Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix, and what is their purpose?
Phusion High-Fidelity PCR Master Mix offers high fidelity and performance for PCR. It consists of Phusion DNA polymerase (1), deoxynucleotides and reaction buffer that has been optimised and also includes MgCl2 (New England Biolabs, 2026). The DNA Polymerase allows for the rapid synthesis of a new DNA strand with high accuracy, as it generates long templates with a single enzyme (Thermo Scientific, 2018). The deoxynucleotides are the essential building blocks for the DNA polymerase to proceed with the synthesis of a new DNA strand, whilst the buffer is what provides the ideal environment for the DNA polymerase to function, resulting in high-yield and fidelity.
References:
2. What are some factors that determine primer annealing temperature during PCR?
The main factor that determines primer annealing temperature during PCR is the calculation of the melting temperature of the primers selected for the amplification. The general rule is to start with an annealing temperature of 3-5°C lower than the lowest melting temperature of the primers (ThermoFisher Scientific, 2019). Another factor that will further determine the temperature is the length of the primers, as they have higher melting temperatures. If the annealing temperature exceeds a temperature of 65°C instead of an optimal temperature of approximately 54°C, there are risks of secondary annealing (Andreas Ebertz, 2022). Furthermore, salt concentration (NA+) further impacts primer annealing as higher levels allow for higher annealing temperature (ThermoFisher Scientific, 2019).
References:
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Polymerase Chain Reaction (PCR) is a laboratory nucleic acid amplification technique used to generate multiple copies of a specific target DNA sequence in vitro. It relies on repeated cycles of thermal denaturation, primer annealing, and extension by a DNA polymerase, leading to exponential amplification of the target DNA section and enabling its detection and analysis. (Khehra et al., 2023).
Protocol(Khehra et al., 2023):
- PCR begins with the extraction of a small nucleic acid sample of DNA or RNA into a reaction tube.
- Denaturation: the DNA is heated to 95°C to break the hydrogen bonds between its complementary base pairs of the double-stranded DNA, separating them into two seperate strands.
- Annealing: the denatured DNA is then cooled down to a temperature of 55°C to 72°C, which allows the binders to bind to each complementary sequence on single-stranded DNA through pairing their 3’ ends to the template strand, providing a start point for DNA synthesis.
- Extension: during the final stage, the temperature is raised to 75°C to 80°C to optimise the DNA polymerase, which promotes strand elongation of the new strand.
DNA polymerase synthesises in a 3’ to 5’ direction, generating new sequences complementary to the template strand. The full process is regulated by the use of a thermal cycler that regulates the time and temperature of each step mentioned above. Conducting multiple cycles results in the amplification of multiple copies of the target DNA within the tube.
Restriction enzyme digestion is a process that involves the cutting of DNA into fragments by restriction enzymes (endonucleases) at specific recognition sequences. This results in DNA fragments that vary in size depending on their location in these recognition sequences. It is usually performed in a microcentrifuge tube with the necessary components: the template DNA, restriction enzymes and Mg2+ under the right conditions (Shen, 2019). There are two possible products depending on the used restriction enzymes; the first one creates staggered cuts (majority of cuts), leaving single-stranded overhangs that will easily anneal to complementary DNA strands (sticky) and the second results in cuts in the middle of the recognition sequence, leaving no overhangs (blunt-end) causing them to ligate less and are therefore harder to clone (QIAGEN, 2013).
Protocol (QIAGEN, 2013):
- Pipette reaction components into a tube (water, DNA, buffer, enzyme)
- Centrifuge the tube.
- Incubate the content in a water bath (usually around 37°C for 1-4 hours).
- In certain applications, it is needed to heat-inactivate the enzyme after digestion ( around 65°C for 20 minutes).
Ultimately, PCR is the amplification of specific DNA sequences to create large amounts of copies, whilst restriction enzyme digestion uses endonucleases to cut DNA in specific sites, meaning they don’t serve the same purpose. PCR is quantity-based, whilst restriction enzyme digestion is used to create smaller DNA fragments. Furthermore, PCR synthesises new DNA strands whilst PCR cuts already existing DNA strands. Lastly, PCR relies on the binding of primers to the DNA strands, whilst restriction enzyme digestion relies on the recognition site of the enzyme.
References:
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences will be appropriate, several factors need to be considered. Firstly, Gibson Assembly are designed to be 20 to 40 nucleotides long (Yu, 2021); it is essential for successful annealing that the primers are designed within that range of base pairs. In addition to this, Gibson Assembly’s optimal temperature melting point typically ranges between 60°C and 70°C, so making sure the primers from PCR also have a similar temperature melting point will ensure a more successful assembly. Additionally, using high-fidelity PCR instead of regular PCR will reduce the risks of mutations during amplification (Eggert et al., 2005). Furthermore, purifying your DNA post-PCR to eliminate any unnecessary byproducts will increase the accuracy of Gibson cloning.
