Subsections of Homework
Week 1 HW: Principles and Practices
1) Describe a biological engineering application or tool you want to develop and why.
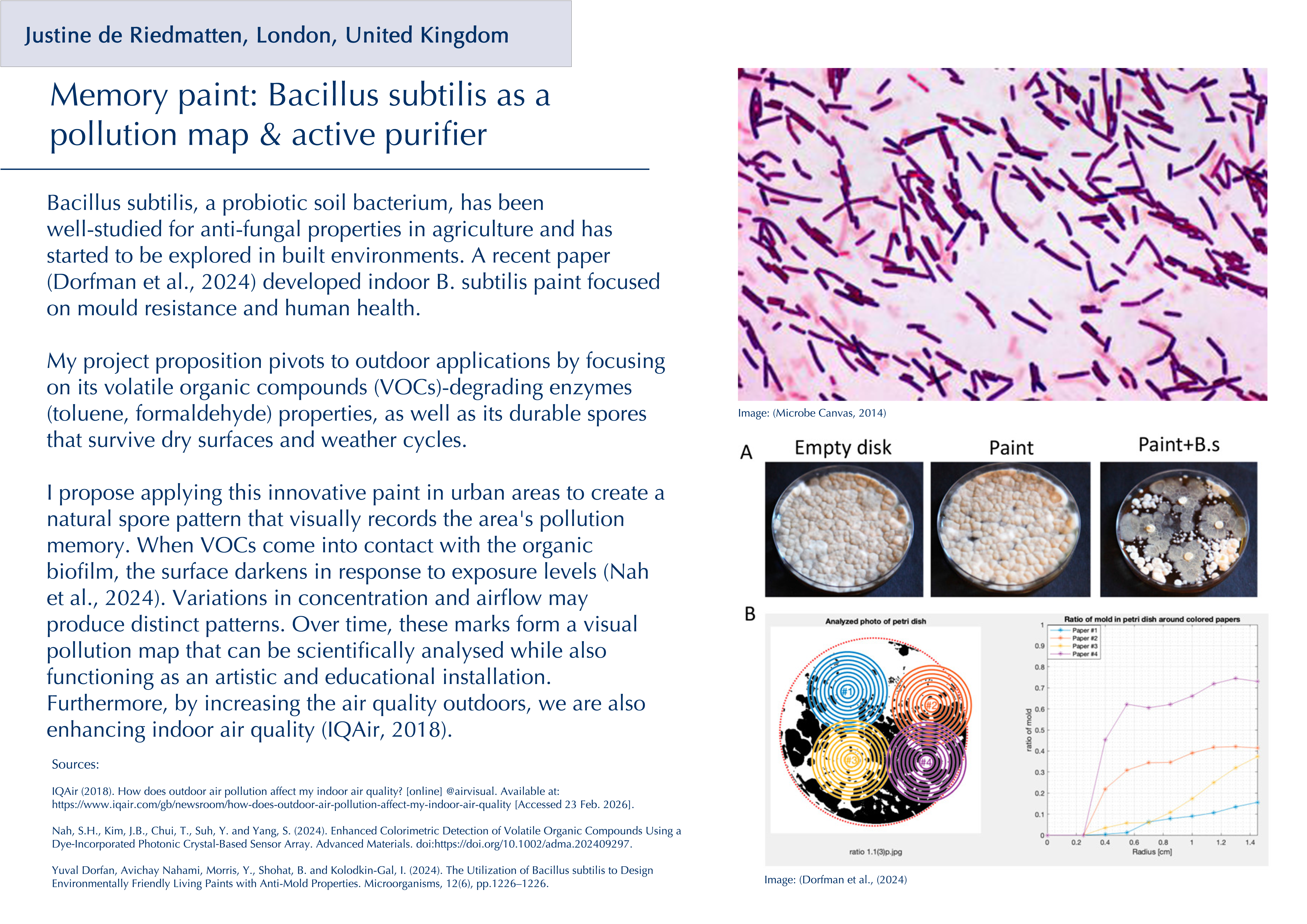
Throughout my previous research as a new biodesigner, I have been particularly drawn to two themes: the intricate relationship between the gut microbiota and its effects on human physical and mental health, and the fascinating world of fungi and their broader implications for planetary health. I have deepened my research into psilocybin and related tryptamine alkaloids (substances that activate serotonin 5-HT receptors), which are already being investigated clinically for neuropsychiatric conditions (Xi et al., 2023), but what especially interests me is emerging evidence that some of their effects may be mediated by the gut microbiota rather than the brain alone (Caspani et al., 2024). There is still a lack of research on the precise impact that serotonergic psychedelics have on the structure and composition of the gut microbiota, even though animal studies suggest possible links (Császár-Nagy et al., 2022). A survey of wild‑type rats has shown changes in the abundance of spore‑forming gut bacteria following oral administration of serotonin, suggesting that serotonergic compounds such as psilocybin could directly or indirectly reshape microbial communities (Fung et al., 2019).
Beyond psilocybin, the traditional use of other psychoactive organisms also points to underexplored antimicrobial and immunomodulatory effects. For instance, the peyote cactus (Lophophora williamsii) has been reported to display activity against multiple penicillin‑resistant strains of Staphylococcus aureus (McCleary et al., 1960). Such examples raise broader questions about how these organisms interact with microbes and the host’s physiology. These kinds of plants, fungi, and compounds have long histories in traditional medicine yet remain only partially understood within Western biomedical frameworks (Doesburg-van Kleffens et al., 2023). I believe that the intersection of psychedelics, microbiota, and immune function is a promising area for synthetic biology yet to be further explored, with potential not only for already studied mental health conditions but also for inflammatory bowel diseases (IBD) (Qureshi et al., 2025). Rather than viewing these compounds solely through a brain‑centric lens, it would be interesting to investigate their therapeutic potential through the gut microbiota as a primary interface rather than a secondary effect.
Psychedelic‑assisted therapy is currently highly regulated, expensive, and accessible to very few people (Rea et Wallace, 2021). From a biodesign and synthetic biology perspective, biosynthesising psilocybin in a bacterial host such as Escherichia coli through recombinant DNA technology (Keller et al., 2025) could potentially provide a more controllable, scalable, and eventually more affordable route to producing psilocybin or related analogues.
In the context of the HTGAA course, I therefore propose to focus on a minimal and safe fragment of this broader concept, with the following objectives:
- Investigate how natural serotonergic psychedelics and related natural compounds might interact with the gut microbiota- immune axis, with a focus on IBD and gut inflammations.
- Design a conceptual framework for microbial psilocybin production in E. coli (Adams et al., 2019) as a platform for future microbiome‑targeted therapies.
- Prototype an E. coli experimental system expressing a heterologous lyase enzyme from a psilocybin-related tryptamine biosynthetic pathway (Abrahms et al., 2025), assembled via PCR-based cloning methods.
- Apply CRISPR‑based editing to modulate the expression of the heterologous enzyme and characterise the resulting change in a readout (through colour or fluorescence) to determine if the right sequence in E.Coli is being targeted.
- Develop a simple in silico pathway model to explore where metabolic bottlenecks might occur in a full psilocybin biosynthesis pathway in E. coli (Irvine et al., 2023).
- Reflect on ethical, regulatory, and accessibility questions around engineering psychoactive compounds and further document how these considerations shape the design of the project.
Reference List:
- Abrahms, Z.N., Sen, A.K. and Jones, J.A. (2025).Pathway engineering for the biosynthesis of psychedelics. Current Opinion in Biotechnology, 94, pp.103314–103314. doi:https://doi.org/10.1016/j.copbio.2025.103314.
- Adams, A.M., Kaplan, N.A., Wei, Z., Brinton, J.D., Monnier, C.S., Enacopol, A.L., Ramelot, T.A. and Jones, J.A. (2019). In vivo production of psilocybin in E. coli. Metabolic Engineering, [online] 56, pp.111–119. doi:https://doi.org/10.1016/j.ymben.2019.09.009.
- Caspani, G., Ruffell, S.G.D., Tsang, W., Netzband, N., Rohani-Shukla, C., Swann, J.R. and Jefferies, W.A. (2024). Mind over matter: the microbial mindscapes of psychedelics and the gut-brain axis. Pharmacological Research, 207, p.107338. doi:https://doi.org/10.1016/j.phrs.2024.107338.
- Császár-Nagy, N., Bob, P. and Bókkon, I. (2022). A Multidisciplinary Hypothesis about Serotonergic Psychedelics. Is it Possible that a Portion of Brain Serotonin Comes From the Gut? Journal of Integrative Neuroscience, 21(5), p.148. doi:https://doi.org/10.31083/j.jin2105148.
- Doesburg-van Kleffens, M., Zimmermann-Klemd, A.M. and Gründemann, C. (2023). An Overview on the Hallucinogenic Peyote and Its Alkaloid Mescaline: The Importance of Context, Ceremony and Culture. Molecules, [online] 28(24), p.7942. doi:https://doi.org/10.3390/molecules28247942.
- Fung, T.C., Vuong, H.E., Luna, C.D.G., Pronovost, G.N., Aleksandrova, A.A., Riley, N.G., Vavilina, A., McGinn, J., Rendon, T., Forrest, L.R. and Hsiao, E.Y. (2019). Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nature Microbiology. doi:https://doi.org/10.1038/s41564-019-0540-4.
- Keller, M.R., McKinney, M.G., Sen, A.K., Guagliardo, F.G., Hellwarth, E.B., Islam, K.N., Kaplan, N.A., Gibbons, W.J., Kemmerly, G.E., Meers, C., Wang, X. and Jones, J.A. (2025). Psilocybin biosynthesis enhancement through gene source optimization. Metabolic Engineering, [online] 91, pp.119–129. doi:https://doi.org/10.1016/j.ymben.2025.04.003.
- McCleary, J.A., Sypherd, P.S. and Walkington, D.L. (1960). Antibiotic activity of an extract of peyote (Lophophora Williamii (Lemaire) Coulter). Economic Botany, 14(3), pp.247–249. doi:https://doi.org/10.1007/bf02907956.
- Qureshi, O., Cowley, J., Pegg, A., Cooper, A.J., Gordon, J., Brady, C.A., Belli, A., Butterworth, S., Upthegrove, R., Andrews, N. and Barnes, N.M. (2025). Are we hallucinating or can psychedelic drugs modulate the immune system to control inflammation? British journal of pharmacology, [online] p.10.1111/bph.70138. doi:https://doi.org/10.1111/bph.70138.
- Xi, D., Berger, A., Shurtleff, D., Zia, F.Z. and Belouin, S. (2023). National Institutes of Health psilocybin research speaker series: State of the science, regulatory and policy landscape, research gaps, and opportunities. Neuropharmacology, [online] 230, p.109467. doi:https://doi.org/10.1016/j.neuropharm.2023.109467.
2) Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Policies Goal: To ensure microbial psilocybin production for microbiome-targeted IBD therapy/medication prioritises safety, equitable access despite microbiome variability, respect for traditional knowledge and transparency to build public trust and prevent harm.
- Risk assessment for gut-specific harm: to safely develop this kind of medication or therapy, it will require a longitudinal patient gut-microbome monitoring prior to and post-dosing to refine safety protocols and the detection of any adverse shifts.
- Accessibility whilst respecting indviduality of the gut microbiome: creating an accessible medication targeting something so unique to the individual will be a challenge. There are now affordable baseline microbiome profiling kits (e.g. Feel Gut - Microbiome Test Kit - 250£) to screen high-risk patients pre-treatment, while developing 2–3 engineered E. coli strains producing dosage variants (low/medium/high) based on symptom severity or gut sensitivity.
- Value of traditional knowledge: crediting and collaborating with communities that have historically used psychedelic organisms for medical approaches through benefit-sharing agreements (BSAs).
- Transparency and accountability: Create traceable audit trails for strains and products to ensure responsibility if misuse occurs and build public trust through open data sharing.
Sources:
- DiliTrust (2025). Understanding Audit Trails: Implementation, Types, and Best Practices. [online] Dilitrust. Available at: https://www.dilitrust.com/audit-trail/ [Accessed 7 Feb. 2026].
- FeelGut (2026). Gut Microbiome Health Test. [online] Feel Gut. Available at: https://feelgut.co.uk/products/gut-microbiome-health-test?gad_source=1&gad_campaignid=21790867659&gbraid=0AAAAAqF2ctBqNuD8W2LgDdGB8xmh-dJkI&gclid=Cj0KCQiAhaHMBhD2ARIsAPAU_D4EnlJcLWOuT0Ol-Gz0qDHbGOnE0a_08EaWu5OmccHvfelHGAJLgeUaAmI0EALw_wcB [Accessed 7 Feb. 2026].
- Ogilvy.com.au (2024). Shared Success: What is benefit sharing and why does it matter? - Insight - MinterEllison. [online] Minterellison.com. Available at: https://www.minterellison.com/articles/what-is-benefit-sharing-and-why-does-it-matter [Accessed 7 Feb. 2026].
- Secretariat of the Convention on Biological Diversity (2011). Convention on Biological Diversity: ABS Theme Access and benefit-sharing. [online] Available at: https://www.cbd.int/abs/infokit/revised/web/factsheet-abs-en.pdf [Accessed 7 Feb. 2026].
3) Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
To meet these policy goals, I propose the following three potential governance actions:
- COLLECTIVE DATABASE:
Call for the development of a collective database where all biologists who have worked on the E. Coli psiloyibin strains would share their protocol and CRISPR history. This calls for the collaboration of universities and researchers worldwide for future research to optimise time by limiting repetitions and consequently reducing costs. The development of controlled access will also be necessary to ensure the information in the database is used ethically and for the correct reasons.- PATIENT GUT-MICROBIOME SCREENING:
Call for the pre-screening of patients’ gut microbiome to ensure appropriate candidates. This also calls for collaboration with a pharmaceutical partner to deliver these kits directly to potential patients or to hospitals and institutions where the samples will be analysed to determine whether they are suitable for treatment of the patient's gut microbiome. This will ensure safety and a reduction of side effects, and better results by than accordingly selecting which strain would benefit the patient the most. There are still some risks considering ‘false negative’ testing, which could therefore make it difficult to fully confirm the appropriate treatment, as well as the potential increase in price of the treatment if the gut-microbiome data were to become a Pharma IP monopoly, limiting access due to often increasing prices (Dosi et al., 2023).
- CROSS COLLABORATION
Call for the implementation of royalty funds for indigenous cultures that have contributed to the development of this project with their knowledge on the medical application of psychedelics to ensure an ethical and transparent development of the project. Furthermore, developing a wider web of collaboration with governments, scientists, hospitals, psychologists, and doctors to meet and discuss/review strains, trials and dosages. This complex cross-collaboration would only be possible with an FDA approval of the usage of such psychedelics to begin with, which would require a long time, many trials and solid evidence of success at a high success rate level (Lamkin, 2022).
References:
- Dosi, G., Marengo, L., Staccioli, J. and Virgillito, M.E. (2023). Big Pharma and Monopoly capitalism: a long-term View.Structural Change and Economic Dynamics, [online] 65, pp.15–35. doi:https://doi.org/10.1016/j.strueco.2023.01.004.
- Lamkin, M. (2021). Prescription Psychedelics: The Road from FDA Approval to Clinical Practice. The American Journal of Medicine, 135(1). doi:https://doi.org/10.1016/j.amjmed.2021.07.033.
4) Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Scale: 1-3 (1: Most effective, 2: Moderately effective, 3: Least effective, or N/A)
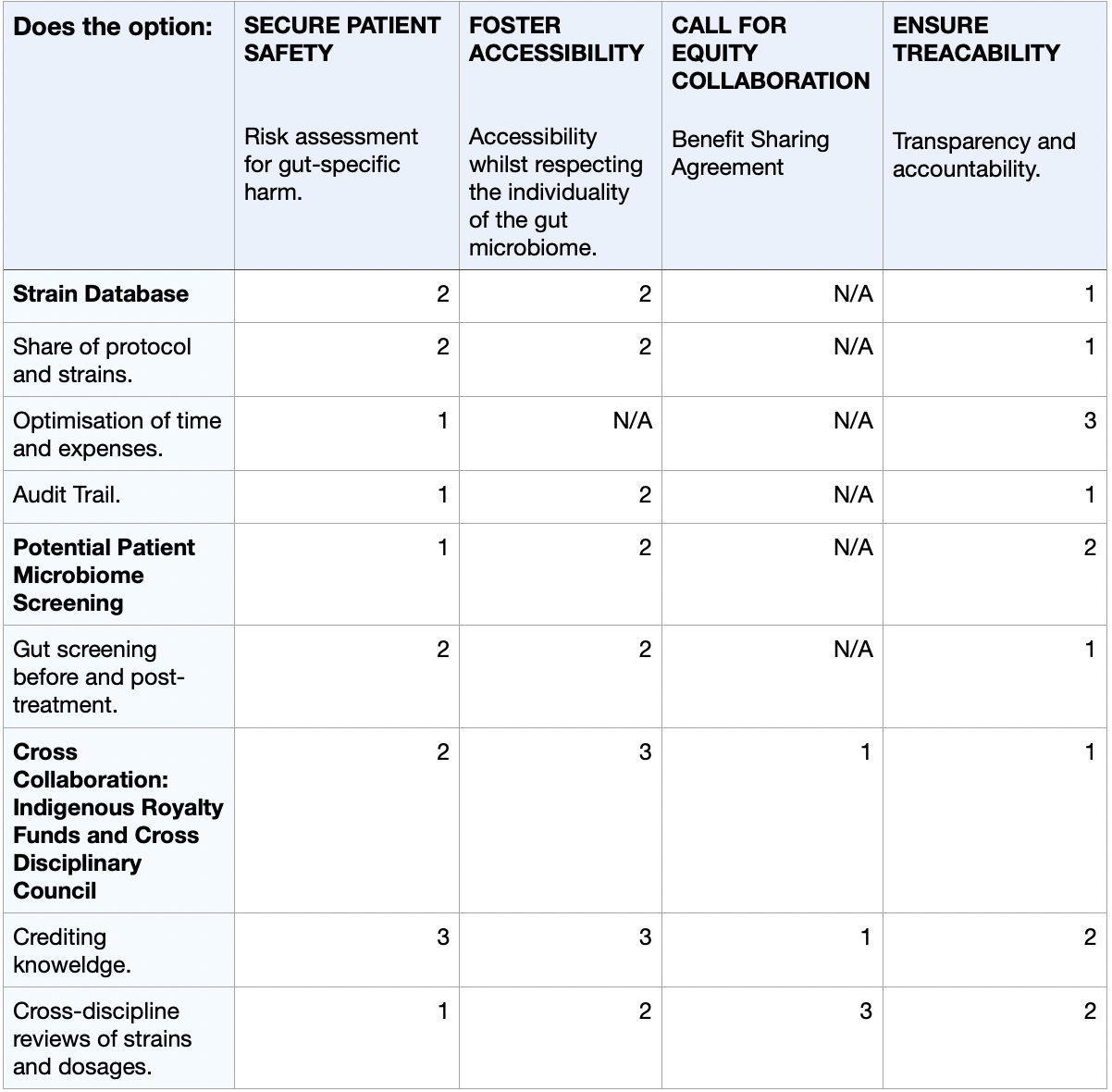
5) Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
I propose the prioritisation of the strain database, as well as the microbiome pre-screening, as the foundational governance actions for the development of microbial psilocybin. Given the strict regulations surrounding the use of psychedelic drugs in the medical world, transparency, feasability and safety are essential for governmental approval. Strategically, developing this project in countries like Switzerland or the Netherlands would offer optimal conditions. In Zurich and Geneva (Switzerland), research on the medical use of psychedelic compounds, such as psilocybin or LSD, in clinical psychiatric trials is continuously growing (Elçi, 2025). Additionally, their biotechnological infrastructure would allow for an ideal environment to conduct the development of such a project. On the other hand, the legal use of psilocybin truffles in the Netherlands should also be considered as a place of interest. Overlooking this as phases, the initial phase should consist of the launch of the strain database for safety and optimisation, followed by mandating affordable gut-microbiome screening. Lastly, validating via in-silico computational models the prediction of human gut-microbiome-psilocybin interactions through pharmacokinetic data (Grogan and Preuss, 2023) to build towards the potential commercialisation of such a product.
List of References:
- Grogan, S. and Preuss, C. (2023). Pharmacokinetics. [online] PubMed. Available at: https://www.ncbi.nlm.nih.gov/books/NBK557744/ [Accessed 9 Feb. 2026].
- Aylin Elçi (2025). Switzerland is home to Europe’s only psychedelics treatment. [online] SWI swissinfo.ch. Available at: https://www.swissinfo.ch/eng/multinational-companies/switzerland-is-home-to-europes-only-psychedelics-treatment/89195943 [Accessed 7 Feb. 2026].
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson:
1)Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate ratio of polymerase is 1:10^6 base pairs, equivalent to one mistake every million bases. In comparison, the human genome consists of approximately 3 billion base pairs (3 x 10^9) (National Human Genome Research Institute, 2026), meaning each replication introduces around 3000 errors (3x10^9 x 10^6), leading to genetic mutations. In synthetic biology, high-fidelity DNA polymerases have proven to have much lower error rates for PCR cloning of long biosynthetic pathways. These polymerases possess 3’ to 5’ exonuclease proofreading domains that detect mismatches through structural perturbations and immediately remove them before continuing synthesis. (Clent Life Science, 2024).
