser(49) (LS) dipeptide essential for type II Sgl lytic function, and a variable C-terminus.(Antillon et al., 2024). Additionally, mutational studies of the MS2 lysis protein L demonstrate that several residues in the central domains are highly conserved across most homologs (Chamakura et al., 2017).
 Figure: Conserved LS and similarity of central domains
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
Figure: Conserved LS and similarity of central domains
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
The figure below analyses the mutational analysis of MS2 L. The sequence is separated into its four domains through the numbered boxes at the bottom of the figure. The Amino acids above the L sequence show single amino acid mutations (missense mutation) that have a lysis defect, but don’t cause protein accumulation. Below the sequence are single amino acid mutations that don’t affect lysis. The amino acids with green asterisks in the L sequence show where a single nucleotide of the sequence could be changed by a stop codon (nonsense mutation). (Chamakura et al., 2017)
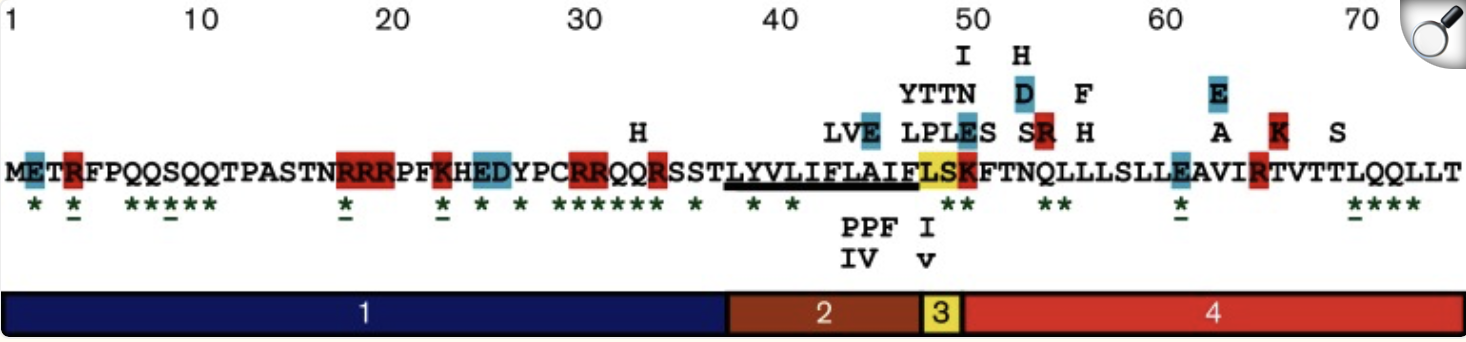 Figure: Mutational Analysis of MS2 L.
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
Figure: Mutational Analysis of MS2 L.
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
References:
Step 2: Approach and Sequence variants
Below is the downloaded top 20 L-protein mutants dataset that I got from the ESM-2 Colab:
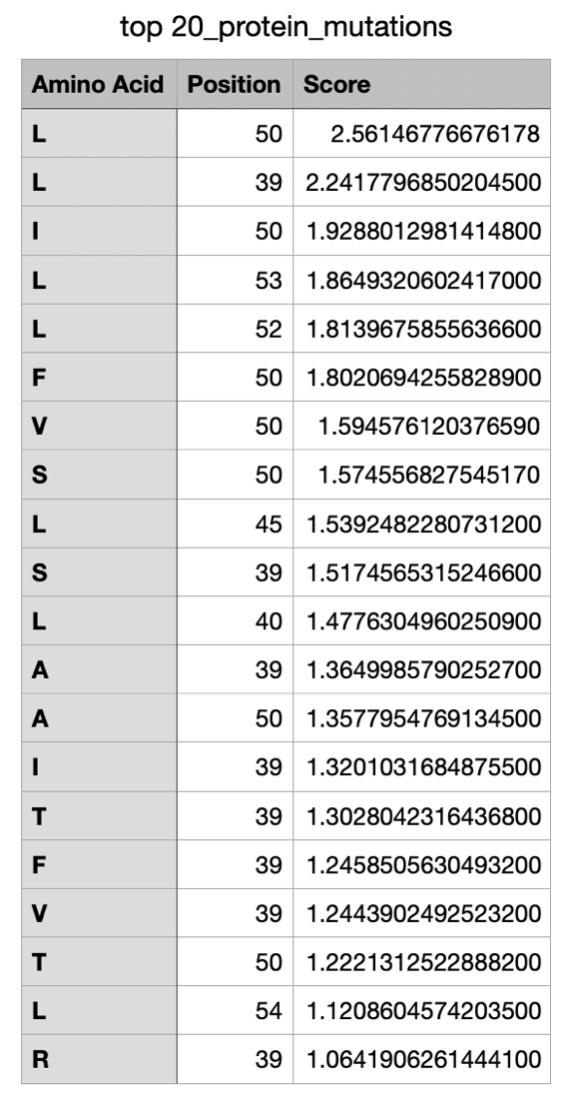
The LLR Scores: A higher log-likelihood ratio is considered to mean the protein mutation of that amino acid in the specific location of the sequence is more supported, whereas a lower LLR score shows a low tolerance for a mutation.