References:
5. How does the plasmid DNA enter the E. coli cells during transformation?
There are two seperate methods of how the plasmid DAN enters the E. Coli cells during transformation:
Chemical Competence method: This method consists of growing E. coli to log phase, which is then chilled on ice and further treated with ice-cold CaCl₂. This binds cell membrane lipids and DNA phosphates, causing the neutralisation of charges, making the negatively charged plasmid DNA less repelled by the membrane. The mix is then heat-shocked at 42°C for 30-90 seconds to temporarily open membrane pores for DNA entry. Lastly, the cell is iced again shortly to reseal the membrane (Froger and Hall, 2007)
Electroporation: The cells ells are mixed with DNA and zapped with a high-voltage pulse (e.g., 2.5 kV). This creates transient membrane holes through dielectric breakdown. Ultimately, the electric field drives charged DNA into the cell before the pores close (Biology LibreTexts, 2021)
References:
6. Describe another assembly method in detail (such as Golden Gate Assembly)
Part 1: Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a DNA cloning technique that allows multiple fragments to be combined in a defined order without leaving extra sequences at the junctions. It uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sites to create specific 4-base overhangs. These overhangs are designed so that each fragment connects only to its correct neighbours. The process takes place in a single reaction mixture that alternates between temperatures, favouring enzyme cutting (37°C) and ligation by T4 DNA ligase (16°C), which aids in correcting incorrect assemblies automatically. To avoid unintended cleavage, DNA sequences are “domesticated” by removing internal enzyme recognition sites. Overall, this method is more efficient than traditional cloning techniques and is especially useful for building complex genetic systems (Bird et al., 2022).
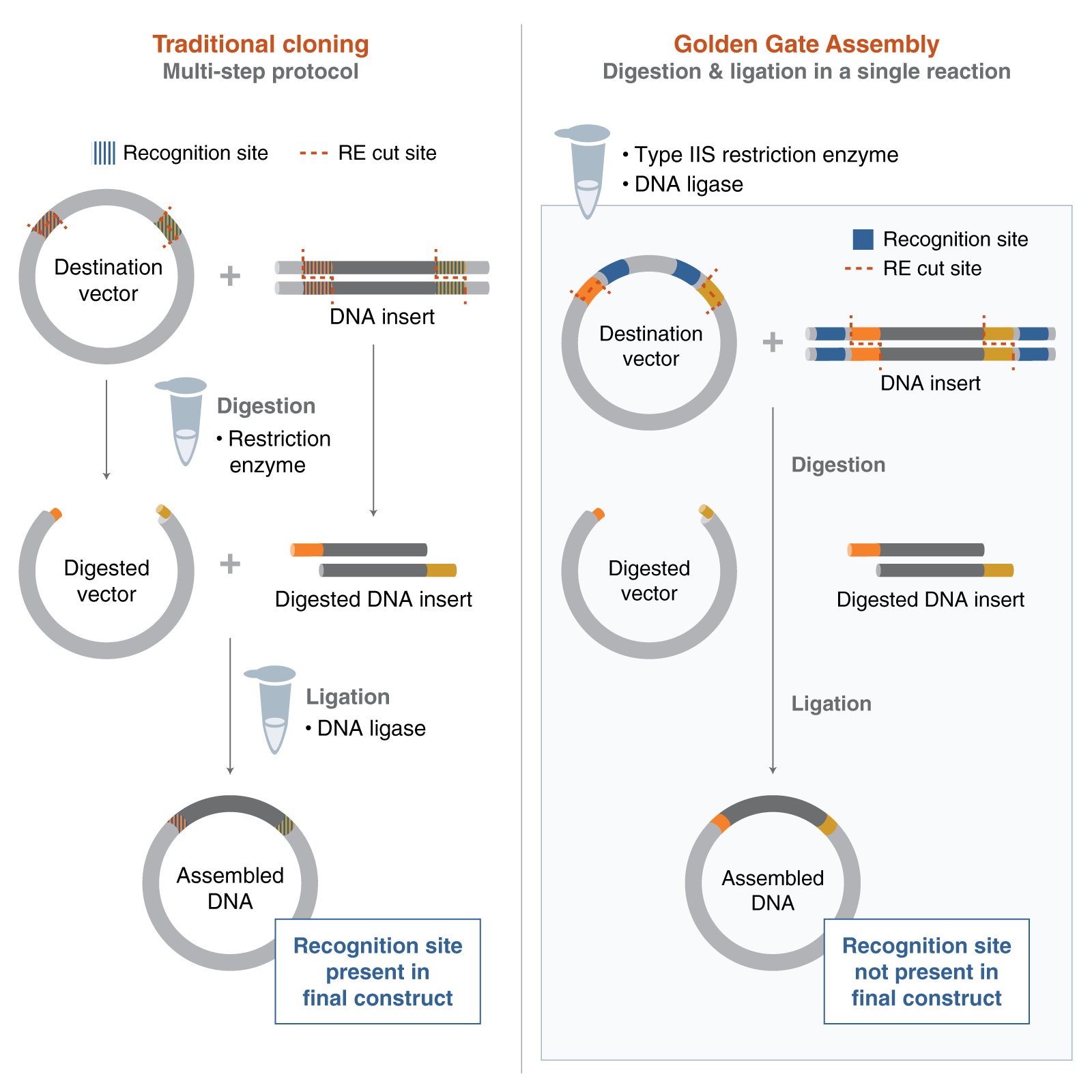 Figure 1: Golden Gate Assembly vs. Traditional Cloning
Image reference: New England Biolabs (2024). Getting Started with Golden Gate Assembly. [online] Neb.com. Available at: https://www.neb.com/en-gb/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoo7ZCRSqsQ1DI7fCFa1awuGBgToH2L_sRbw7czYQBuj4tPjf8lh [Accessed 20 Mar. 2026].