2)How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice, what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
There are astronomically many possible ways to code for an average human protein. For example, a 300-amino-acid protein may have 10^100 different synonymous codon combinations (Rajbanshi and Guruacharya, 2025). However, most fail in practice because cells prefer specific codons (codon usage bias), which causes rare codons to slow down the tRNA process. Consequently, this creates nonuniform ribosome decoding rates on mRNAs and in turn disrupts the contranslational protein folding process, which is essential for proper protein function (Liu et al., 2021).
References:
- Bates, S. (2019). Base Pair. [online] Genome.gov. Available at: https://www.genome.gov/genetics-glossary/Base-Pair [Accessed 8 Feb. 2026].
- Clent Life Science (2024). High-fidelity DNA Polymerases & When to use them. [online] Clent Life Science. Available at: https://clentlifescience.co.uk/high-fidelity-dna-polymerases-and-when-to-use-them/ [Accessed 8 Feb. 2026].
- Liu, Y., Yang, Q. and Zhao, F. (2021). Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annual Review of Biochemistry, 90(1), pp.375–401. doi:https://doi.org/10.1146/annurev-biochem-071320-112701.
- Rajbanshi, B. and Guruacharya, A. (2025). codonGPT: reinforcement learning on a generative language model enables scalable mRNA design. Nucleic Acids Research, [online] 53(22). doi:https://doi.org/10.1093/nar/gkaf1345.
Homework Questions from Dr. LeProust:
1)What’s the most commonly used method for oligo synthesis currently?
Solid-phase phosphoramidite is currently the most commonly used method for oligo synthesis. It is a cyclical four-step phosphoramidite synthesis method (McLaughlin, 2025), which was developed in 1981 by Marvin Caruthers.
2) Why is it difficult to make oligos longer than 200nt via direct synthesis?
Making oligos longer than 200 nucleotides via direct synthesis is difficult due to cumulative yield losses. During this synthesising method, one nucleotide is added at a time. Although there is a high success rate (99%), each added nucleotide will contribute to yield loss (ATDBio Ltd, 2005). At the 200th nucleotide added (0.99^200), the overall yield production will have a new success rate of 0.135%, resulting in extremely low yield and consequently resulting in a waste of expensive reagents and purification time (Mühlegger,2025).
3) Why can’t you make a 2000bp gene via direct oligo synthesis?
As mentioned in the last question, chemical DNA synthesis adds only one nucleotide at a time, at a 99% success rate. Taking into consideration that we only use one strand of DNA for the synthesis, 0.99^2000 would reduce the success rate by 0.0002% yield for a 2000bp gene. Considering this extremely low amount, it would make it practically impossible for the yield to be detected for the purification process.
References:
- ATDBio Ltd (2005). ATDBio - Solid-phase oligonucleotide synthesis. [online] atdbio.com. Available at: https://atdbio.com/nucleic-acids-book/Solid-phase-oligonucleotide-synthesis [Accessed 8 Feb. 2026].
- Glen Research (2026). Glen Report 21.211 - TECHNICAL BRIEF – Synthesis of Long Oligonucleotides. [online] Glenresearch.com. Available at: https://www.glenresearch.com/reports/gr21-211 [Accessed 8 Feb. 2026].
- McLaughlin, L. (2025). What Is Oligonucleotide Synthesis? Phosphoramidite oligonucleotide synthesis. [online] Biotechnologyreviews.com. Available at: https://www.biotechnologyreviews.com/p/what-is-oligonucleotide-synthesis [Accessed 8 Feb. 2026].
- Michael Mühlegger (2025). Oligonucleotide manufacturing – challenges & solutions. [online] Single Use Support: Pionneering Biopharma. Available at: https://www.susupport.com/blogs/biopharmaceutical-products/oligonucleotide-manufacturing-challenges-solutions [Accessed 8 Feb. 2026].
Homework Question from George Church:
1) What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The essential amino acids are: Histidine (His), Isoleucine (Ile), Leucine (Leu), Lysine (Lys), Methionine (Met), Phenylalanine (Phe), Threonine (Thr), Tryptophan (Trp), Valine (Val), and Arginine (ARG). (Lopez and Mohiuddin, 2024).
The Lysine Contingency was a genetically engineered fail‑safe created by Dr Henry Wu in the late 1980s to prevent Jurassic Park’s dinosaurs from synthesising lysine. The idea was that park staff would have to supplement the animals with lysine to keep them alive, and if any dinosaur escaped Isla Nublar, it would die without this dietary supply. This was intended as a strategy to protect outside ecosystems. However, this design was fundamentally flawed. In 1997, Dr Sarah Harding showed that the dinosaurs could obtain enough lysine from their environment through their normal diet, making the contingency ineffective in practice. Since all animals already depend on dietary lysine in the first place, the Contingency never provided the precise control the park claimed (Jurassic-Pedia, 2024).
I agree with Dr Sorkin’s view that, even though these dinosaurs were cloned, they still deserved rights as living animals. Humans chose to recreate this extinct species, so it seems ethically wrong that we should also reserve the right to let them die for our convenience or as a simplistic safety measure. This raises a wider question about how much power humans should have over organisms they create or modify. In my view, these ethical discussions should happen before such cloning work begins, not after problems appear. Overall, the Lysine Contingency feels poorly thought through and aimed at solving a problem that should never have been framed that way in the first place.
References:
- Lopez, M.J. and Mohiuddin, S.S. (2024). Biochemistry, Essential Amino Acids. [online] PubMed. Available at: https://www.ncbi.nlm.nih.gov/books/NBK557845/ [Accessed 8 Feb. 2026].
- Jurassic-Pedia (2024). Lysine Contingency (S/F) / (S/F-T/G) – Jurassic-Pedia. [online] Jurassic-Pedia. Available at: https://www.jurassic-pedia.com/lysine-contingency-sf/ [Accessed 9 Feb. 2026].
Week 2 HW: Dna-Read-Write-and-Edit
Part 1: Benchling & In-silico Gel Art
I simulated the Restriction Enzyme Digestion in Benchling to create a design. I found it initially difficult to visualise patterns or images with the 7 restriction enzymes. I therefore decided to mix certain enzymes in the same wells to generate more DNA fragments and explore shapes further.

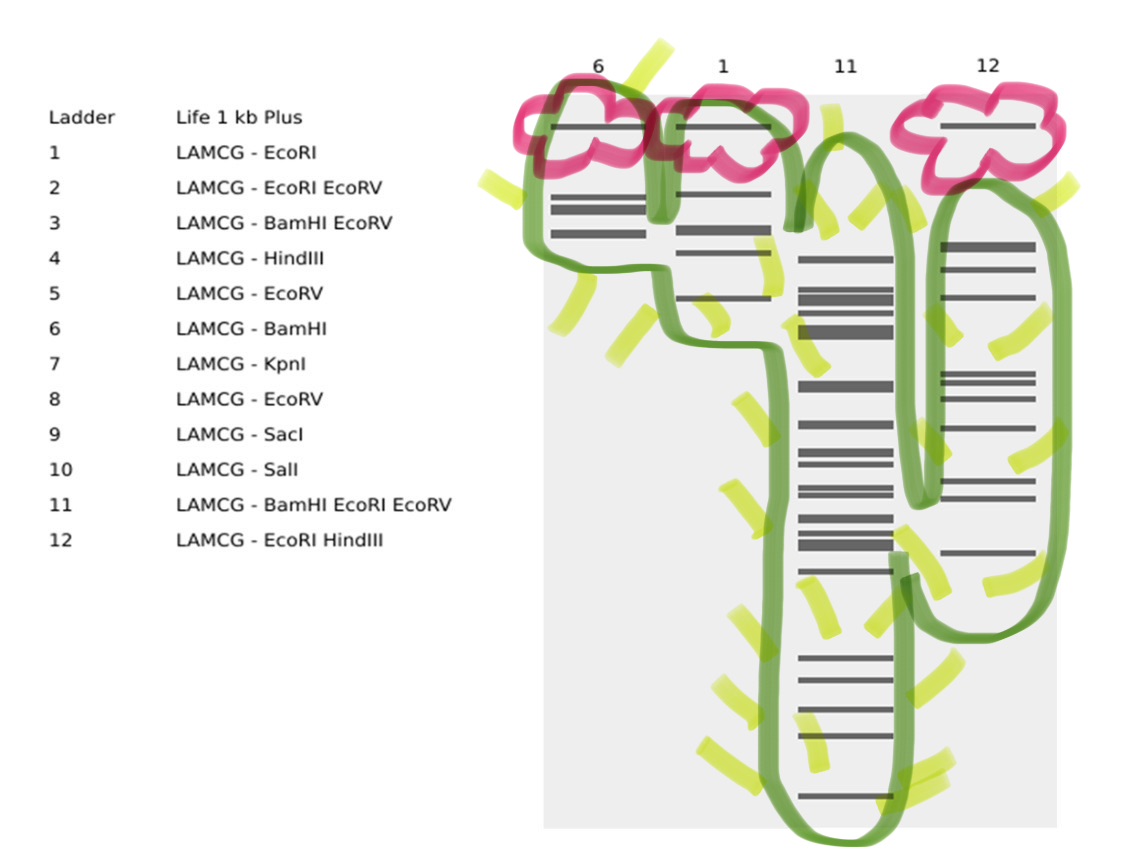
Part 3: DNA Design Challenge
3.1 Which protein have you chosen and why? Using one of the tools described in the recitation (NCBI, UniProt, Google), obtain the protein sequence for the protein you chose.Name of protein: psiH (tryptamine 4-monooxygenase)
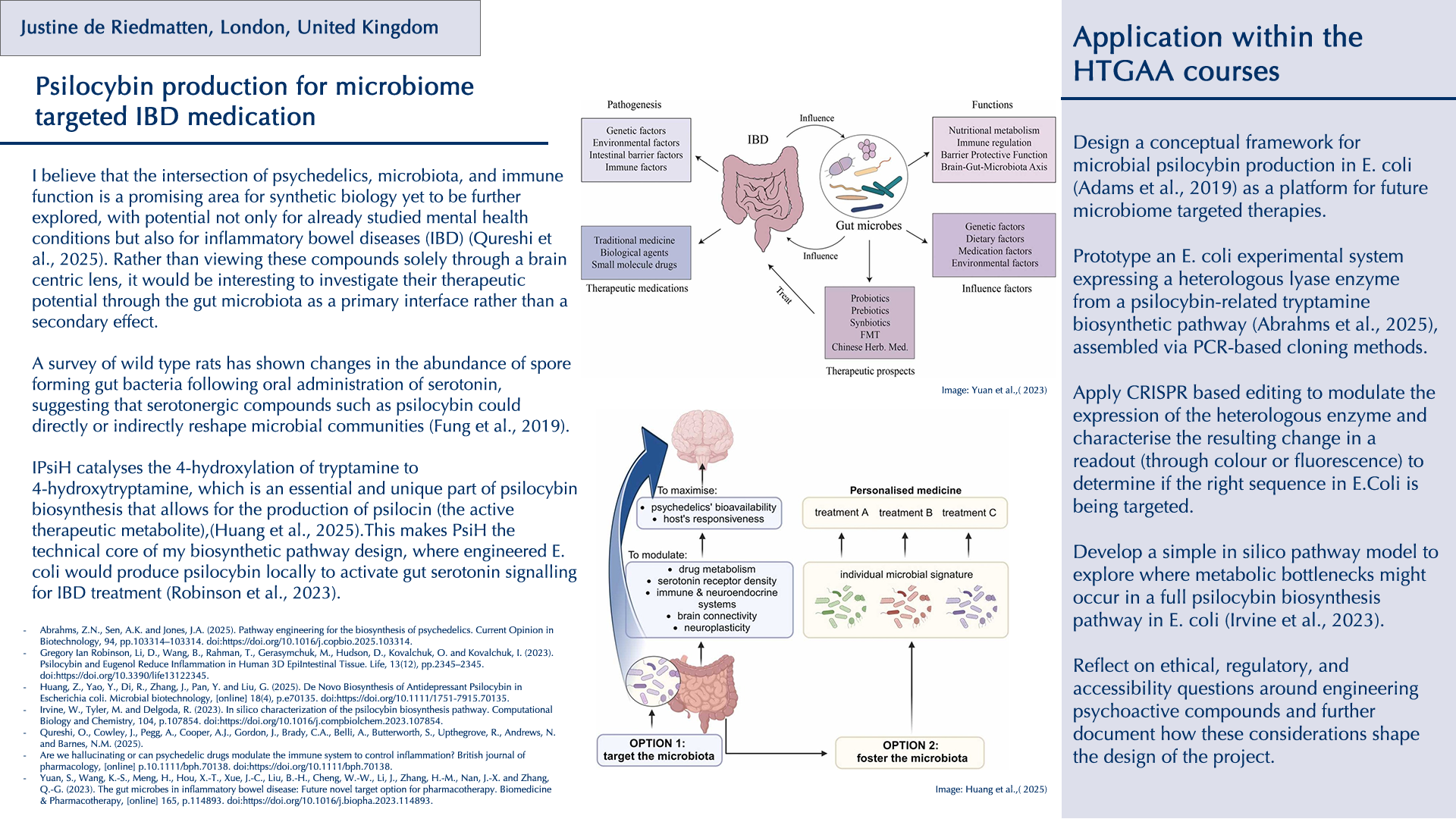
I chose this specific protein as it relates to my project idea from homework 1. PsiH catalyses the 4-hydroxylation of tryptamine to 4-hydroxytryptamine, which is an essential and unique part of psilocybin biosynthesis that allows for the production of psilocin (the active therapeutic metabolite). This P450 enzyme (psiH) acts as a critical rate‐limiting step of psilocybin production. Furthermore, it needs exact heme binding and substrate fit, which is rare in nature and tough to engineer in E. coli (Huang et al., 2025). This makes PsiH the technical core of my biosynthetic pathway design from Homework 1, where engineered E. coli would produce psilocybin locally to activate gut serotonin signalling for IBD treatment (Robinson et al., 2023).
Refrences:
Huang, Z., Yao, Y., Di, R., Zhang, J., Pan, Y. and Liu, G. (2025). De Novo Biosynthesis of Antidepressant Psilocybin in Escherichia coli.Microbial biotechnology, [online] 18(4), p.e70135. doi:https://doi.org/10.1111/1751-7915.70135.Gregory Ian Robinson, Li, D., Wang, B., Rahman, T., Gerasymchuk, M., Hudson, D., Kovalchuk, O. and Kovalchuk, I. (2023). Psilocybin and Eugenol Reduce Inflammation in Human 3D EpiIntestinal Tissue.Life, 13(12), pp.2345–2345. doi:https://doi.org/10.3390/life13122345.Amino Acid Protein Sequence:
MIAVLFSFVIAGCIYYIVSRRVRRSRLPPGPPGIPIPFIGNMFD
MPEESPWLTFLQWGRDYNTDILYVDAGGTEMVILNTLETITDLLEKRGSIYSGRLEST
MVNELMGWEFDLGFITYGDRWREERRMFAKEFSEKGIKQFRHAQVKAAHQLVQQLTKT
PDRWAQHIRHQIAAMSLDIGYGIDLAEDDPWLEATHLANEGLAIASVPGKFWVDSFPS
LKYLPAWFPGAVFKRKAKVWREAADHMVDMPYETMRKLAPQGLTRPSYASARLQAMDL
NGDLEHQEHVIKNTAAEVNVGGGDTTVSAMSAFILAMVKYPEVQRKVQAELDALTNNG
QIPDYDEEDDSLPYLTACIKELFRWNQIAPLAIPHKLMKDDVYRGYLIPKNTLVFANT
WAVLNDPEVYPDPSVFRPERYLGPDGKPDNTVRDPRKAAFGYGRRNCPGIHLAQSTVW
IAGATLLSAFNIERPVDQNGKPIDIPADFTTGFFRHPVPFQCRFVPRTEQVSQSVSGP
Source:National Library of Medicine (2026). Psilocybe cubensis strain FSU 12409 putative monooxygenase (psiH) gene - Nucleotide - NCBI. [online] Nih.gov. Available at: https://www.ncbi.nlm.nih.gov/nuccore/MF000993 [Accessed 16 Feb. 2026].3.2 DNA Reverse Translation:
atgattgcggtgctgtttagctttgtgattgcgggctgcatttattatattgtgagccgc
cgcgtgcgccgcagccgcctgccgccgggcccgccgggcattccgattccgtttattggc
aacatgtttgatatgccggaagaaagcccgtggctgacctttctgcagtggggccgcgat
tataacaccgatattctgtatgtggatgcgggcggcaccgaaatggtgattctgaacacc
ctggaaaccattaccgatctgctggaaaaacgcggcagcatttatagcggccgcctggaa
agcaccatggtgaacgaactgatgggctgggaatttgatctgggctttattacctatggc
gatcgctggcgcgaagaacgccgcatgtttgcgaaagaatttagcgaaaaaggcattaaa
cagtttcgccatgcgcaggtgaaagcggcgcatcagctggtgcagcagctgaccaaaacc
ccggatcgctgggcgcagcatattcgccatcagattgcggcgatgagcctggatattggc
tatggcattgatctggcggaagatgatccgtggctggaagcgacccatctggcgaacgaa
ggcctggcgattgcgagcgtgccgggcaaattttgggtggatagctttccgagcctgaaa
tatctgccggcgtggtttccgggcgcggtgtttaaacgcaaagcgaaagtgtggcgcgaa
gcggcggatcatatggtggatatgccgtatgaaaccatgcgcaaactggcgccgcagggc
ctgacccgcccgagctatgcgagcgcgcgcctgcaggcgatggatctgaacggcgatctg
gaacatcaggaacatgtgattaaaaacaccgcggcggaagtgaacgtgggcggcggcgat
accaccgtgagcgcgatgagcgcgtttattctggcgatggtgaaatatccggaagtgcag
cgcaaagtgcaggcggaactggatgcgctgaccaacaacggccagattccggattatgat
gaagaagatgatagcctgccgtatctgaccgcgtgcattaaagaactgtttcgctggaac
cagattgcgccgctggcgattccgcataaactgatgaaagatgatgtgtatcgcggctat
ctgattccgaaaaacaccctggtgtttgcgaacacctgggcggtgctgaacgatccggaa
gtgtatccggatccgagcgtgtttcgcccggaacgctatctgggcccggatggcaaaccg
gataacaccgtgcgcgatccgcgcaaagcggcgtttggctatggccgccgcaactgcccg
ggcattcatctggcgcagagcaccgtgtggattgcgggcgcgaccctgctgagcgcgttt
aacattgaacgcccggtggatcagaacggcaaaccgattgatattccggcggattttacc
accggcttttttcgccatccggtgccgtttcagtgccgctttgtgccgcgcaccgaacag
gtgagccagagcgtgagcggcccg
Source
The Sequence Manipulation Suite (2024).Reverse Translate. [online] www.bioinformatics.org. Available at: https://www.bioinformatics.org/sms2/rev_trans.html [Accessed 16 Feb. 2026].3.3 Codon optimisation
In your own words, describe why you need to optimise codon usage. Which organism have you chosen to optimise the codon sequence for and why?
Although different codons can code for the same amino acid, each species/organism has a bias for its codon preferences. This is done by changing/optimising the DNA codon sequence (not the amino-acid sequence) of the protein in order to match the codon preferences of the host organism (Cheema et al., 2022). If I were to take a human gene and insert it into a bacterium, it might use certain codons that the bacterium wouldn’t/would rarely use. This, in turn, makes the translation process slower or incomplete, resulting in a low protein yield (Creative BioLabs,2025). Therefore, by optimising the sequence with a specific host, I can make the translation process faster and more reliable.
For this specific exercise, I chose to optimise the psiH protein for E. Coli. I made this choice because E. coli is one of the most commonly used hosts for genetic engineering due to its rapid culture rate, simple nutritional needs and well-understood genetics (Adamczyk and Reed, 2017). Additionaly it is relatively cheap to culture
(Francis and Page,2010). In relation to Homework 1, during the time of this course, E. coli is an appropriate host for prototyping the psilocybin pathway to conceptually extend toward microbiome-targeted therapies.
sources
Creative BioLabs (2025). Codon Optimization and Its Impact on mRNA Translation Efficiency. [online] Creative-biolabs.com. Available at: https://ribosome.creative-biolabs.com/codon-optimization-and-its-impact-on-mrna-translation-efficiency.htm [Accessed 17 Feb. 2026].Cheema, N., Georgios Papamichail and Dimitris Papamichail (2022). Computational tools for synthetic gene optimization. Elsevier eBooks, pp.171–189. doi:https://doi.org/10.1016/b978-0-12-824469-2.00018-x.Adamczyk, P.A. and Reed, J.L. (2017). Escherichia coli as a model organism for systems metabolic engineering. Current Opinion in Systems Biology, [online] 6, pp.80–88. doi:https://doi.org/10.1016/j.coisb.2017.11.001.Francis, D.M. and Page, R. (2010). Strategies to optimize protein expression in E. coli. Current protocols in protein science, [online] Chapter 5(1), p.Unit 5.24.1-29. doi:https://doi.org/10.1002/0471140864.ps0524s61.What technologies could be used to produce this protein from your DNA? Describe in your own words how the DNA sequence can be transcribed and translated into a protein.
A variety of technologies could be used to produce the psiH protein from its DNA. One of the most suitable that we have discussed in lectures would be Gibson Assembly. It would allow us to stitch together multiple DNA fragments (promoter, Ribosome binding site, my optimised DNA sequence of psiH, terminator…) inside the plasmid of the E. Coli without the use of restriction enzymes. You may also use external platforms like Twist Bioscience to order your full plasmid by giving them the appropriate optimised sequence.