Now continuing through the Colab, I managed to get the top 10 of the mutations and added the experimental data as a cvs file to also gather the top 10 mutations from the data:
Top 10 from Colab:

Experimental data:
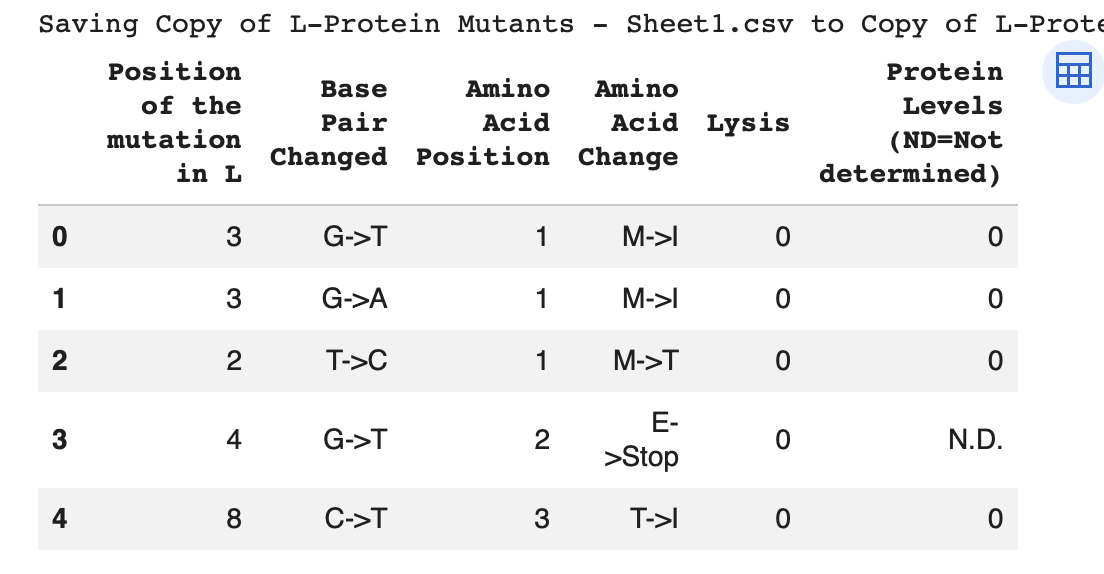
Top 10 of experimental Data:

Step 3: Filtering and Ranking sequences
To determine the location of the mutations, it is important to identify the soluble N-terminus (1–39 aa) and the transmembrane domain (40–75 aa) (Mezhyrova et al., 2023). As shown in the tables above, all experimental and generated mutations are located within these regions of the sequence.
By comparing the experimental data with the mutations generated in Colab, it is possible to identify the most likely mutations present in both datasets. The highest LLR-scored mutations are mainly located in the N-terminal region, particularly at position 29, with one mutation also found in the transmembrane domain at position 50. Since higher LLR scores indicate greater mutational tolerance, these mutations appear to be the most tolerated. The three mutations that appear in both datasets are:
References:
Step 4: Submit 5 mutated sequences
*Note: in all the structure images below, the top protein is the original, and the bottom one is the mutated structure*
Mutation 1: K50L
Mutated sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
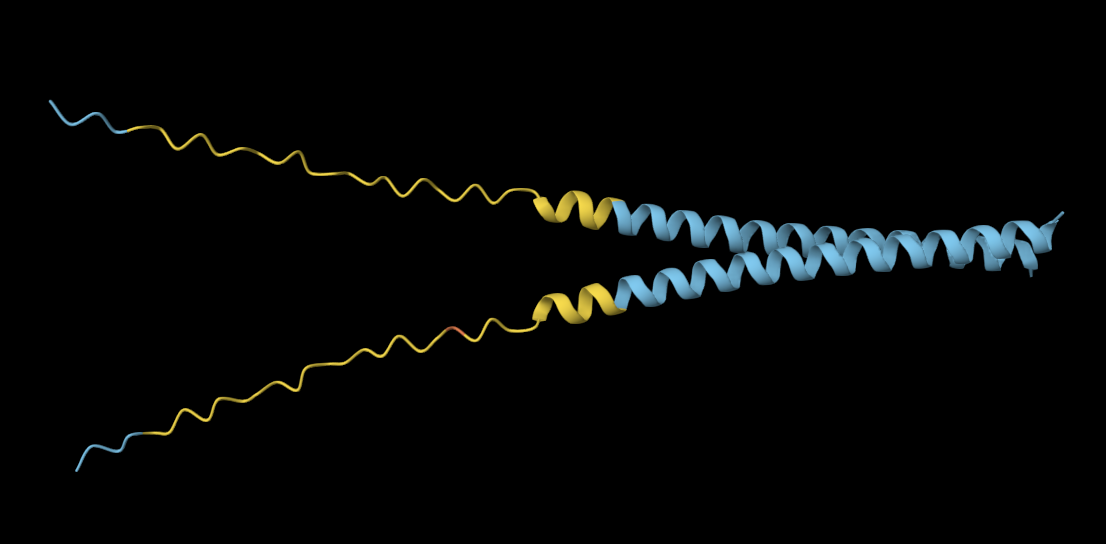
The mutated sequence seems slightly more elongated and compacted than the original sequence.