Figure 1: Golden Gate Assembly vs. Traditional Cloning
Image reference: New England Biolabs (2024). Getting Started with Golden Gate Assembly. [online] Neb.com. Available at: https://www.neb.com/en-gb/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoo7ZCRSqsQ1DI7fCFa1awuGBgToH2L_sRbw7czYQBuj4tPjf8lh [Accessed 20 Mar. 2026].
References:
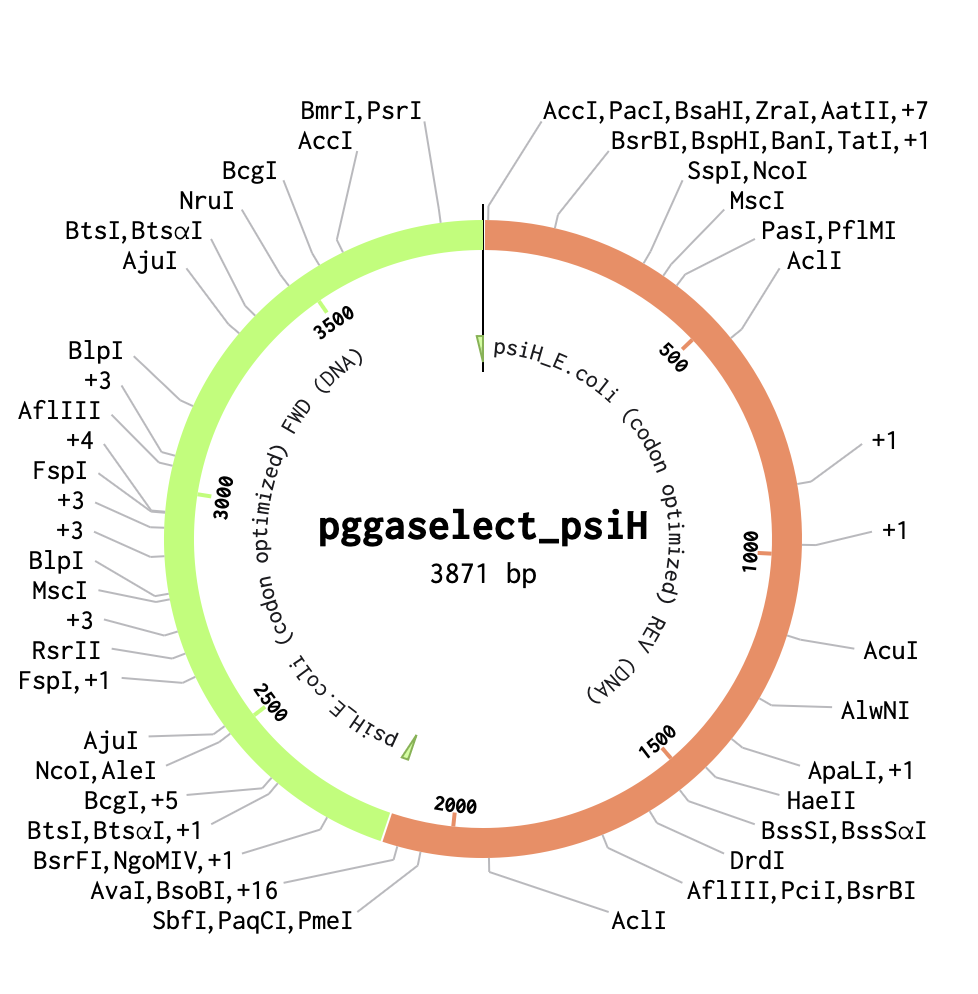
Part 2: Model this assembly method with Benchling or Asimov Kernel!
To start, I selected the pGGA backbone (a common domesticated vector used for Golden Gate assembly) to use in my construct. After obtaining the sequence and adding it to Benchling as a circular plasmid map, I inserted the PsiH codon-optimised enzyme sequence from a previous assignment. This enzyme, involved in psilocybin biosynthesis, aligns with the concept for my Project 1 final design.
PGGAselect: 2020bp DNA Full sequence and details of the cloning vector are available at: https://www.neb.com/en/-/media/nebus/page-images/tools-and-resources/interactive-tools/dna-sequences-and-maps/text-documents/pggaselectgbk.txt?rev=5882d0f571c34713a43c67e6d64c25ea&hash=5C1F89A2A1E3E98F94CA667DE322DDF0