The DNA sequence may be transcribed and translated into a protein with the following steps:
Transcription: The RNA Polymerase will bind to the promoter sequence of the DNA. The double helix of the DNA unwinds and allows for the RNA polymerase to create a complementary mRNA strand following the base-pairing rules.Translation: Ribosomes then bind to the mRNA at the RBS near the start codon (ATG). tRNA molecules then match their anticodons to the mRNA codons, resulting in specific amino acids. Lastly, the ribosomes will continue to read through the sequence until a stop codon is reached, causing the release of the protein chain.Sources:
Nature Education (2014). The Information in DNA Determines Cellular Function via Translation | Learn Science at Scitable. [online] Nature.com. Available at: https://www.nature.com/scitable/topicpage/the-information-in-dna-determines-cellular-function-6523228/ [Accessed 15 Feb. 2026].Webster, M.W. and Weixlbaumer, A. (2021). The intricate relationship between transcription and translation. Proceedings of the National Academy of Sciences, [online] 118(21). doi:https://doi.org/10.1073/pnas.2106284118.Part 4: Prepare a Twist DNA Synthesis Order
Building my DNA Insert Sequence
I began by optimising my psiH translated protein DNA sequence in Benchling with a linear topology and optimising it for E. coli. I then added the reading direction (forward), and the given DNA sequences highlighted in the homework (Promoter, RBS, Coding Sequence, 7x His Tag, Stop Codon, Terminator).
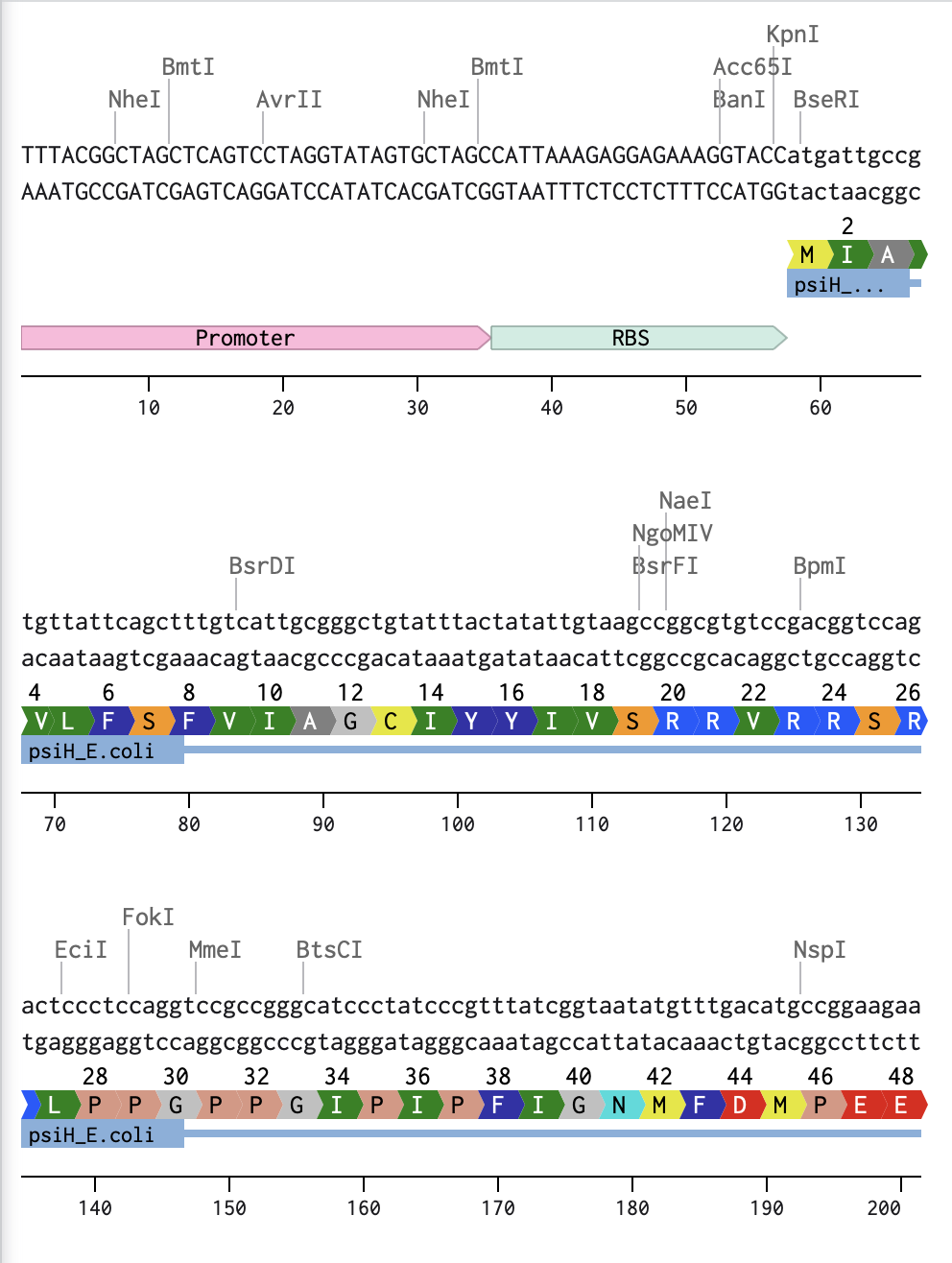
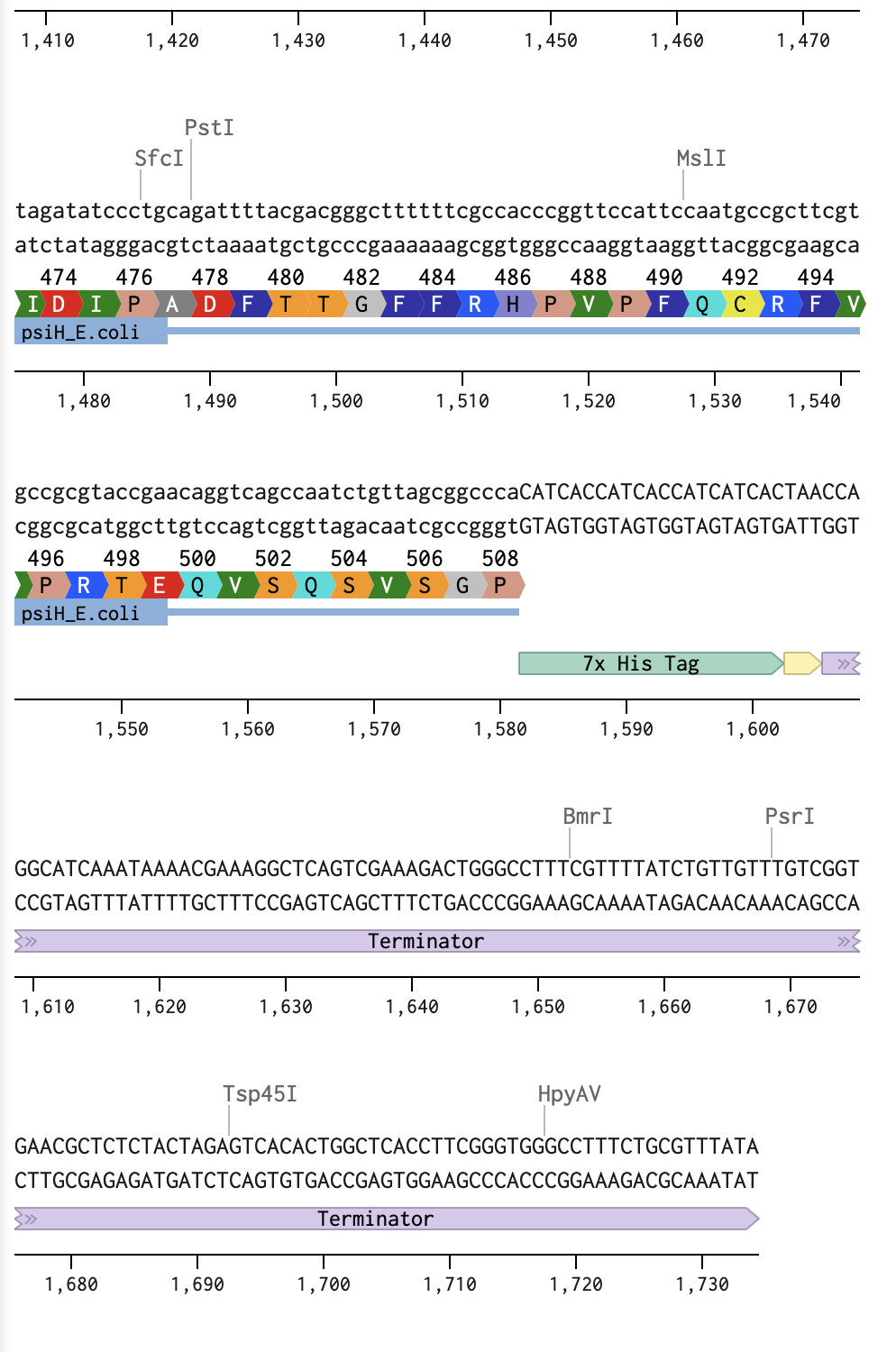
Linear Map of the entire sequence:
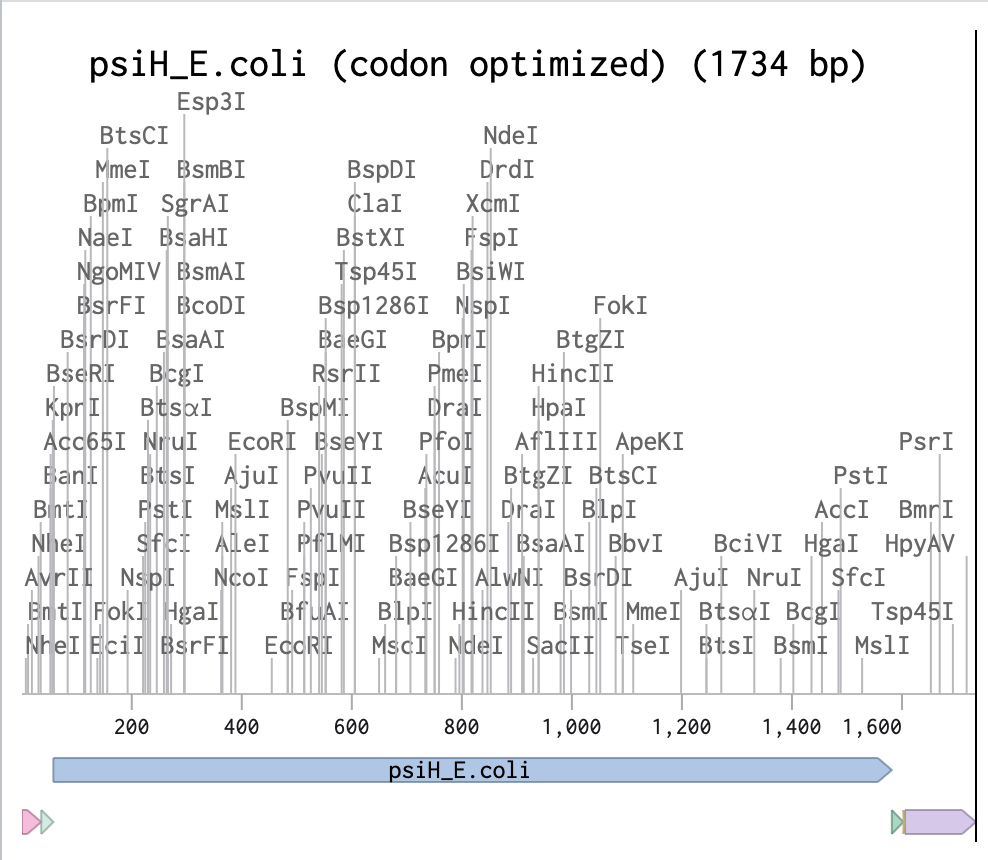
Final Sequence Benchling link!
Building my Full Plasmid Sequence
After downloading my insert sequence (expression cassette) as a FASTA file and uploading it into my Twist account, selecting an appropriate vector (pTwist Amp High Copy), I was able to download the full plasmid sequence (GenBank). I then imported the GenBank file of my plasmid back into Benchling.
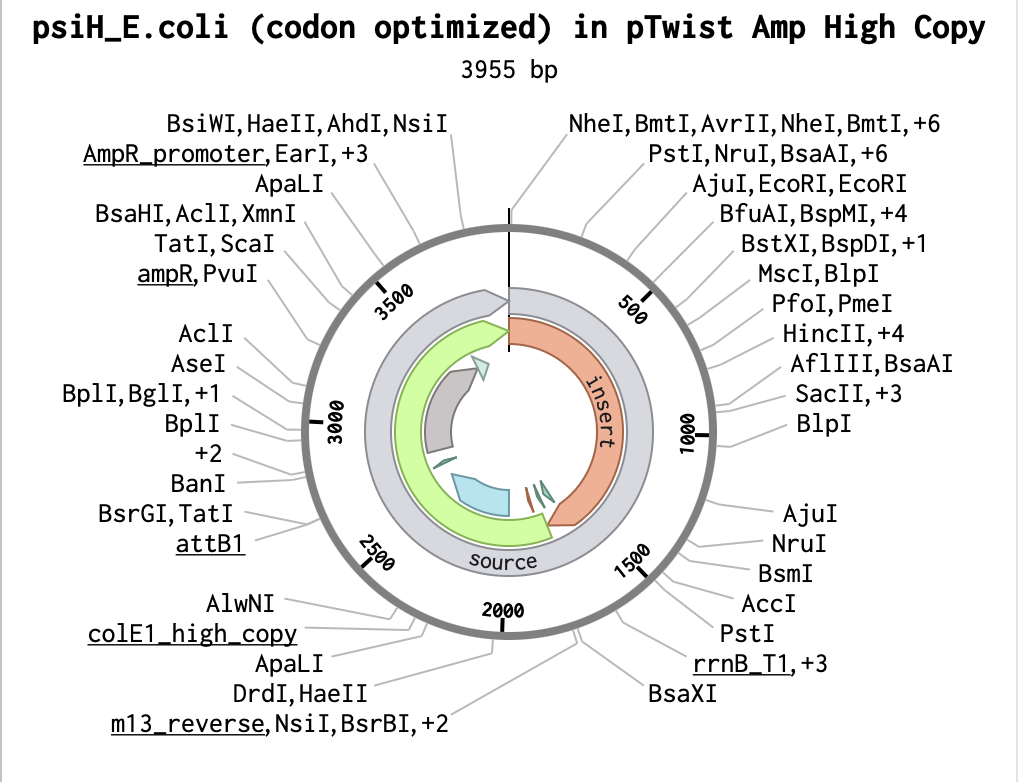
Part 5: DNA Read/Write/Edit
5.1 DNA Read
1) What DNA would you want to sequence (e.g., read) and why?
I want to sequence the 3955 bp E. coli plasmid containing the codon-optimised PsiH gene (tryptamine 4-monooxygenase from Psilocybe cubensis) that I developed with Twist Bioscience technology for exercise 4. This would allow for the verification of the construct (Gibson Assembly) of the plasmid, to verify that there are no mutations in the critical heme-binding site essential for the following steps of psilocibin synthesis. Ultimately, the goal would be to create a baseline sequence for future IBD therapy-scale engineering (Adams et al., 2019).
Sources
Adams, A.M., Kaplan, N.A., Wei, Z., Brinton, J.D., Monnier, C.S., Enacopol, A.L., Ramelot, T.A. and Jones, J.A. (2019). In vivo production of psilocybin in E. coli. Metabolic Engineering, [online] 56, pp.111–119. doi:https://doi.org/10.1016/j.ymben.2019.09.009.1) In the lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA, and why?
I initially considered Sanger sequencing due to its high accuracy (~99.9%) and effectiveness for targeted validation of small DNA regions. However, my E. coli psiH plasmid has 3955 bp, which exceeds Sanger’s read length of ~800 bp per reaction. Full coverage would require multiple reads; consequently, needing the development of multiple primers, which creates a time-consuming and costly process (AAT Bioquest, 2024).
I therefore continued my research and thought Oxford Nanopore Technologies' MinION device was a more appropriate fit (Oxford Nanopore Technologies, 2024). This technology generates long reads (>20 kb) capable of sequencing my entire 3955 bp plasmid in 1-2 reads with >99% accuracy (Brown et al., 2023). This approach would allow for the verification of the complete PsiH integration in the plasmid, promoter/RBS/terminator junctions and detect assembly errors.
Sources
AAT Bioquest (2024). What are the limitations of the Sanger Sequencing method?W | AAT Bioquest. [online] Aatbio.com. Available at: https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-limitations-of-the-sanger-sequencing-method [Accessed 15 Feb. 2026].Brown, S.D., Dreolini, L., Wilson, J.F., Miruna Balasundaram and Holt, R.A. (2023). Complete sequence verification of plasmid DNA using the Oxford Nanopore Technologies’ MinION device.BMC Bioinformatics , 24(1). doi:https://doi.org/10.1186/s12859-023-05226-y.Oxford Nanopore Technologies (2024). Plasmidsaurus redefine the gold standard: whole-plasmid sequencing with Oxford Nanopore. [online] Oxford Nanopore Technologies. Available at: https://nanoporetech.com/blog/plasmidsaurus-redefine-the-gold-standard-whole-plasmid-sequencing-with-oxford-nanopore [Accessed 17 Feb. 2026].1) Is your method first-, second-, or third-generation or other? How so?
Oxford Nanopore Technologies' MinION is a 3rd generaation sequencer. This means that it can read much longer sequences than any 1st or 2nd generation sequencing technologies (Hilt and Ferrieri, 2022). Additionally, it is one of the rare sequencing technologies that allows for real-time analysis. (Oxford Nanopore Technologies,2021)
Sources
Hilt, E.E. and Ferrieri, P. (2022). Next Generation and Other Sequencing Technologies in Diagnostic Microbiology and Infectious Diseases. Genes, 13(9), p.1566. doi:https://doi.org/10.3390/genes13091566.Oxford Nanopore Technologies (2021). How Nanopore Sequencing Works. [online] Oxford Nanopore Technologies. Available at: https://nanoporetech.com/platform/technology [Accessed 17 Feb. 2026].2) What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Input: Purified plasmid DNA from my engineered E. coli (3955 bp PsiH construct)
Preparation
Grow Escherichia coli strain DH5α (Huan et al., 2025)Introduce the plasmid (containing the optimised psiH gene) through an overnight culture.Extraction of the plasmid to obtain the pure PsiH-plasmid DNA out of E. coli cells.Measuring DNA through NanoDrop (spectrophotometer): usually around 1-2 µL of sample to measure concentration and purity. (Thermo Fisher Scientific, 2026).For Nanoprre preparation, add motor proteins (acting as enzymes that control the speed and direction of DNA as it moves through the nanopore). (Oxford Nanopore Technologies, 2025)Sources
Huang, Z., Yao, Y., Di, R., Zhang, J., Pan, Y. and Liu, G. (2025). De Novo Biosynthesis of Antidepressant Psilocybin in Escherichia coli. Microbial biotechnology, [online] 18(4), p.e70135. doi:https://doi.org/10.1111/1751-7915.70135.Oxford Nanopore Technologies (2025). How Oxford Nanopore sequencing works. [online] Oxford Nanopore Technologies. Available at: https://nanoporetech.com/blog/how-oxford-nanopore-sequencing-works [Accessed 17 Feb. 2026].Thermo Fisher Scientific (2026). NanoDrop Microvolume Spectrophotometers - US. [online] www.thermofisher.com. Available at: https://www.thermofisher.com/uk/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/uv-vis-spectrophotometry/instruments/nanodrop.html [Accessed 17 Feb. 2026].3) What are the essential steps of your chosen sequencing technology? How does it decode the bases of your DNA sample (base calling)?
Miniprep of plasmid: isolates and purifies plasmid DNA from bacterial culture. Includes a colour buffer system for a visual quality check to ensure my extraction worked successfully.Quantify DNA (spectrophotometer)Library preparation: Addition of barcode and motor proteins.Priming of the Nanopore flow cell: removing air and addition of bufferLoad the library onto the MinION flow cell.Start running through the software and wait for sequencing (usually around 24-48 hours).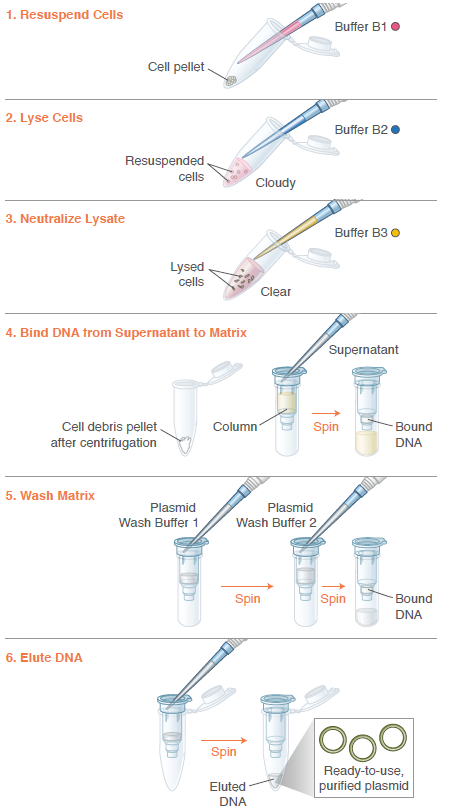 Image: Instructions from Monarch Spin Plasmid Miniprep Kit, 2026.
Image: Instructions from Monarch Spin Plasmid Miniprep Kit, 2026.