Mutation 2: C29L
Mutated sequence: METRFPQQSQQTPASTNRRRPFKHEDYPLRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
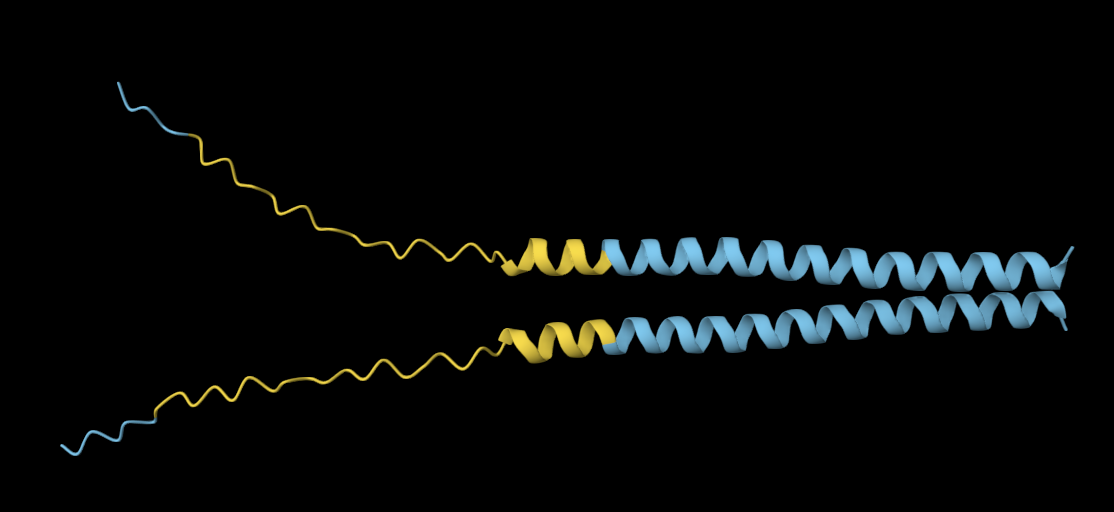
My observations reveal no noticeable structural differences between the original and mutated sequences.
Mutation 3: Y39L
Mutated sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
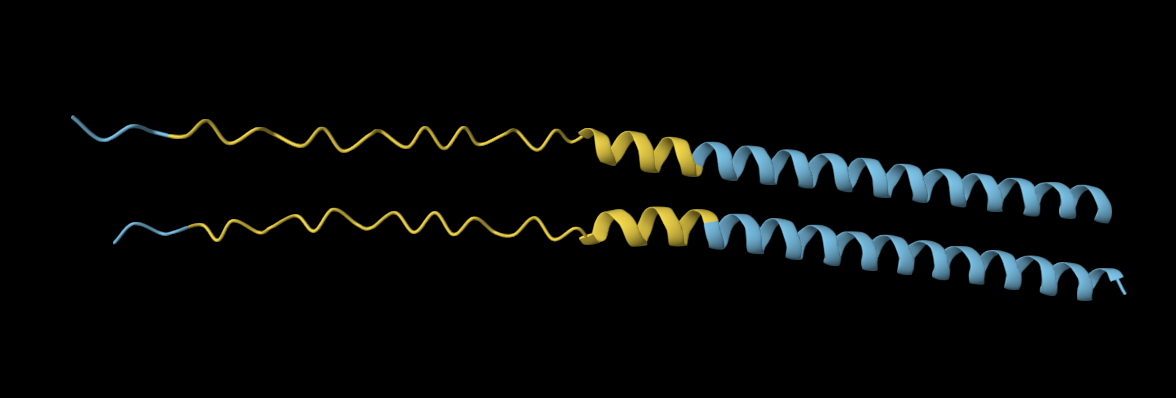
The original sequence exhibits more elongated yet compacted alpha helices compared to the mutated sequence.