Sources
New England Biolabs (2025). [online] Neb.com. Available at: https://www.neb.com/en-gb/products/t1110-monarch-spin-plasmid-miniprep-kit [Accessed 17 Feb. 2026].PANDORA-ID-NET Consortium (2021). Oxford Nanopore flow cell priming and loading tutorial. [online] YouTube. Available at: https://www.youtube.com/watch?v=IknVaEnuDz0 [Accessed 17 Feb. 2026].4) What is the output of your chosen sequencing technology?
Output: A FASTQ file containing the complete 3955 bp sequence of my optimised E.coli PsiH plasmid.
Sources
Oxford Nanopore Technologies plc. (2020). Output Structure - Oxford Nanopore Output Specifications. [online] Github.io. Available at: https://nanoporetech.github.io/ont-output-specifications/latest/minknow/output_structure/ [Accessed 19 Feb. 2026].5.2 DNA Write
I will synthesise the recombinant DNA of the engineered E. Coli as it contains the introduced plasmid from the psilocybin producing fungi.
5.2 DNA Write
(i)What DNA would you want to synthesise (e.g., write) and why?
I would like to synthesise the codon-optimised PsiH gene. This is an essential step for the development of this project, as fungal codons usually don’t express well in bacteria (Naqvi et al., 2016). Synthesising it will allow for a higher level of psiH with the ultimate goal of psilocin production.
Sources
Naqvi, S.H.Z., Cord‐Landwehr, S., Singh, R., Frank, B., Kolkenbrock, S. and Moerschbacher, B.M. (2016). A Recombinant Fungal Chitin Deacetylase Produces Fully Defined Chitosan Oligomers with Novel Patterns of Acetylation. Applied and Environmental Microbiology, 82(22), pp.6645–6655. doi:https://doi.org/10.1128/aem.01961-16.(ii)What technology or technologies would you use to perform this DNA synthesis, and why?
I would use the services of Twist Bioscience to ensure a correct synthesis with no PCR errors, and be ready for the Gibson Assembly of my plasmid.
1) What are the essential steps of your sequencing methods?
Twist uses a Phosphoramidite synthesis method. These are the four essential steps:
Coupling: the first phosphoramidite in the chain is attached to the surface with a catalysed condensation reaction.Oxidation: the phosphite triester is unstable, so it is converted to a phosphate to improve the sequenceCoupling: the next phosphoramidite is coupled to the available -OH on the previous deblocked molecule.Capping: Sometimes the coupling is not 100% efficient, and therefore, the coupling fails. To stop this, an unreactive group is added, blocking further extension.Repetition: Oxidation is repeated to extend the oligonucleotide molecule in a desired sequence.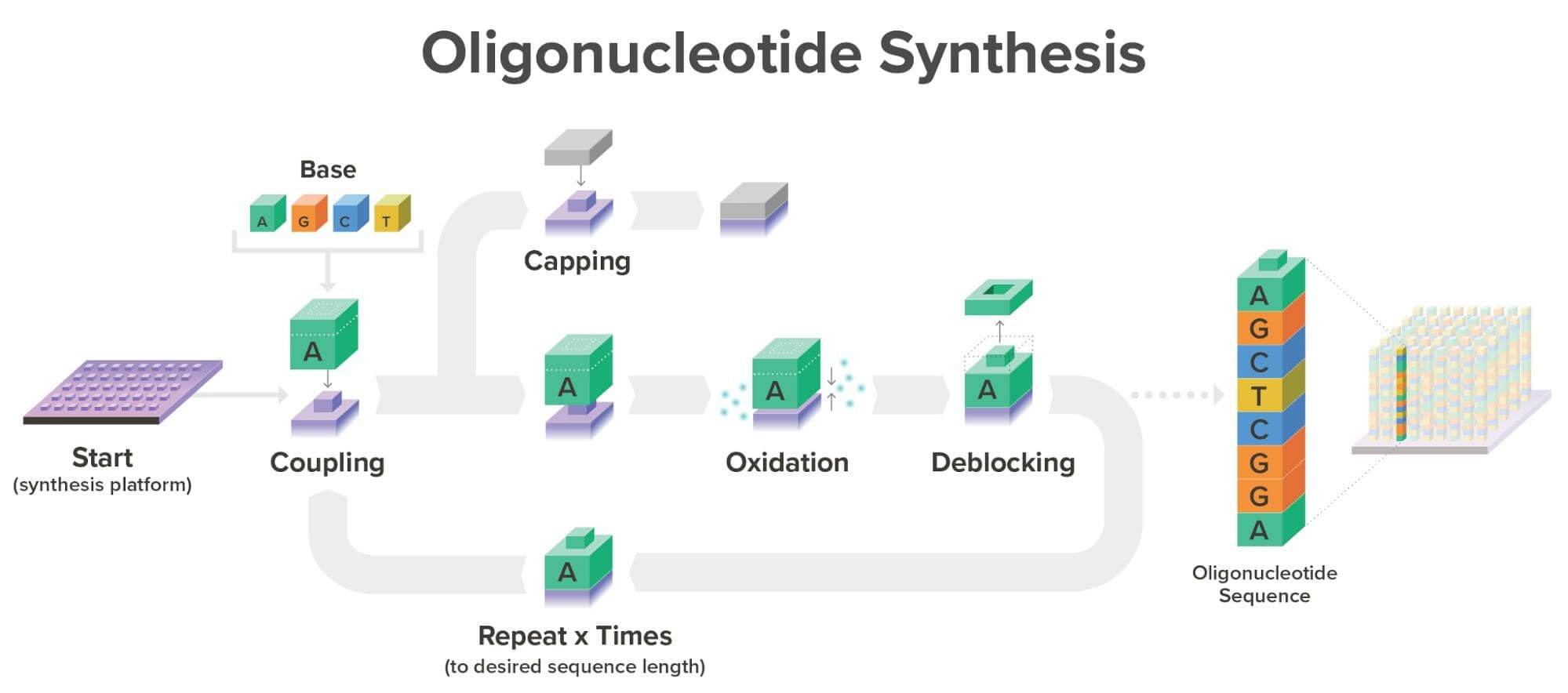 Image: Twist Bioscience Website, 2026.
Image: Twist Bioscience Website, 2026.
Sources
Twist Bioscience (2018). Phosphoramidite Chemistry for DNA Synthesis | Twist Bioscience. [online] www.twistbioscience.com. Available at: https://www.twistbioscience.com/blog/science/simple-guide-phosphoramidite-chemistry-and-how-it-fits-twist-biosciences-commercial [Accessed 18 Feb. 2026].2) What are the limitations of your sequencing method (if any) in terms of speed, accuracy, and scalability?
Phosphoramidite synthesis makes short DNA pieces (200-1500 bp) (Hughes and Ellington,2017), but Twist Bioscience assembles them into my full 2155 bp PsiH gene with error correction anyway, so it wouldn’t affect the actual synthesis process of my DNA. However, another issue I would like to raise is the use of hazardous reagents in reactions, washing and purification processes of this type of synthesis, which raises significant safety and environmental issues which shouldn’t be undermined (Gao et al., 2025).
Sources
Gao, N., Yu, A., Yang, W., Zhang, X., Shen, Y. and Fu, X. (2025). Enzymatic de novo oligonucleotide synthesis: Emerging techniques and advancements. Biotechnology Advances, [online] 82, p.108604. doi:https://doi.org/10.1016/j.biotechadv.2025.108604.Hughes, R.A. and Ellington, A.D. (2017). Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harbor Perspectives in Biology, [online] 9(1), p.a023812. doi:https://doi.org/10.1101/cshperspect.a023812.5.3 DNA Edit
What DNA would you want to edit and why?
I’d like to edit the PsiH gene via mutagenesis. This would allow for the optimisation of tryptamine binding, which in turn will enhance enzyme activity in E. coli without needing host genome changes (Huang et al., 2025).
Sources
Huang, Z., Yao, Y., Di, R., Zhang, J., Pan, Y. and Liu, G. (2025). De Novo Biosynthesis of Antidepressant Psilocybin in Escherichia coli. Microbial biotechnology, [online] 18(4), p.e70135. doi:https://doi.org/10.1111/1751-7915.70135.(ii)What technology or technologies would you use to perform these DNA edits and why?
I would use the site-directed mutagenesis method to examine the relationship between function and structure of my selected protein (Zhang et al., 2009). To do so, I would amplify my Twist PsiH plasmid with mutant primers in order to enhance the tryptamine binding in E.coli host.
Sources
Zhang, B., Zhang, X., An, X., Ran, D., Zhou, Y., Lu, J. and Tong, Y. (2009). An easy-to-use site-directed mutagenesis method with a designed restriction site for convenient and reliable mutant screening. Journal of Zhejiang University SCIENCE B, 10(6), pp.479–482. doi:https://doi.org/10.1631/jzus.b0820367.1) How does your technology of choice edit DNA? What are the essential steps?
PCR site-directed mutagenesis edits PsiH using custom primers carrying my mutation (one base change to improve tryptamine binding) to amplify the full Twist plasmid during PCR, producing nicked circular copies with the incorporated alteration.
2) What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Design psiH primers with mutations.Use PCR to amplify the full plasmid.The DpnI enzyme is used to digest parental DNA, removing the original psiH plasmid.Verify the mutant through sequencing.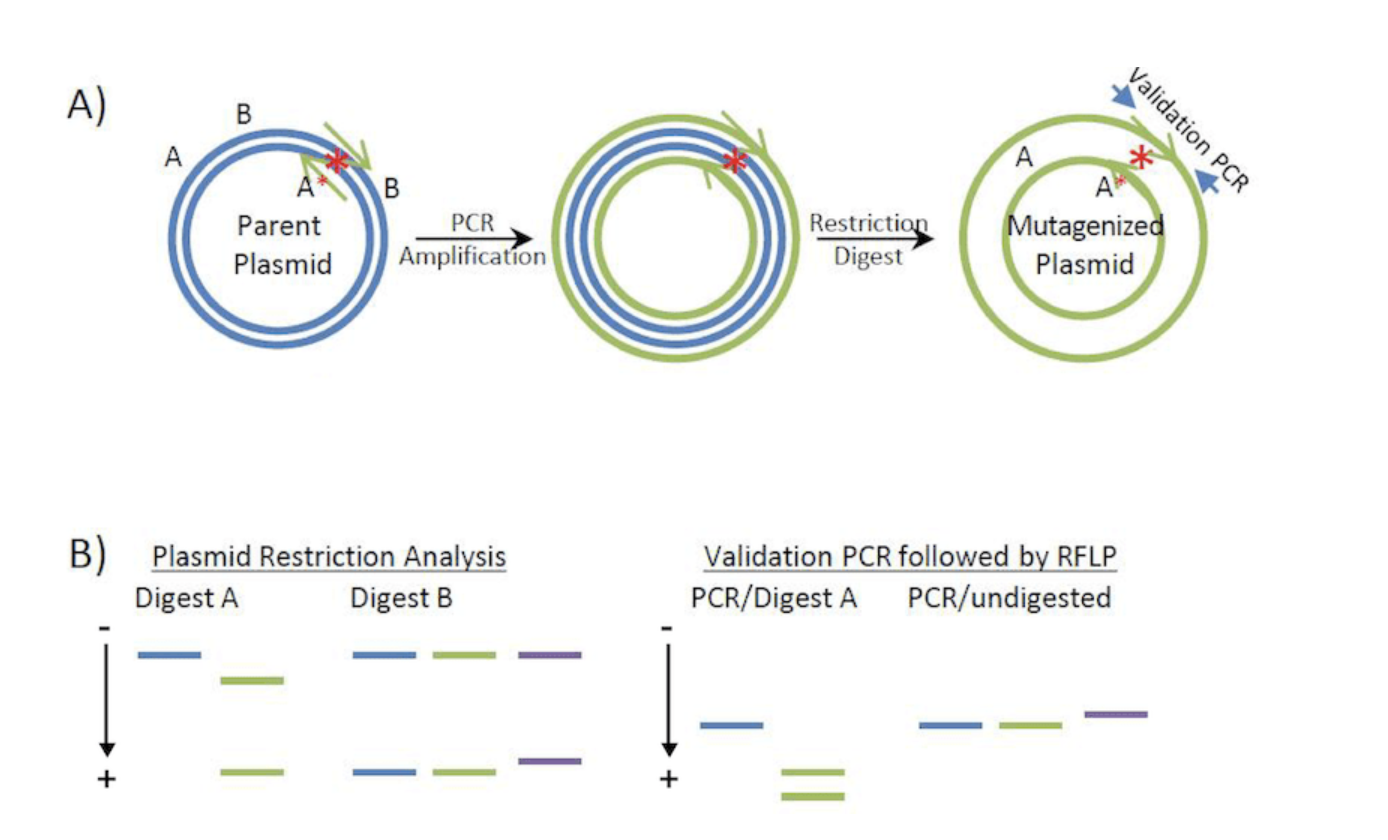 image: From Addgene.org
image: From Addgene.org
Sources
Kristian Laursen (2016). Site-directed mutagenesis by PCR. [online] Addgene.org. Available at: https://blog.addgene.org/site-directed-mutagenesis-by-pcr [Accessed 20 Feb. 2026].3) What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Considering that I am only editing a small part of my DNA, using this method has a relatively high precision rate for a single alteration. Prior steps, though, like a mistake in the primer preparation, which will cause wrong mutations (Alvarez, 2024), as well as a very large plasmid, will cause a drop in accuracy (Jacobs et al., 2011).
Sources
Alvarez, D. (2024). 5 Common Challenges in Site-Directed Mutagenesis and How to Overcome Them. [online] TeselaGen Biotechnology. Available at: https://teselagen.com/blog/challenges-site-directed-mutagenesis/ [Accessed 21 Feb. 2026].Jacobs, J.S., Hong, X. and Eberl, D.F. (2011). A mesmerising new approach to site-directed mutagenesis in large transformation-ready constructs. Fly, [online] 5(2), pp.162–169. doi:https://doi.org/10.4161/fly.5.2.15092.Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork
1) Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
My OpenTron design is inspired by the nudibranch (sea slug) from the Mollusca phylum.
 Image source: Siewert, I. (2014). Nudibranch - Marine Life in Thailand. [online] Diving in Phuket Thailand. Available at: https://www.diving-thailand-phuket.com/nudibranch-marine-life-thailand/.
Image source: Siewert, I. (2014). Nudibranch - Marine Life in Thailand. [online] Diving in Phuket Thailand. Available at: https://www.diving-thailand-phuket.com/nudibranch-marine-life-thailand/.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
After following the instructions, I downloaded the Python script from the GUI website to insert it into my Google Drive code. The code seemed to be all correct, but I encountered an error when it came to the proteins available on the GUI website and the actual colours in the code. I therefore used the Gemini function available in Google Colab to debug my code when it couldn’t recognise some of the colours. Gemini helped identify missing arguments in the load_labware function and suggested remapping custom protein names to standard colours for visualisation. This is why my final image colours don't match those on the OpenTrons art website.
Final image:
Post Lab-Questions
1) Find and describe a published paper that utilises the Opentrons or an automation tool to achieve novel biological applications.
I found a 2025 paper named 'Real‑time AI‑driven quality control for laboratory automation: a novel
computer vision solution for the opentrons OT‑2 liquid handling robot'that was
I found a 2025 paper named: Real‑time AI‑driven quality control for laboratory automation: a novel
computer vision solution for the Opentrons OT‑2 liquid handling robot (Khan et al., 2025), which I found particularly interesting.
This paper explores a novel AI-driven computer vision model called YOLOv8 (object detection model), created for the enhancement of quality that limits the accuracy and reliability of liquid handling robots like Opentrons OT-2. Combining these two systems would allow for the precise detection of pippette tips as well as liquid volumes, consequently providing real-time feedback on potential errors like incorrect placement or the absence of pipette tips as well as liquid levels. The paper highlights that the results obtained with their model present an effective, accessible and affordable solution for the improvement of laboratory automation for both academic and research laboratories.
In the introduction, the authors discuss the advantages ( enhancing reproducibility, efficiency and safety) but also disadvantages (high costs, protocol variability and limited expertise) of automation as well as how using it in life sciences remains limited compared to its high success rates in other industries like manufacturing and food production. The paper, therefore, addresses this gap through their YOLOv8 model in combination with the Opentrons OT-2 liquid handling robot.
The paper continues into the Methodology section, which was divided into 4 sections:
Data collection: taking images of pipette tips and liquid volumes using a camera in the OT-2 robot under a variety of lab conditions.Image annotation: used the Computer Vision Annotation Tool for the labelling of pipette tips and liquid volumes for object detection.Model Training: trained the YOLOv8 on the annotated dataset.Real-time Integration and Error Detection: using the trained YOLOv8 in a server-client setup for real-time error detection and feedback of the OT-2 pipetting system.The following are the experimental results and analysis, where they began by evaluating the YOLOv8 performance and concluding that the trend indicates the model had progressively enhanced its ability to recognise and, in turn, categorise objects more accurately throughout the training. They continued by looking at the readings from the detected tips and liquids by testing the YOLOv8 system through the conduction of 50 experiments. The experiments consisted of running the OT-2 robot in real time, and intentionall exclude certain pipette tips. The tests resulted in a 98% accuracy of identifying missing tips and their exact location. In addition, the authors tested the measurement of liquids within the pipette tips by conducting 100 experiments using a variety of liquid quantities. Their results showed a 95% accuracy in the assessment of liquid within the tips, which suggested that it was an effective approach but that there is still room for improvement. The authors suggest the implementation of liquid segmentation techniques to do so.
In the discussion section, the authors mainly highlight the potential of their technology as an affordable alternative to costly proprietary solutions and how it aids in the improvement of the quality of produced results, which consequently increases the throughput of the laboratory.
Lastly, in the conclusion and future direction section, the paper describes the potential of the YOLOv8 coupled with OT-2 in the advamncement in life science laboratory automation. The authors also state the limitations of their study, such as their experiment focusing on specific pipette types and liquid volumes. They suggest that future works should address these by expanding the dataset to include a wider range of experimental conditions as well as additional liquid handling tasks. Furthermore, they aim to further develop the YOLOv8 AI computer to detect challenges such as air bubbles, which also affect liquid handling accuracy.
Paper Reference:
Khan, S.U., Møller, V.K., Frandsen, R.J.N. and Mansourvar, M. (2025). Real-time AI-driven quality control for laboratory automation: a novel computer vision solution for the Opentrons OT-2 liquid handling robot. Applied Intelligence, 55(6). doi:https://doi.org/10.1007/s10489-025-06334-3.
2) Write a description about what you intend to do with automation tools for your final project.
For my final project (if I continue the direction of my initial project proposition), I would use a Python script to automate E. coli transformation for screening Psilocybe-derived psiH pathway variants.(Bryant, 2022)
The automation tools I would need/ want to use apart from Python:
Opentrons OT-2 robot: handles precise pipetting of cells, plasmids, and media across all the wells simultaneously(Opentrons,2026).Thermocycler module: automates heat shock for DNA uptake into E. coli cells. (Bryant, 2022).Ginkgo Nebula service: for providing a library of codon-optimised (Saras, 2023) psiH variant plasmids in the correct well format for the OT-2 robot.Custom 3D-printed inserts made in PrusaSlicer (Pamidi et al., 2024). Could be used for the cultivation or automation process of the project.As a biodesign student, I want to combine industrial automation (OT-2) with hands-on fabrication challenges. The 3D modelling is a new territory for me, but pushing these design boundaries alongside molecular automation is core to biodesign practice. Additionally, since lab access through my node is uncertain and equipment availability unknown, I'll prioritise developing the Python script and 3D printed components, as these I can prototype independently while planning for potential laboratory access.
Sources
Bryant, J.A., Kellinger, M., Longmire, C., Miller, R. and R Clay Wright (2022). AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots. Synthetic biology, [online] 8(1). doi:https://doi.org/10.1093/synbio/ysac032.Saras, N. (2023). Arbor® Biotechnologies. [online] Arbor Biotechnologies®. Available at: https://arbor.bio/arbor-biotechnologies-announces-collaboration-with-ginkgo-bioworks-to-advance-the-discovery-and-development-of-precision-gene-editors/ [Accessed 22 Feb. 2026].Opentrons (2026). OT-2 Robot - Opentrons. [online] Opentrons.com. Available at: https://opentrons.com/products/ot-2-robot?sku=999-00111 [Accessed 22 Feb. 2026].Pamidi, A.S., Spano, M.B. and Weiss, G.A. (2024). A Practical Guide to 3D Printing for Chemistry and Biology Laboratories. Current Protocols, 4(10). doi:https://doi.org/10.1002/cpz1.70036.Final Project Ideas

Week 4 HW: Protein Design I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average, an amino acid is ~100 Daltons)There are a variety of meats depending on the kind. Let’s take an average of about 20% of protein by mass of meat (Day, 2016). This would therefore mean that 500g of meat would contain roughly 100g of protein. The average molecular weight of an amino acid residue is 100 Daltons, which translates to about 100g per mole of the amino acid unit (100g/100g/mol = 1 mole of amino acid units) (ProPrep, 2019). 1 mole contains 6.022 x 10^23 (Avogadro’s number) amount of particles (The ChemTeam, 2026). Therefore, if you consume 500g of meat, you are also ingesting approximately 6 x 10^23 amino acid residues.
References
Day, L. (2016). Protein: Food Sources. Encyclopedia of Food and Health, pp.530–537. doi:https://doi.org/10.1016/b978-0-12-384947-2.00576-6.Dawson, J. (2021). Biological Macromolecules and Amino Acids.ecampusontario.pressbooks.pub. [online] Available at: https://ecampusontario.pressbooks.pub/bioc2580/chapter/bioc2580-lecture-1-biological-macromolecules-amino-acids/ [Accessed 28 Feb. 2026].ProPrep (2019).How can molecular weight conversion be achieved from dalton to g/mol, and why is this unit of measurement significant in molecular biology? [online] Proprep.com. Available at: https://www.proprep.com/questions/how-can-molecular-weight-conversion-be-achieved-from-dalton-to-gmol-and-why-is-this-unit-of-measurem [Accessed 1 Mar. 2026].The ChemTeam (2026). Welcome To Zscaler Directory Authentication. [online] Chemteam.info. Available at: https://www.chemteam.info/Mole/MolarMass.html.12.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
There are several reasons why eating another animal does not make us become that animal, and they centre on two key concepts. First, what makes us human rather than another organism is the DNA in our own cells and how our genes are regulated (Brown, 2002). Our genome encodes for specific human proteins, cells, and tissues, and this blueprint does not change just because we eat another species. Second, chemical digestion breaks food down into small building blocks before absorption. When a human consumes an organism with a different genotype, they do not absorb that organism’s genome. Instead, digestive enzymes break down animal proteins into individual amino acids and degrade the animal’s DNA into nucleotides (Patricia and Dhamoon, 2022). Because virtually all life is built from the same basic set of around 20 natural amino acids, what really matters is how these building blocks are arranged in the polypeptide chain. The human body uses the amino acids it absorbs to make human proteins according to its own genetic instructions.
References
Brown, T.A. (2002). The Human Genome. [online] Nih.gov. Available at: https://www.ncbi.nlm.nih.gov/books/NBK21134/ [Accessed 1 Mar. 2026].Patricia, J.J. and Dhamoon, A.S. (2022). Physiology, Digestion. [online] National Library of Medicine. Available at: https://www.ncbi.nlm.nih.gov/books/NBK544242/ [Accessed 1 Mar. 2026].3. Why are there only 20 natural amino acids?
Ultimately, a set of fundamental physicochemical imperatives (molecular properties such as solubility, acidity, and basicity) dictates the prebiotic selection of the canonical 20 amino acid types. The guiding principle was parsimony, where nature retained only the simplest structures with high functional value, avoiding redundancy and unnecessary complexity (Moldoveanu and David, 2022). Energy efficiency also played a key role. The study ‘Why twenty amino acid residue types suffice(d) to support all living systems (2018) used quantum chemistry and chemoinformatics to analyse a large panel of candidate chemicals, followed by statistical analysis of their complexity and property scores. The results showed that the 20 canonical amino acids were the most likely to form under prebiotic conditions and had optimal physicochemical properties (Bywater, 2018).
References
Bywater, R.P. (2018). Why twenty amino acid residue types suffice(d) to support all living systems. PLOS ONE, 13(10), p.e0204883. doi:https://doi.org/10.1371/journal.pone.0204883.Moldoveanu, S. and David, V. (2022). Characterization of analytes and matrices. Essentials in Modern HPLC Separations, 2, pp.179–205. doi:https://doi.org/10.1016/b978-0-323-91177-1.00003-x.4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can make other non-natural amino acids, known as non-proteinogenic or non-canonical amino acids (ncAAs), which have been routinely synthesised and engineered in labs to be added into proteins to alter certain physicochemical and biological properties for an optimal protein (e.g., reactivity), which has been a key development for enzymology and drug discovery (Adhikari et al., 2021). To design ncUAAs, you need to analyse their structure and determine what must remain the same and what may be altered. The core structure has a central alpha carbon that is bonded to an amino group, a carboxyl group, a hydrogen atom, and an R group (custom side chain). The R group can be hydrophobic, polar, or charged. The size, shape and potential for covalent interactions determine how each amino acid residue interacts with its environment (Whiteburn, 2024). It is also the area that needs to be altered/engineered to create an ncUAA.
References
Adhikari, A., Bhattarai, B.R., Aryal, A., Thapa, N., KC, P., Adhikari, A., Maharjan, S., Chanda, P.B., Regmi, B.P. and Parajuli, N. (2021). Reprogramming natural proteins using unnatural amino acids.RSC Advances , 11(60), pp.38126–38145. doi:https://doi.org/10.1039/d1ra07028b.Whitburn, T. (2024). Exploring Biological Molecules: Amino Acids, Protein Structure, and Function. [online] Online A-level Biology Tutor. Available at: https://www.alevelbiologytutor.com/tutoring-blog/2024/3/1/exploring-biological-molecules-understanding-amino-acids-protein-structure-and-function [Accessed 27 Feb. 2026].5.Where did amino acids come from before enzymes that make them, and before life started?
Amino acids on prebiotic Earth formed through non-biological chemical reactions (abiogenesis), both from space delivery and surface synthesis (Cowing, 2023). Meteorites (carbonaceous chondrites) delivered large quantities of abiotically formed amino acids like glycine, alanine, α-amino-n-butyric acid, isovaline, and β-alanine, which landed in early oceans (Strasdeit, 2009). Atmospheric sparks from electric discharges (lightning) and volcanic ash-gas clouds also synthesised amino acids in water droplets (Cowing, 2025). Over time, wet-dry cycles in lagoons or tidal pools concentrated these amino acids and polymerised them into peptides, creating the building blocks for early life.
References
Cowing, K. (2023). How Were Amino Acids Formed Before The Origin Of Life On Earth? - Astrobiology. [online] Astrobiology. Available at: https://astrobiology.com/2023/04/how-were-amino-acids-formed-before-the-origin-of-life-on-earth.html [Accessed 1 Mar. 2026].Cowing, K. (2025). Microlightning In Water Droplets May Have Sparked Life On Earth - Astrobiology. [online] AstrobiologyAvailable at: https://astrobiology.com/2025/03/microlightning-in-water-droplets-may-have-sparked-life-on-earth.html [Accessed 1 Mar. 2026].Strasdeit, H. (2009). Prebiotic amino acid chemistry on the early Earth. EPSC Abstracts, [online] 4. Available at: https://meetingorganizer.copernicus.org/EPSC2009/EPSC2009-70.pdf [Accessed 1 Mar. 2026].6.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Most amino acids come in at least two forms, whose structures are mirror images of each other and are referred to as the right-handed or left-handed optical isomers. Natural proteins use L-amino acids and form right-handed alpha helices, whereas D-amino acids are the mirror image, therefore producing the left-handed helices (enantiomers).
(BOC Sciences, 2025).
References
BOC Sciences (2025). D-Amino Acids. [online] Bocsci.com. Available at: https://aapep.bocsci.com/amino-acids/d-amino-acids-3260.html [Accessed 1 Mar. 2026].7. Can you discover additional helices in proteins?
There may be additional helices in proteins, such as secondary-structure helices, that may be discovered. For example, the Pi-Helices, often described as rare or uncommon, are found in approximately 15% of proteins and are often overlooked despite their important functions (Cooley et al., 2010).
References
Cooley, R.B., Arp, D.J. and Karplus, P.A. (2010). Evolutionary Origin of a Secondary Structure: π-Helices as Cryptic but Widespread Insertional Variations of α-Helices That Enhance Protein Functionality. Journal of Molecular Biology, 404(2), pp.232–246. doi:https://doi.org/10.1016/j.jmb.2010.09.034.8. Why are most molecular helices right-handed?
Most protein α-helices are right-handed because L-amino acids adopt dihedral angles that minimise steric clashes between side chains and the backbone. Left-handed alpha-helices are possible on the Ramachandran plot (2D visualisation of backbone dihedral angles for amino acid residues in proteins) but suffer more side chain-backbone collisions for L-amino acids, making them energetically unfavourable and rare in nature (Robinson and Afzal, 2014).
References
Robinson, S.W., Afzal, A.M. and Leader, D.P. (2014). Bioinformatics: Concepts, Methods, and Data. Handbook of Pharmacogenomics and Stratified Medicine, pp.259–287. doi:https://doi.org/10.1016/b978-0-12-386882-4.00013-x.9. Why do β-sheets tend to aggregate?
Beta-Sheets tend to aggregate because their exposed edges promote intermolecular hydrogen bonding that readily pairs with other beta-strands from separate proteins and misfolded chains. These associations form stable cross-beta structures, often aggregating into amyloid fibrils linked to diseases such as Alzheimer’s. (Richardson and Richardson, 2002).
References
Richardson, J.S. and Richardson, D.C. (2002). Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proceedings of the National Academy of Sciences, [online] 99(5), pp.2754–2759. doi:https://doi.org/10.1073/pnas.052706099.Part B: Protein Analysis and Visualization
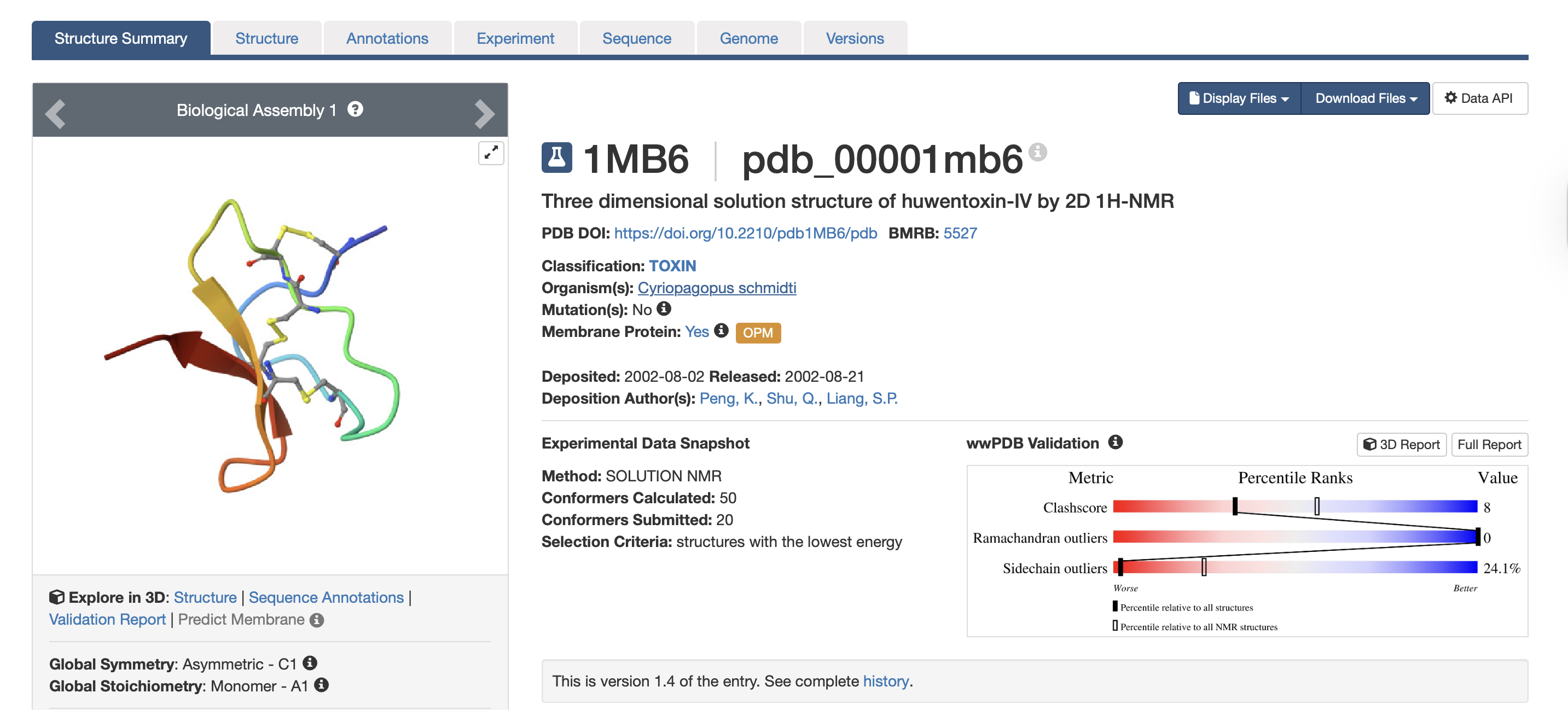
1. Briefly describe the protein you selected and why you selected it.
For this exercise, I’m looking at Huwentoxin-IV (HwTx-IV) from Chinese bird spider venom (Cyriopagopus schmidti). It is a 35-amino-acid peptide that blocks pain signals by hitting the NaV1.7 channels (Sermadiras et al., 2013). These channels are voltage-gated sodium channels expressed in certain neurons, where they play an important role in the generation and transmission of pain-related information (Fouillet et al., 2017). This fits into one of my project propositions for the development of an analgesic cream for the treatment of arthritis pain as a non-opioid painkiller alternative. Understanding its 3D structure shows me exactly where the disulfide bonds need to form and which face binds to the NaV1.7 pain receptor, so when I clone it into E. coli, I will reduce the risks of misfolds in the protein or inactive peptide (Weiss et al., 2022). Essentially, the structure serves as the blueprint for developing a topical formulation for my design proposition.
References
Sermadiras, I., Revell, J., Linley, J.E., Sandercock, A. and Ravn, P. (2013). Recombinant Expression and In Vitro Characterisation of Active Huwentoxin-IV. PLoS ONE, 8(12), p.e83202. doi:https://doi.org/10.1371/journal.pone.0083202.Fouillet, A., Watson, J.F., Piekarz, A.D., Huang, X., Li, B., Priest, B.T., Nisenbaum, E.S., Sher, E. and Ursu, D. (2017). Characterisation of Nav1.7 functional expression in rat dorsal root ganglia neurons by using an electrical field stimulation assay. Molecular Pain, 13, p.174480691774517-174480691774517. doi:https://doi.org/10.1177/1744806917745179.Weiss, K., Racho, J. and Riemer, J. (2022). Compartmentalized disulfide bond formation pathways. Redox Chemistry and Biology of Thiols, pp.321–340. doi:https://doi.org/10.1016/b978-0-323-90219-9.00020-0.2. Identify the amino acid sequence of your protein.
Full precursor sequence:
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVD
DNSKGEERECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQIGK
(National Library of Medicine, 2026)
The bold section of the amino acid sequence is the active section of the protein, as seen below on the RCSB PDB website, highlighted in purple:
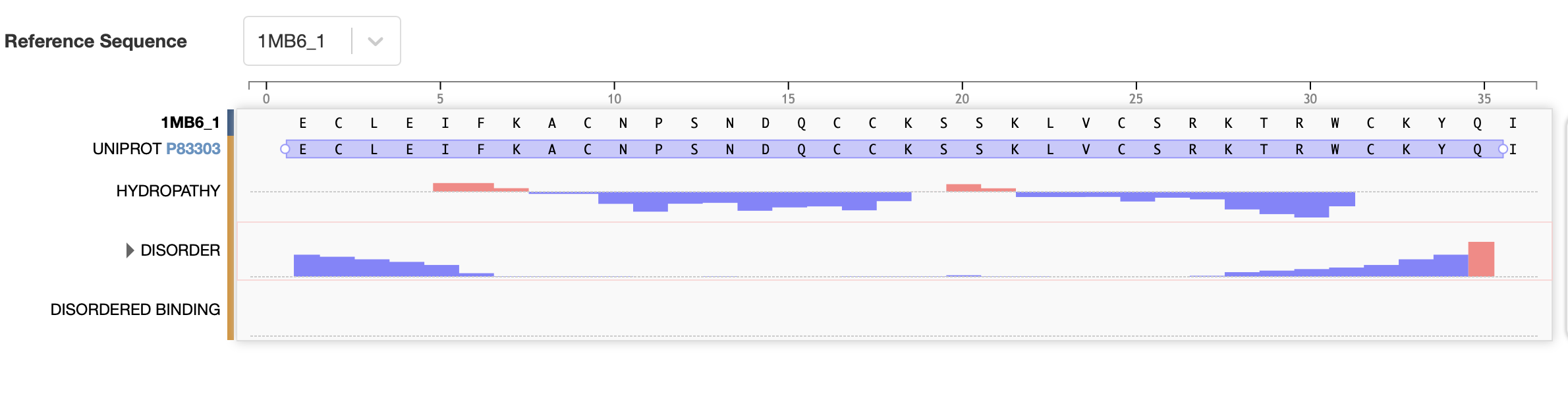
References
PDB (2017). RCSB PDB - 1MB6:Three-dimensional solution structure of huwentoxin-IV by 2D 1H-NMR. [online] Rcsb.org. Available at: https://www.rcsb.org/structure/1MB6#entity-1 [Accessed 2 Mar. 2026].National Library of Medicine (2026). Ornithoctonus huwena huwentoxin-IV precursor, mRNA, complete cds - Nucleotide - NCBI. [online] Nih.gov. Available at: https://www.ncbi.nlm.nih.gov/nuccore/30575583 [Accessed 2 Mar. 2026].- How long is it? What is the most frequent amino acid?
The full precursor sequence is 89aa long (UniProt, 2026) whilst the active protein is 35aa (PDB, 2017). Additionally, the most frequent amino acid is Cysteine (C) that appears 6 times in the active protein.
Below is the Python Code as well as the initial error I got, which was fixed through Gemini Built-In Tool:
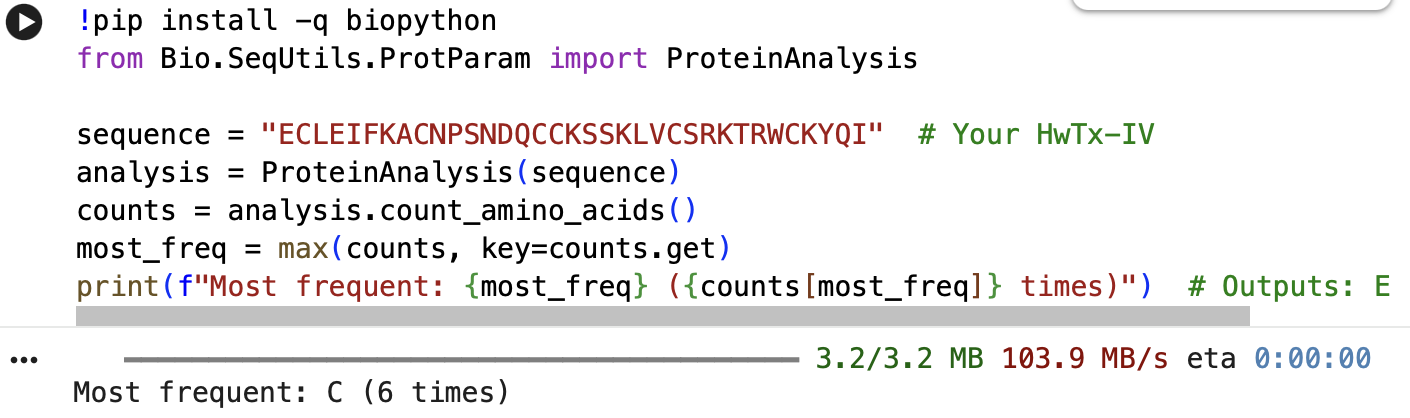
- How many protein sequence homologs are there for your protein?
When focusing specifically on the Huwentoxin-IV protein from Cyriopagopus schmidti (C. schmidti)and restricting the search by taxonomy, I identified 13 homologous sequences sharing between 42.0% and 73.8% sequence similarity with the selected protein.
When all taxonomic groups were included in the analysis, the number of homologous sequences increased immensely to 193 across spider species. Among these, five sequences showed the highest similarity to Huwentoxin-IV, ranging from 89.5% to 90.7%. Notably, these highly similar sequences were derived from Cyriopagopus hainanus, another species classified as a Chinese bird spider. (Uniprot: Blast Tool, 2026)
References
UniProt (2026). UniProt Blast. [online] UniProt. Available at: https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260302-171309-0587-87679188-p1m/overview [Accessed 2 Mar. 2026].- Does your protein belong to any protein family?
Yes, it belongs to the neurotoxin 10 (Hwtx-1) family, furthermore to the 22 (Htx-4) subfamily. I found this information through the Family & Domains section of my selected protein (UniProt, 2026).
3.Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? A good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)The structure was solved in August 2002 by authors Peng, K., Shu, Q., Liang, S.P. (PDB, 2017). In terms of resolution, the RCSB entry does not clearly indicate the resolution for the isolated toxin structure. However, according to the study “Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na⁺ channels in nanodisc” by Gao et al. (2020), the structure of HWTX-IV bound to human Nav1.7 was obtained at an overall resolution of 3.2 Å, with the local resolution of the toxin improving from approximately 6 Å to approximately 4 Å. In structural biology, lower resolution values indicate higher structural quality. While a resolution of 2.70 Å would generally be considered good quality, the reported resolutions of 3.2–3.5 Å are moderate rather than high. Nevertheless, these resolutions are sufficient for visualising overall toxin docking.
References
PDB (2017). RCSB PDB - 1MB6: Three-dimensional solution structure of huwentoxin-IV by 2D 1H-NMR. [online] Rcsb.org. Available at: https://www.rcsb.org/structure/1MB6#entity-1 [Accessed 2 Mar. 2026].Gao, S., Valinsky, W.C., On, N.C., Houlihan, P.R., Qu, Q., Liu, L., Pan, X., Clapham, D.E. and Yan, N. (2020). Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na+ channels in nanodisc. Proceedings of the National Academy of Sciences, [online] 117(25), pp.14187–14193. doi:https://doi.org/10.1073/pnas.1922903117.- Does your protein belong to any structure classification family?
Yes, it belongs to the inhibitor cystine knot structural family (Peng et al., 2002).