Mutation 4: C29R
Mutated sequence: METRFPQQSQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
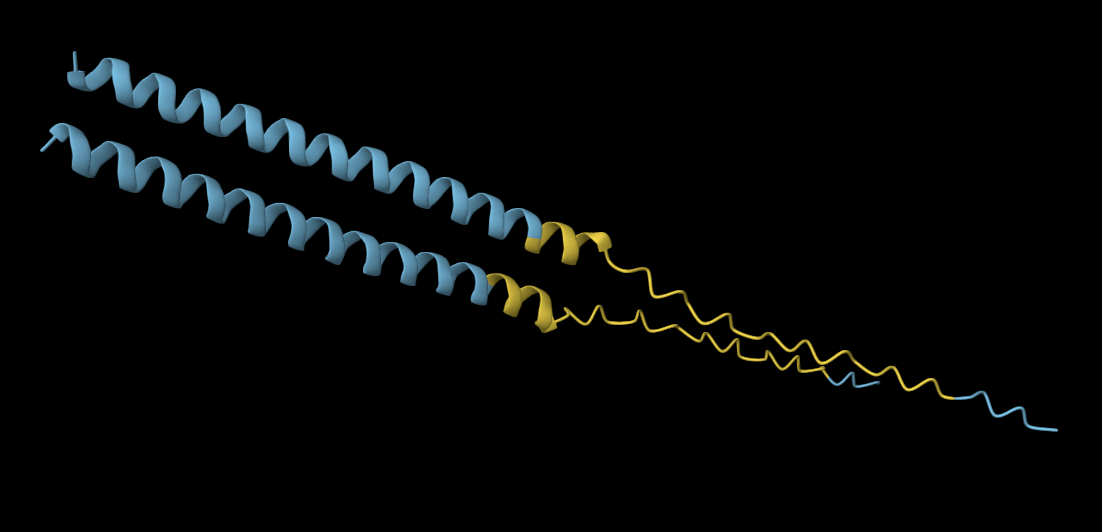
The mutated sequence produces a slightly more elongated structure compared to the more compact alpha-helical fold of the original protein. This suggests the mutation C29S in the N-terminus slightly impacts secondary structure without fully destabilising the fold.
Mutation 5: C29S
Mutated sequence: METRFPQQSQQTPASTNRRRPFKHEDYPSRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
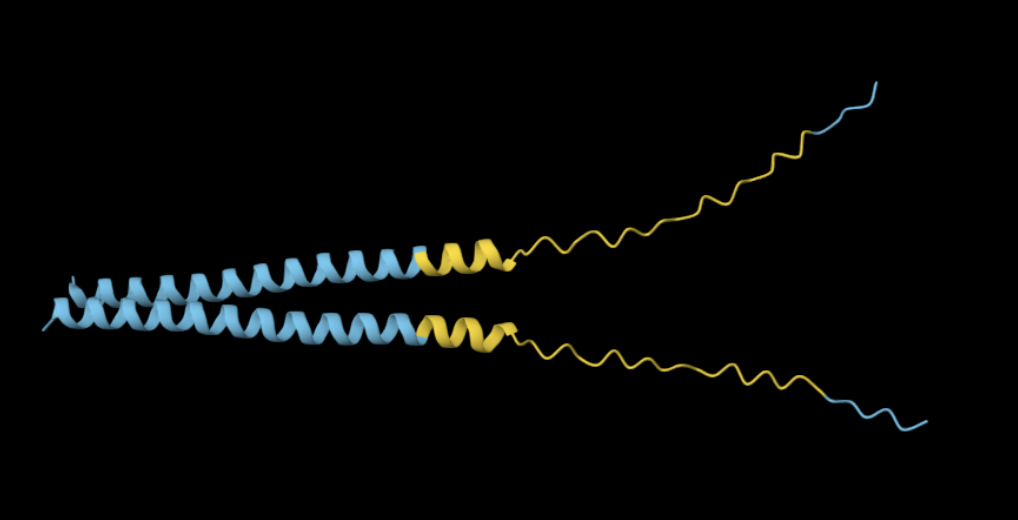
Similarly to the previous mutation in the same location, the mutated sequence produces a slightly more elongated structure compared to the more compact alpha-helical fold of the original protein.