References
Peng, K., Shu, Q., Liu, Z. and Liang, S. (2002). Function and Solution Structure of Huwentoxin-IV, a Potent Neuronal Tetrodotoxin (TTX)-sensitive Sodium Channel Antagonist from Chinese Bird Spider Selenocosmia huwena. Journal of Biological Chemistry, 277(49), pp.47564–47571. doi:https://doi.org/10.1074/jbc.m204063200.4. Open the structure of your protein in any 3D molecule visualization software:
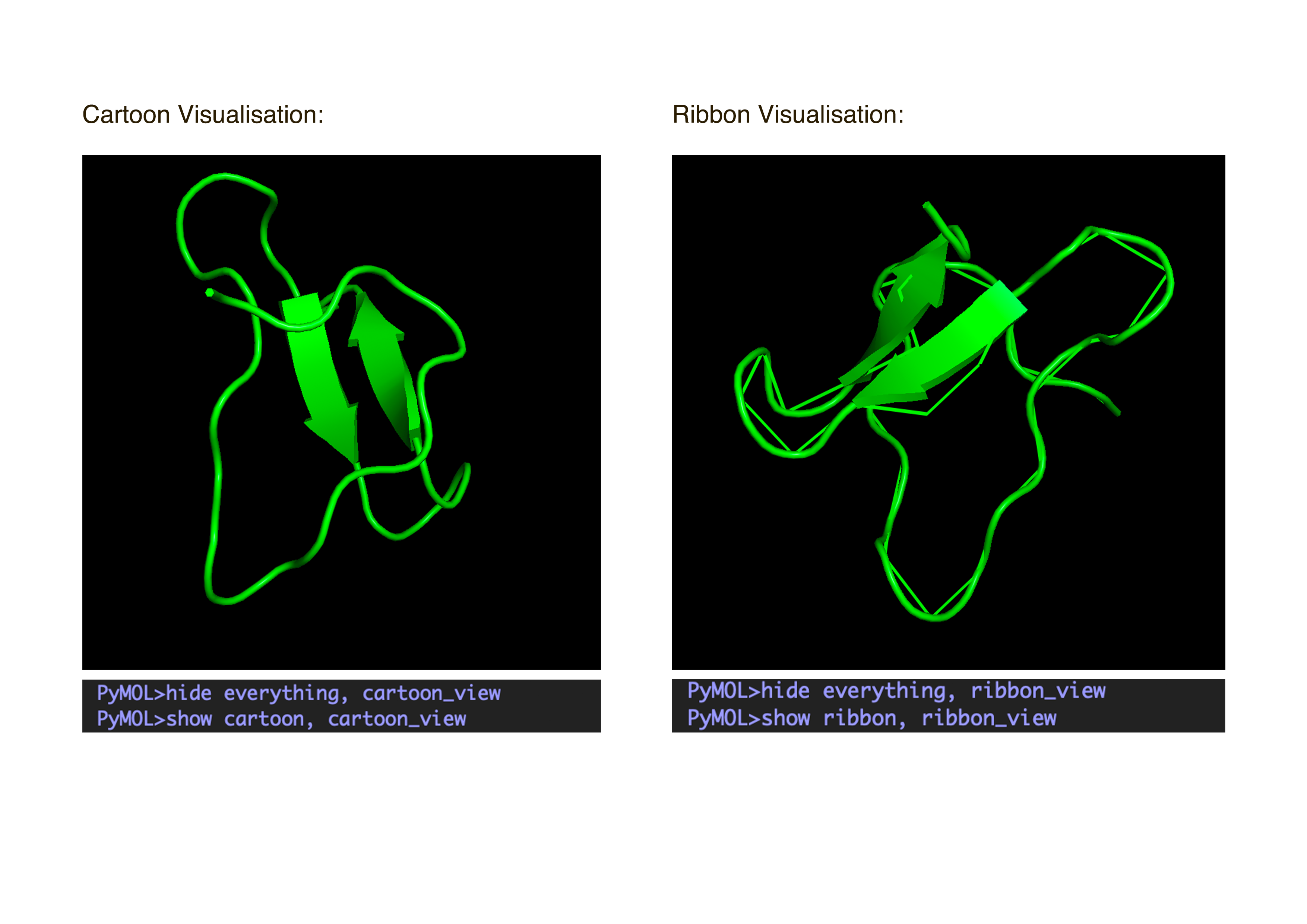
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.After downloading my protein structure from RCSB PDB as a .cif file, I was able to open it in PyMol. I then played around with the commands on the right in the ’S’ section to alter the visual structure of my selected protein.

- Color the protein by secondary structure. Does it have more helices or sheets?
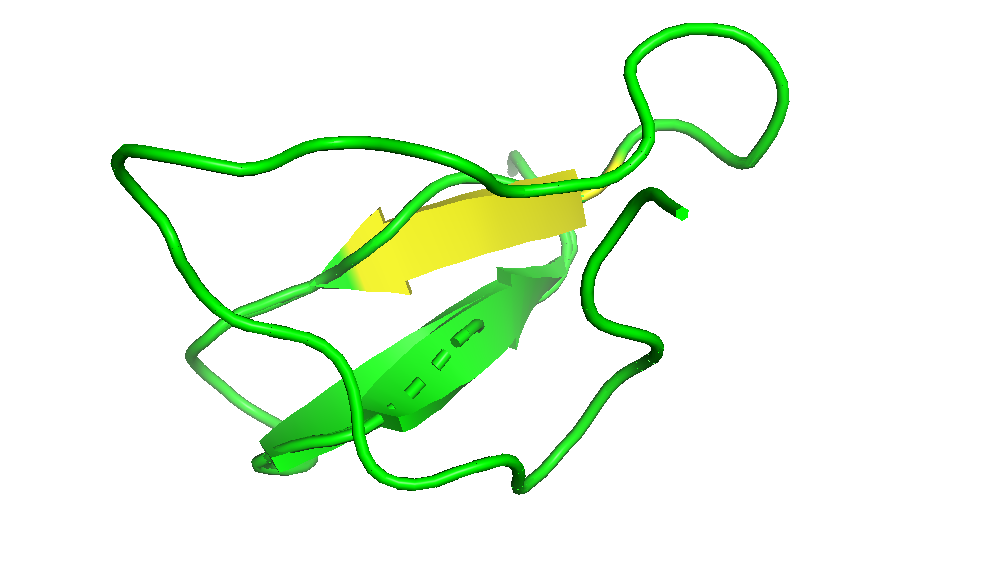
I coloured the protein by secondary structure in the cartoon representation. The green represents coils (loops), the yellow arrows represent sheets, and red represents helices. No red helices appear in the structure, while yellow sheet arrows are present. To verify this, I used the PyMOL count_items function to count residues assigned to each secondary structure type. The results showed 0 residues classified as helices and 6 residues classified as β-sheets (based on Cα atom counts). Therefore, the protein contains sheets but no helices. The remaining residues (274 atoms total) are classified as loops. Below is the following code used to colour and to verify this through PyMOL’s counting function:
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Colour Code: Yellow = hydrophobic, Green = polar (uncharged), Blue = positively charged, Red = negatively charged.

ANALYSIS: The positively charged residues (blue and red) are strongly hydrophilic and seem to be concentrated more on the outer loops than the centre. The polar residues are usually also hydrophilic and appear spread throughout the protein structure. The hydrophobic residues (yellow) appear to be seen in the core of the sheet, but are also spread out around the full structure.
- Visualise the surface of the protein. Does it have any “holes” (aka binding pockets)?

ANALYSIS: Across the whole surface of the protein, only a single small cavity is apparent and could be identified as a binding pocket. Overall, the protein surface is relatively smooth and compact, which would explain why the protein doesn’t have any other molecules, as it may be limiting interactions by providing an unstable environment for binding.
Part C: Using ML-Based Protein Design Tools
1. Choose your favorite protein from the PDB
C.1 Protein Language Modeling
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
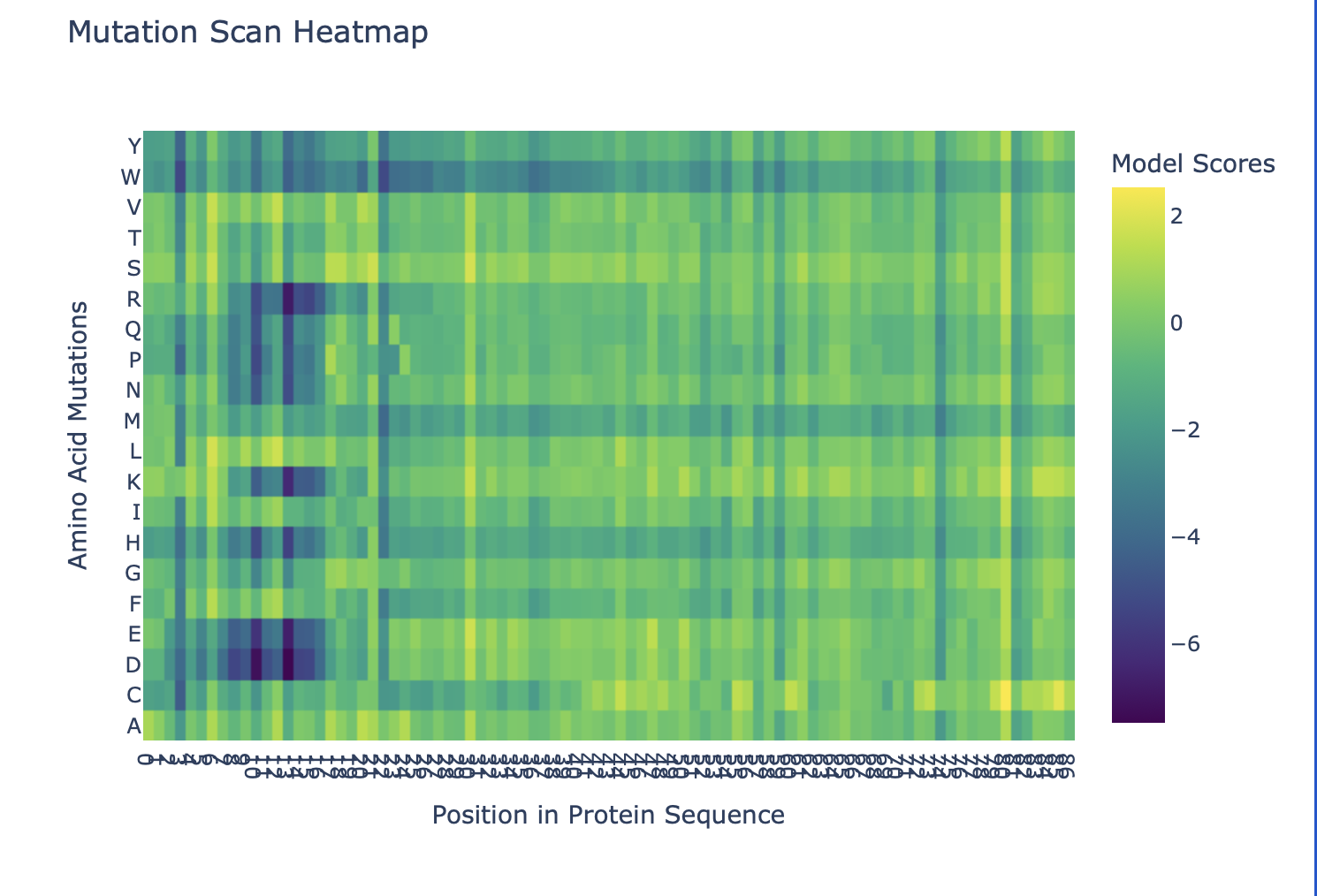
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Overall, the heatmap shows many positions with predominantly high scores (yellow/green), indicating mutation-tolerant sites, which contradicts the idea that smaller proteins would be more susceptible to mutations, disrupting protein function. Positions 8–16 stand out with a high concentration of blue (-4/-6), suggesting high mutation intolerance here. W mutations seem to be specifically prone to mutations wherever they are located in the sequence implyging implying that mutations in these amino acids could critically disrupt core stability or binding despite the region's flexibility.
C.2 Latent Space
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
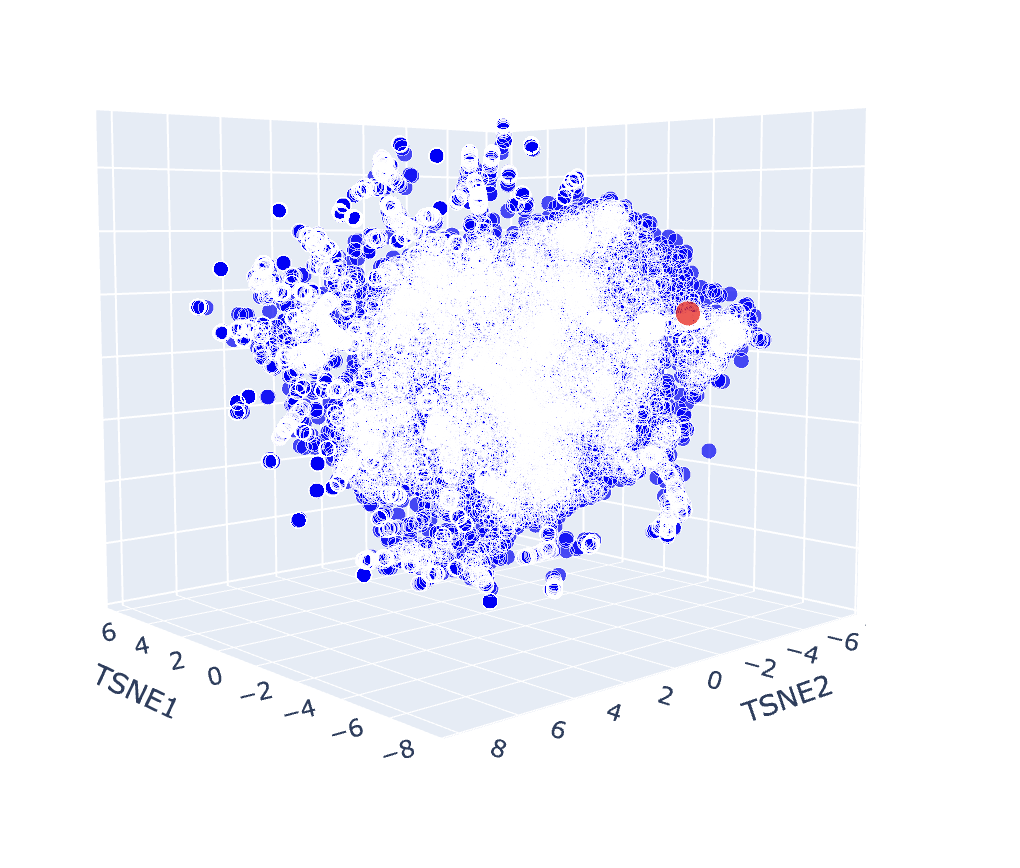
b. Analyse the different formed neighbourhoods: do they approximate similar proteins?
The latent space map groups proteins into neighbourhoods where closer points share similar sequences, structures, or functions. For example, the circled group in the image below includes proteins from soil bacteria like Bacillus subtilis and Enterococcus faecalis, which have overlapping membrane and stress-response features, showing that the map captures biological similarity.
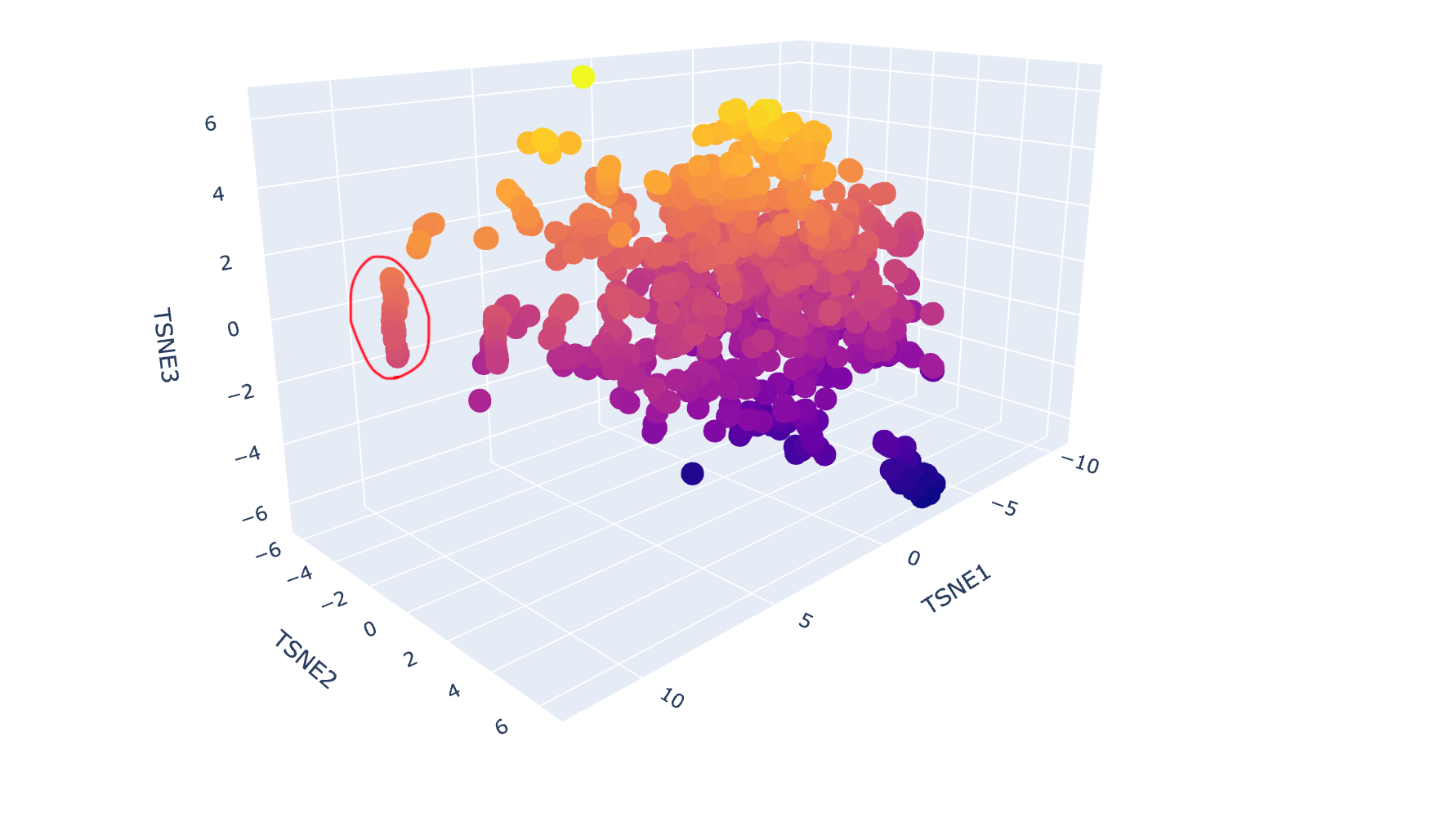
c. Place your protein in the resulting map and explain its position and similarity to its neighbours.
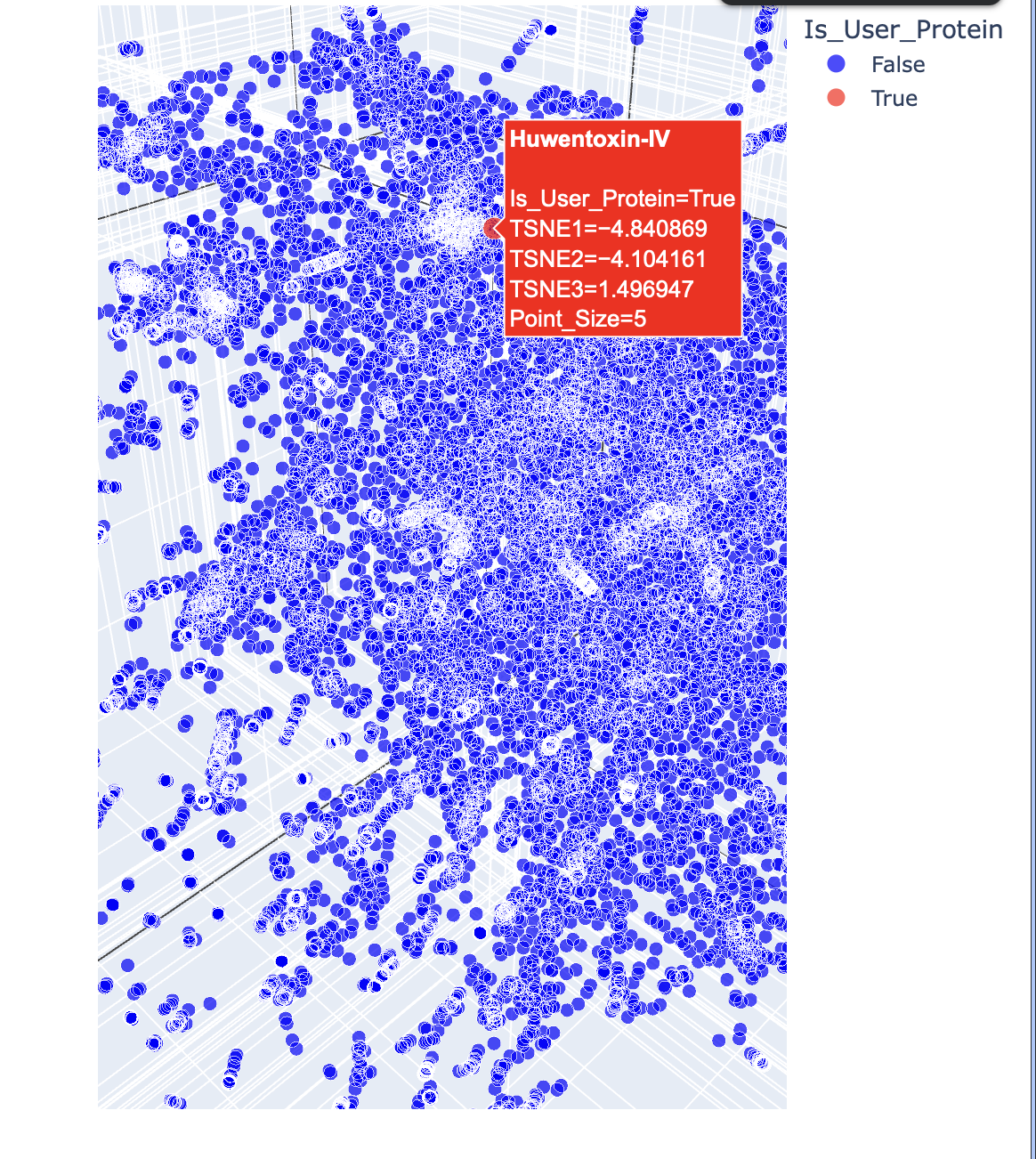
Below are the names and structures of three proteins in the same neighbourhood as my selected protein:
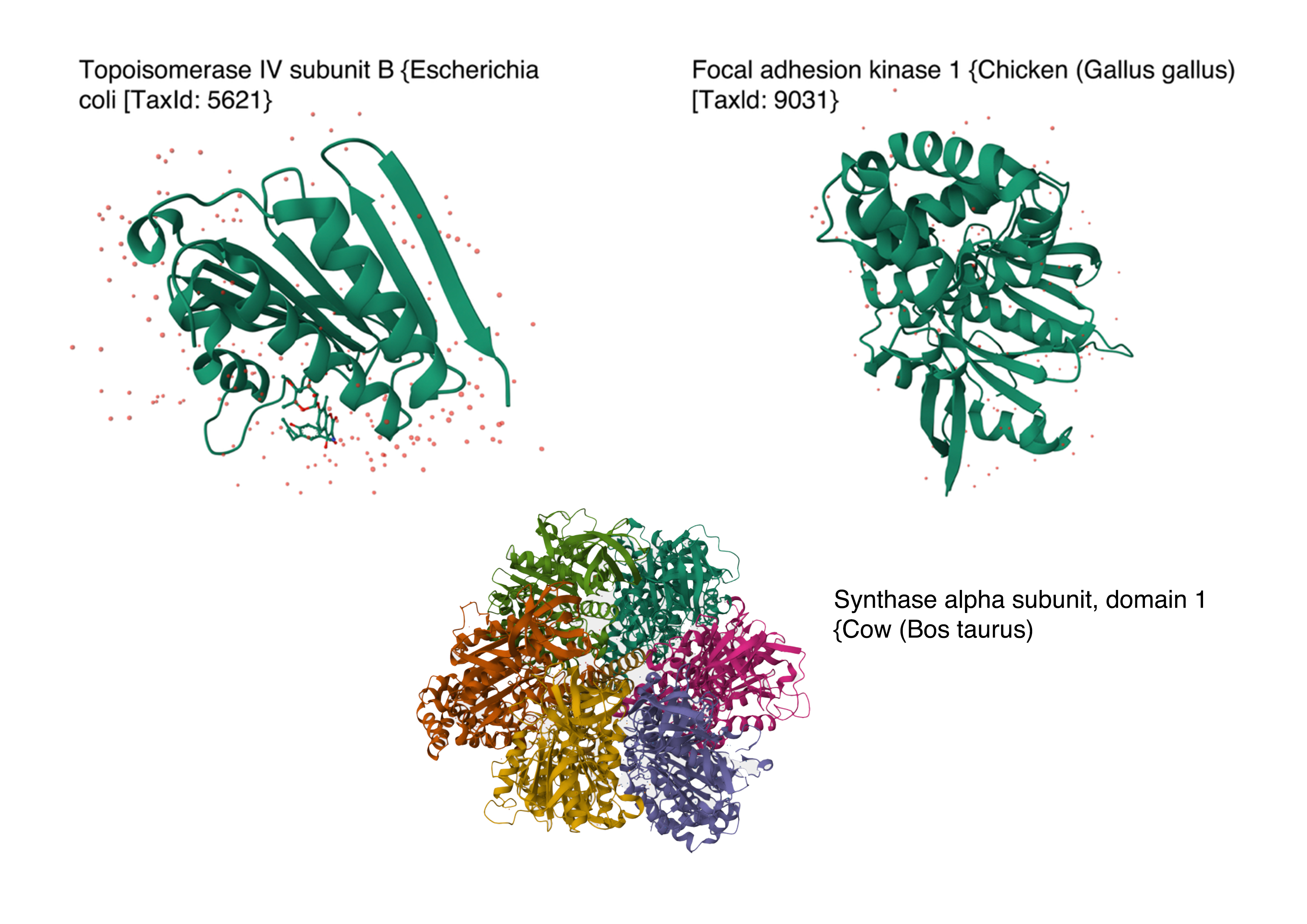 Image references:
Image references:
- Topoisomerase IV subunit B and Focal adhesion kinase 1 from RCSB PDB Website
- Synthase alpha sunbunit, domain 1 from Uniprot Website
In the protein latent space, Huwentoxin-IV from Cyriopagopus schmidti sits in a region populated more by regulatory and interaction-focused proteins than by purely structural ones. Its closest neighbours include an ATPase domain from topoisomerase IV, along with fragments of chicken focal adhesion kinase and a synthase alpha subunit. Despite their differences in size, fold, and cellular context, these proteins all seem to rely on chemically specialised surfaces to influence the behaviour of other macromolecules. This suggests that the model is picking up on less global structural similarity and more on shared biophysical features, such as molecular interactions and regulation.
C.2 Protein Folding
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
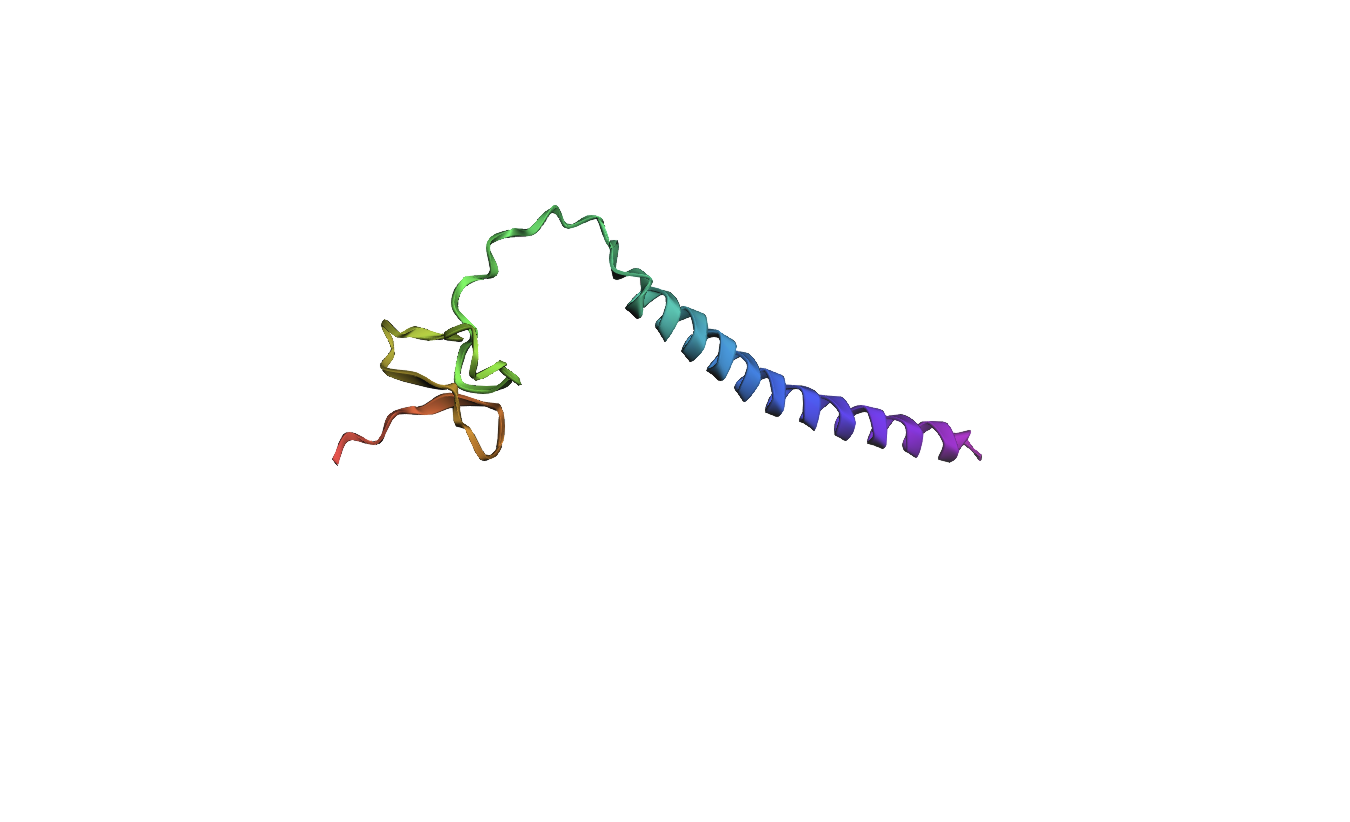
The 35aa active part of the 89aa Huwentoxin-IV mini-protein, which I looked at in my Part B homework:
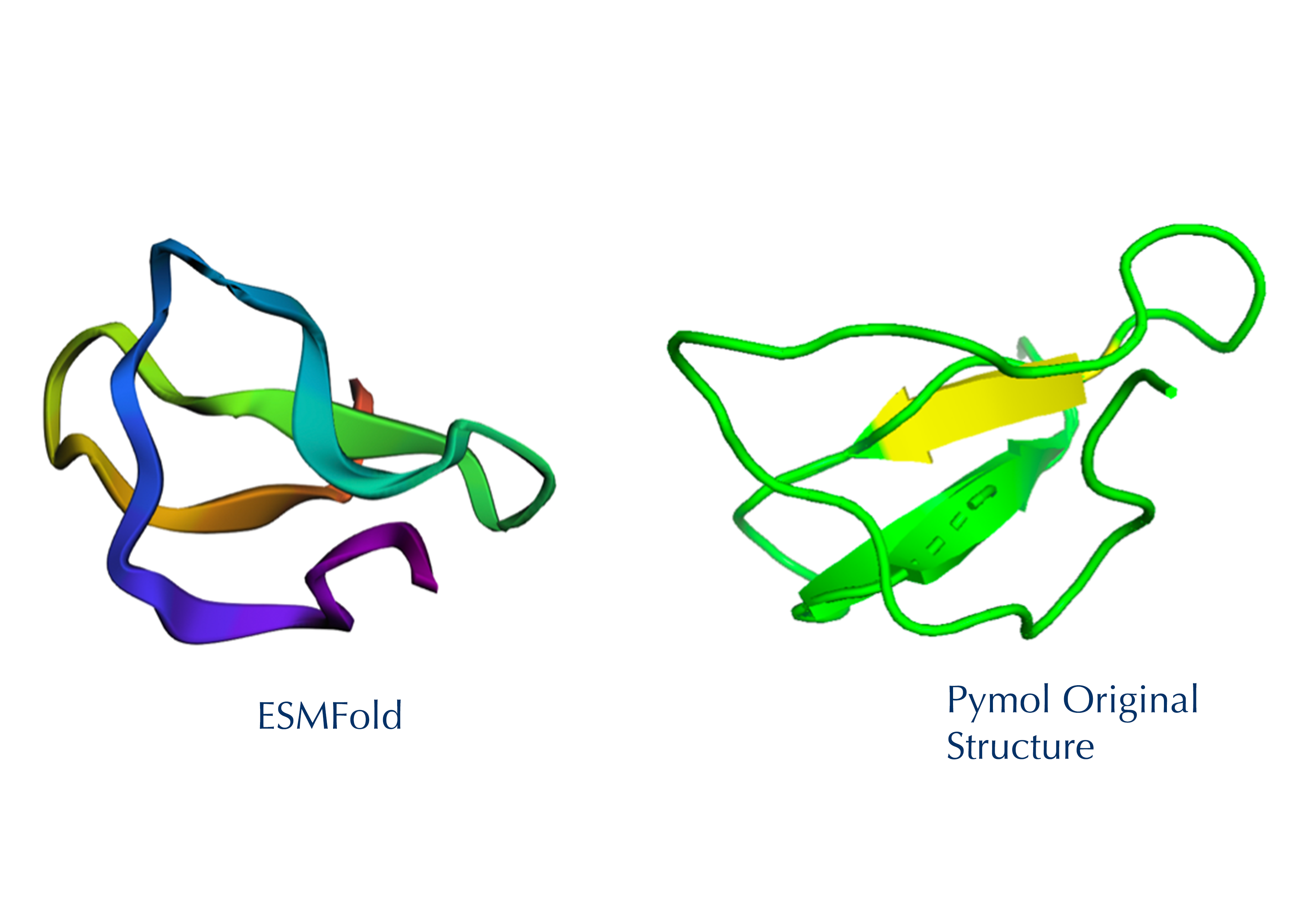
Yes, the predicted coordinates from ESMFold appear to match the original structure quite well. The overall beta sheet-rich fold is preserved, and the main secondary structure elements align closely with the original 35-aa active region of Huwentoxin-IV. While there may be minor differences in loop orientation or flexible regions, the core remains essentially the same.
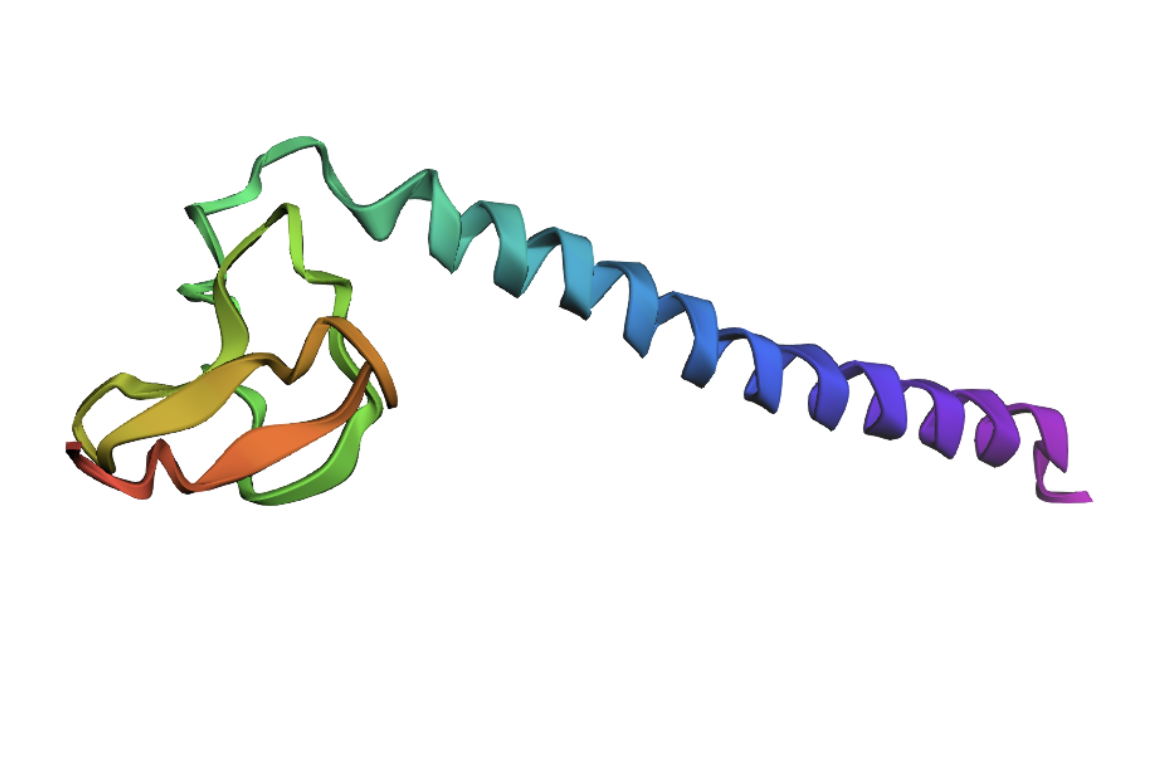
I decided to also look at the full precursor protein, since I had not done so in Section B because of some initial confusion. Most papers mainly mention and highlight the 35-amino-acid mature toxin, and although the 89-aa precursor is listed on the PDB mini-protein page, opening the .cif file in PyMOL only shows the structure of the active toxin region. Since this is also the main focus of my final project idea (Option 3), it makes sense that this is where most researchers have concentrated their attention. Looking at the full precursor, we can see that what was previously a structure dominated by beta sheets now also includes a long alpha helix, which is therefore not part of the active toxin site.
2.Try changing the sequence: first try some mutations, then large segments. Is your protein structure resilient to mutations?
Processor Sequence:
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQIGK
(Bold: active protein)
MUTATION 1
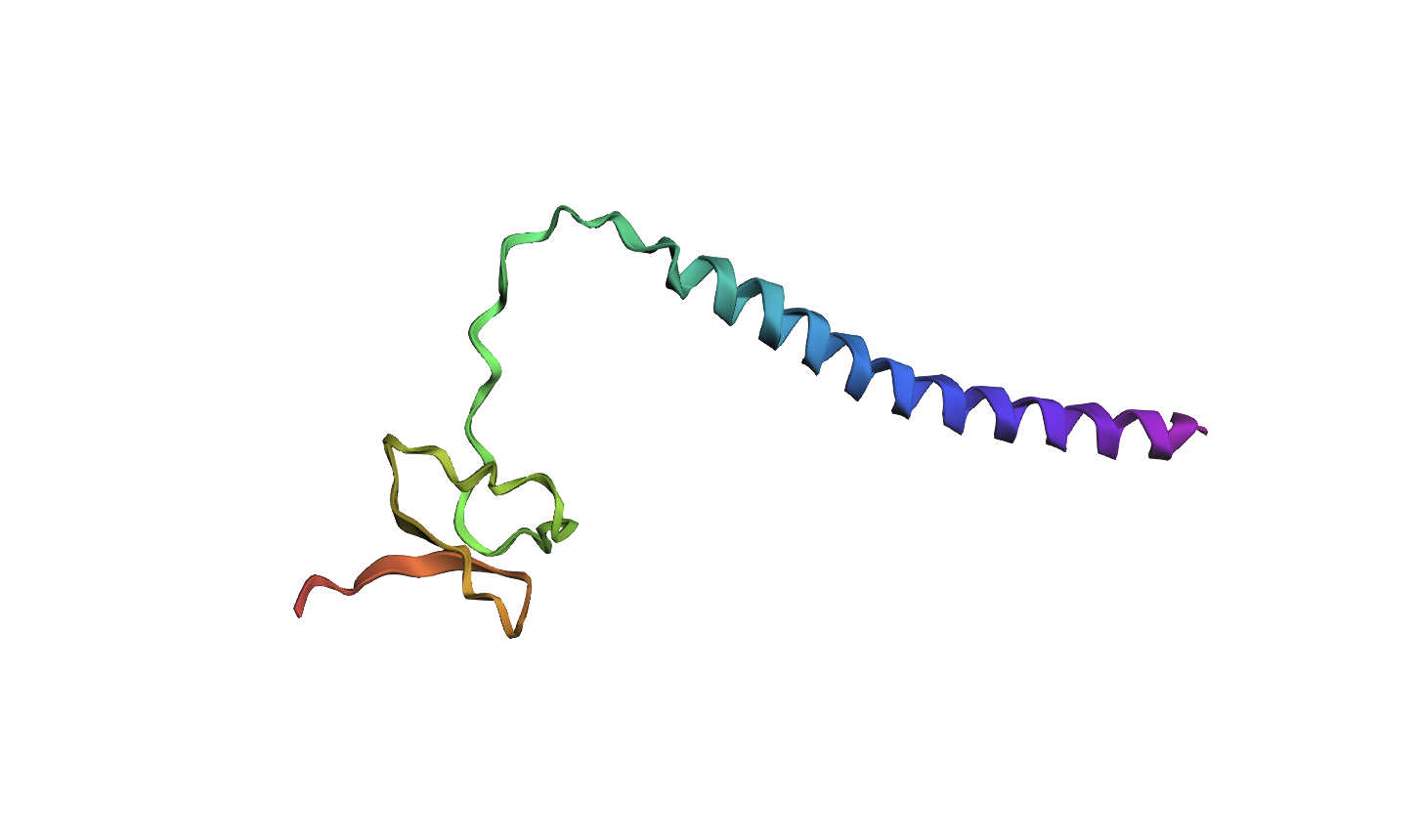
The active part of the protein, located at 50-85 in the full precursor sequence, Amino acids (Tryptophan) W, Histidine (H) and Methionine (M) seem to have lower tolerance to mutations throughout that section of the sequence. I want to see if altering one of these specific amino acids in the active protein will produce a visible change in the structure.
Mutated Sequence 1: Changed Lysine (K) to Tryptophan (W) on 57aa
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWACNPSNDQCCKSSKLVCSRKTRWCKYQIGK
Analysis: It appears that even changing a singular amino acid in the active site of the protein has already significantly altered the local structure.
MUTATION 2
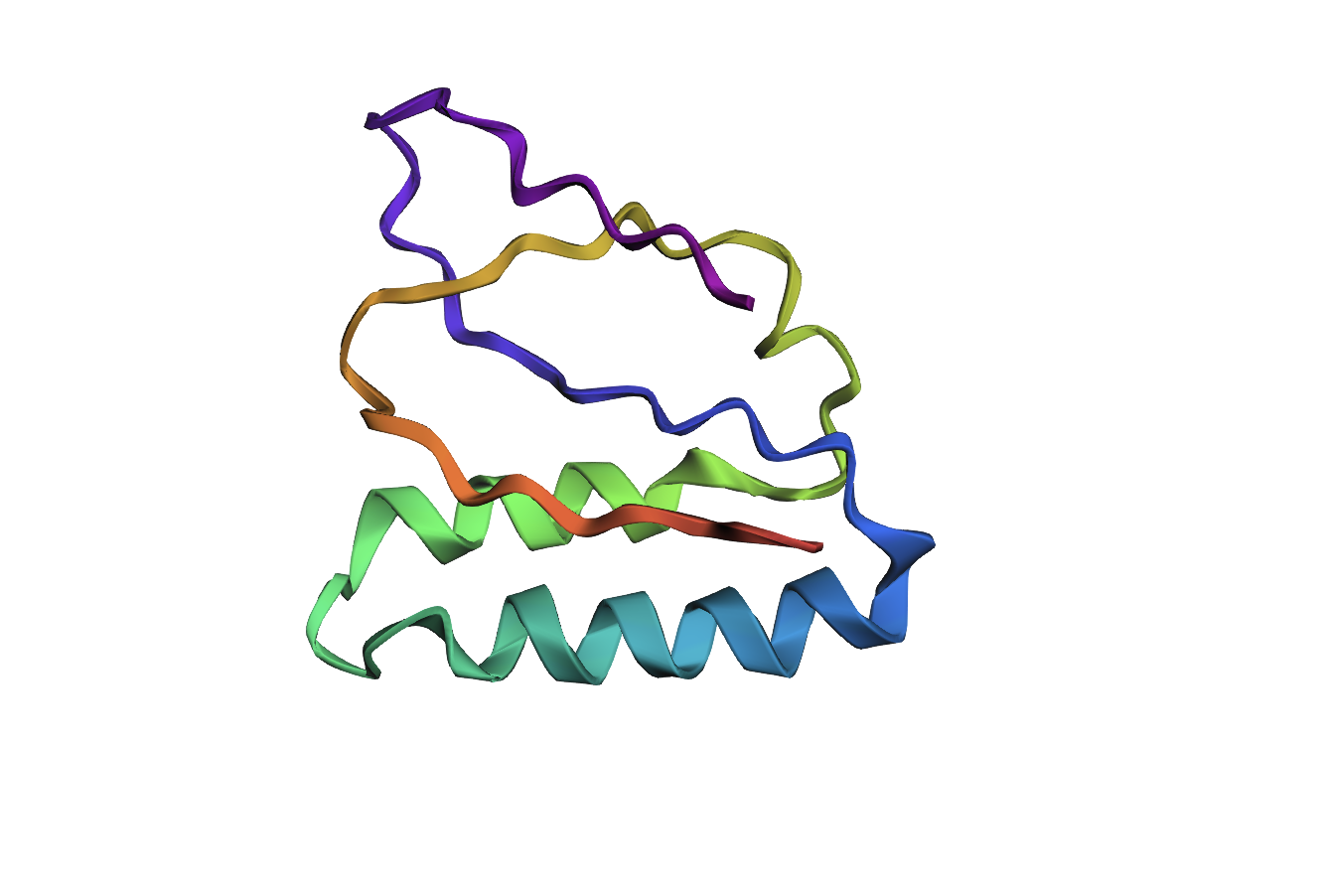
Secondly, I changed 3 amino acids in the protein’s active region, once again focusing on those at higher risk of mutation.
Mutated Sequence 2: Changed KAC to WHM on 57-59aa
MVNMKASMFLALAGLVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
Analysis: Similar to the previous mutation, altering amino acids in the active site continues to modify the local structure. However, the active-site fold remains broadly similar across both mutations. Relative to the original structure, the beta strands seem to become progressively more separated, suggesting that increasing the number of mutations may gradually reduce the compactness of the native fold.
MUTATION 3
Additionally, I wanted to focus on the active protein as it also showed interesting results on the mutation heat map. Specifically, in amino acids E and D towards the start of the sequence (positions 11 and 13), which are located in the alpha helix section of the protein, show significantly low numbers on the scale (-6), suggesting high mutational intolerance. I therefore predict this would alter the structure/position of the helix.
Mutated sequence 3: Changed MFLALAGL to EDEDDEED on 8-16aa
MVNMKASEDEDDEEDVLLFVVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
Analysis: The mutation caused a major alteration in the protein’s overall structure, leading to significant distortion of the alpha helices and beta sheets and a noticeable change in the overall folding pattern.
MUTATION 4
Lastly, I also wanted to look at changing a larger section of my sequence and opted for the first 20 amino acids, as overall, the mutation heat map suggests a high tolerance to mutations. Therefore, I predict that altering this part of the sequence shouldn’t alter the overall structure too much.
Mutated Sequence 4: Changed MVNMKASMFLALAGLVLLFV to EDWWHMELFALALSQKICVD on 1-20aa
EDWWHMELFALALSQKICVDVCYASESEEKEFSNELLSSVLAVDDNSKGEERECLEIFWHMNPSNDQCCKSSKLVCSRKTRWCKYQIGK
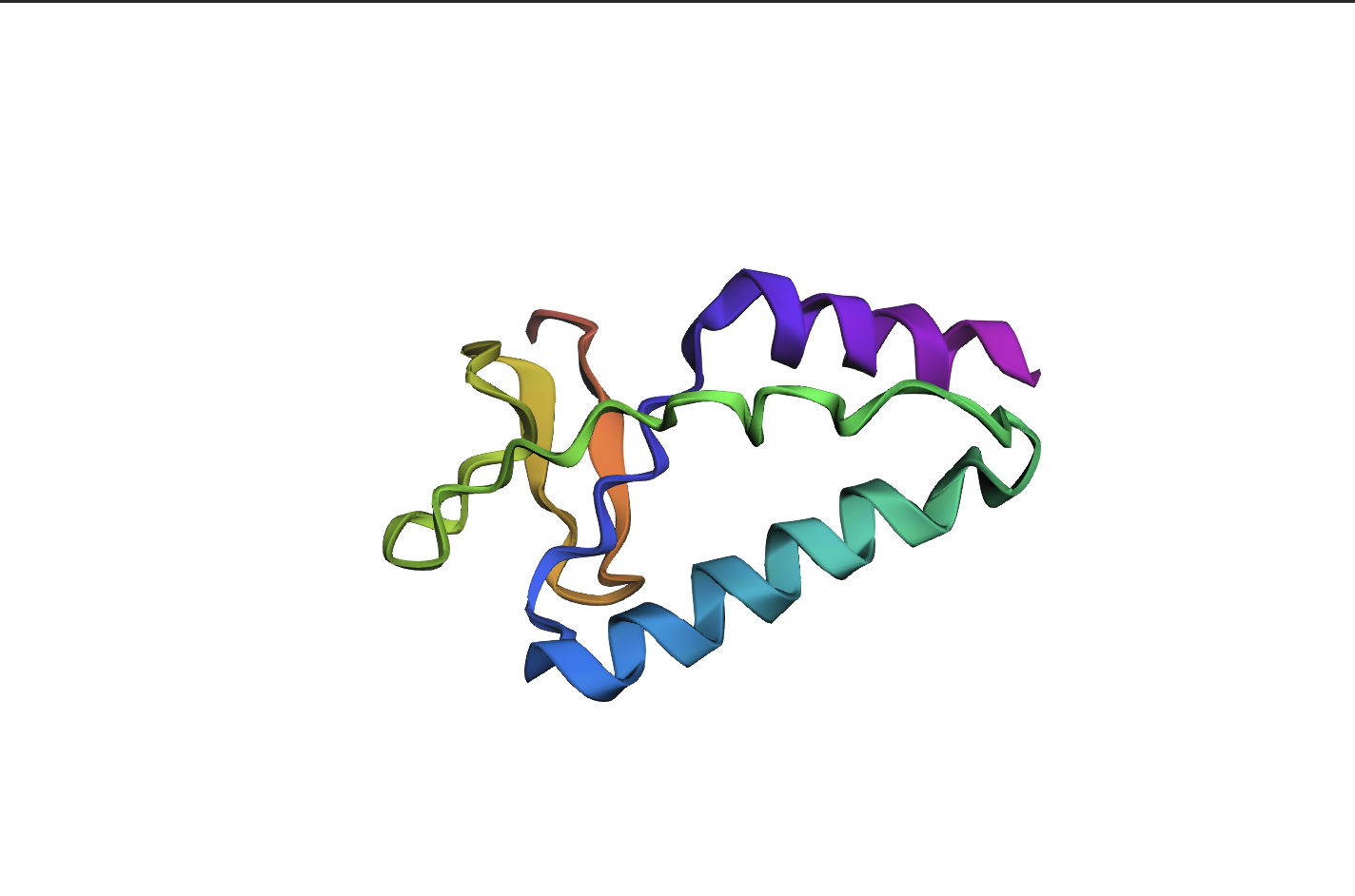
Analysis: The mutation in the first 20 amino acids resulted in a significant alteration of the overall protein structure, with major disruptions to the alpha helices and beta sheets. This indicates that, despite predictions from the mutation heat map, this region is less tolerant to sequence changes than expected.
Conclusion: Overall, the protein shows very low resistance to mutations, likely due to its small size and peptide nature, which makes structural stability more sensitive to sequence changes.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.

Using the backbone from Huwatoxin (PDB: 1MB6, active 35aa region) in ProteinMPNN, I generated a new sequence candidate:
EPKGENTPCTEENQNCDKEKNVECSPEKGACAPP
It scored 1.1293, lower than the original backbone’s 2.3287, showing it’s a plausible but less optimal fit to the structure. The sequence recovery of 0.2286 (23%) indicates low similarity to the wild-type (~9% identity), meaning a substantial redesign. Compared to the original’s mutation heat map, this designed sequence appears much more mutation-tolerant, suggesting improved structural robustness for engineering.
Heat map of the generated sequence candidate:
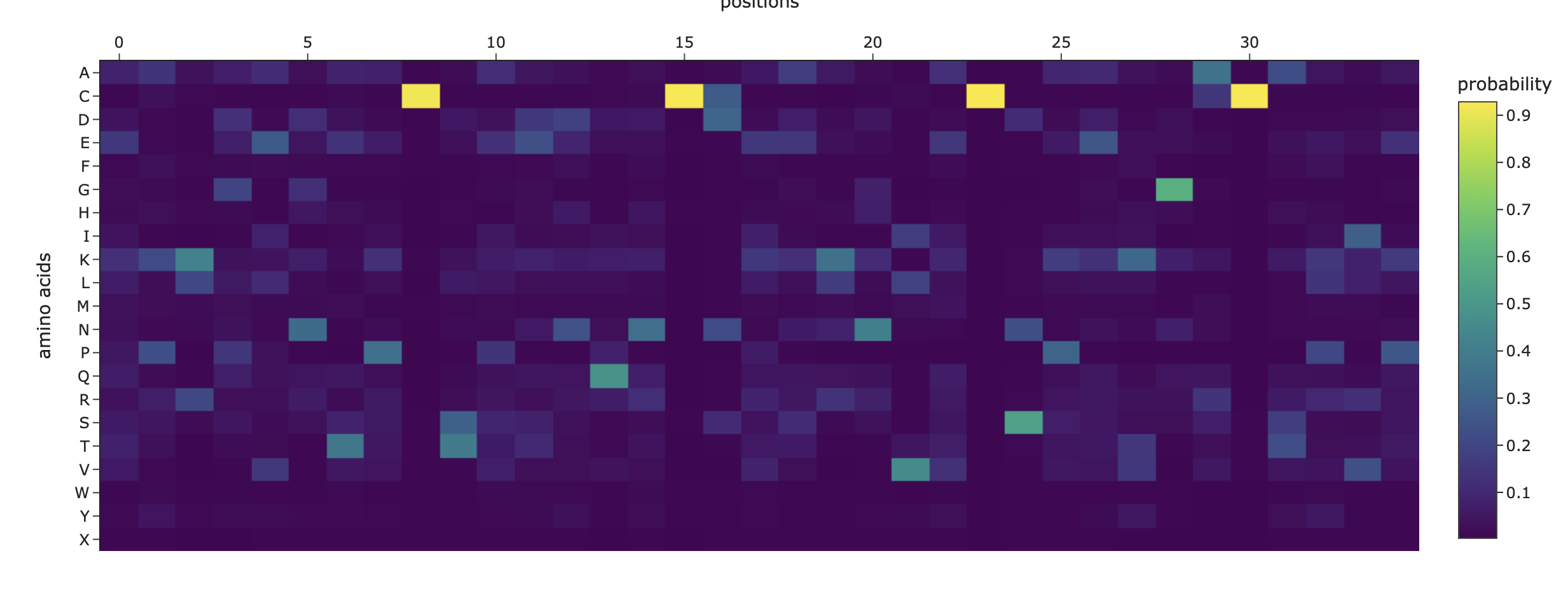
2. Input this sequence into ESMFold and compare the predicted structure to your original.
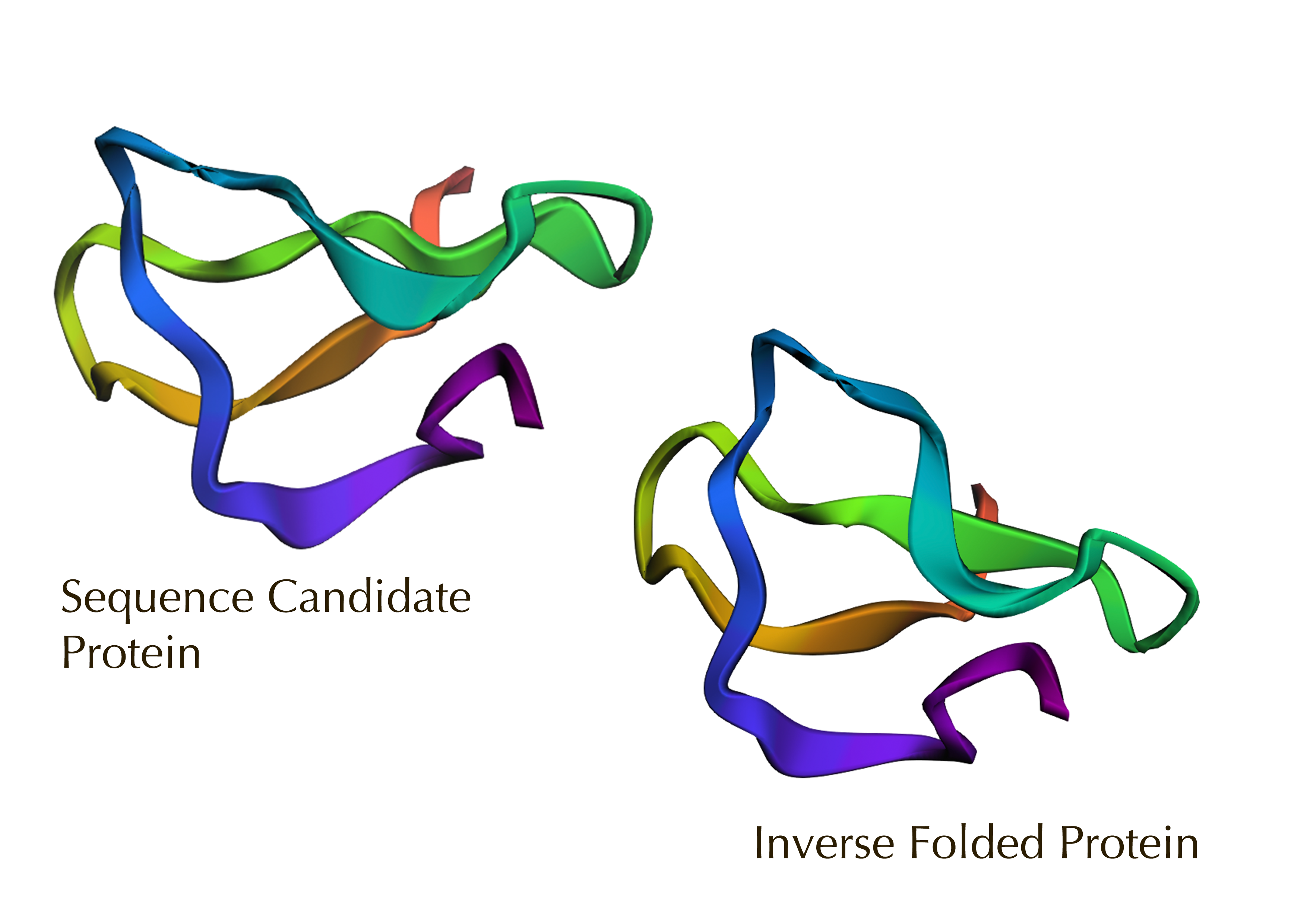
The sequence candidate model appears more looped and flexible, while the inverse-folded model on a fixed backbone is more compact and helical. Overall, this confirms a high fidelity visual overlap despite sequence changes, validating the design’s structural stability.
Part D. Group Brainstorm on Bacteriophage Engineering
Group of Commited Listeners LifeFabs: Sara Gaviria Escobar, Ruben Janssen, Justine de RiedmattenProposal: Engineering the MS2 Lysis Protein L to Enhance Stability Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis; this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been widely used in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
To use computational protein design tools to engineer possible stable variants of the MS2 L-protein.
To identify variants where structure is preserved but higher stability is shown
Analyse if the variants can fold like the original protein and if they interact correctly with DnaJ, its chaperone
MethodsObtain an initial protein backbone for MS2 L-protein using ESMFold.Obtain alternative sequences to the protein using ProteinMPNN.Mutate the alternative sequences to the protein using ESM-2.Model the variants using ESMFold and analyse if the folding is maintained in comparison to the initial protein (MS2 L-protein). Create a ranking based on the confidence metric.sAssess the top 3 variants’ interactions with DnaJ using AlphaFold-Multimer to predict 3D structures of protein complexes (co-folding multiple chains)Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.References
Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28.Mylon, S. E., Rinciog, C. I., Schmidt, N., Gutierrez, L., Wong, G. C., & Nguyen, T. H. (2010). Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir, 26(2), 1035-1042.Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 lysis of Escherichia coli depends on host chaperone DnaJ. Journal of Bacteriology, 199(12), 10-1128.Mondal, A., Singh, B., Felkner, R. H., De Falco, A., Swapna, G. V. T., Montelione, G. T., … & Perez, A. (2024). A Computational Pipeline for Accurate Prioritization of Protein‐Protein Binding Candidates in High‐Throughput Protein Libraries.Angewandte Chemie International Edition, 63(24), e202405767.Week 5: Protein Design II
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.Human SOD1 Sequence (154AA per monomer - 308AA):
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQMATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutation: The alanine to valine mutation at codon 4 (A4V) of SOD1 causes a rapid and progressive form of amyotrophic lateral sclerosis (ALS). In Uniprot, however, it appears that the sequence starts with Methionine, making the mutation from Alanine to Valine actually at codon 5.
Mutation A4V (one of the monomers):
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
References:
Saeed, M., Yang, Y., Deng, H-X., Hung, W-Y., Siddique, N., Dellefave, L., Gellera, C., Andersen, P.M. and Siddique, T. (2009). Age and founder effect of SOD1 A4V mutation causing ALS. Neurology, 72(19), pp.1634–1639. doi:https://doi.org/10.1212/01.wnl.0000343509.76828.2a.Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
 3. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
3. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
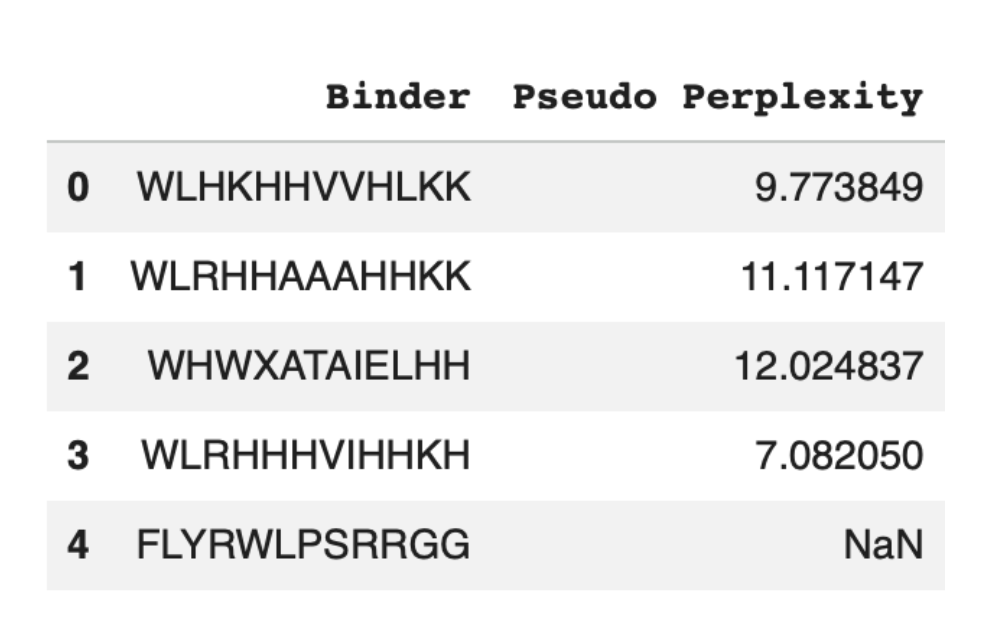
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Asked for the built-in Gemini AI feature on the Colab for another code to get the pseudo perplexity of the known SOD1-binding peptide:
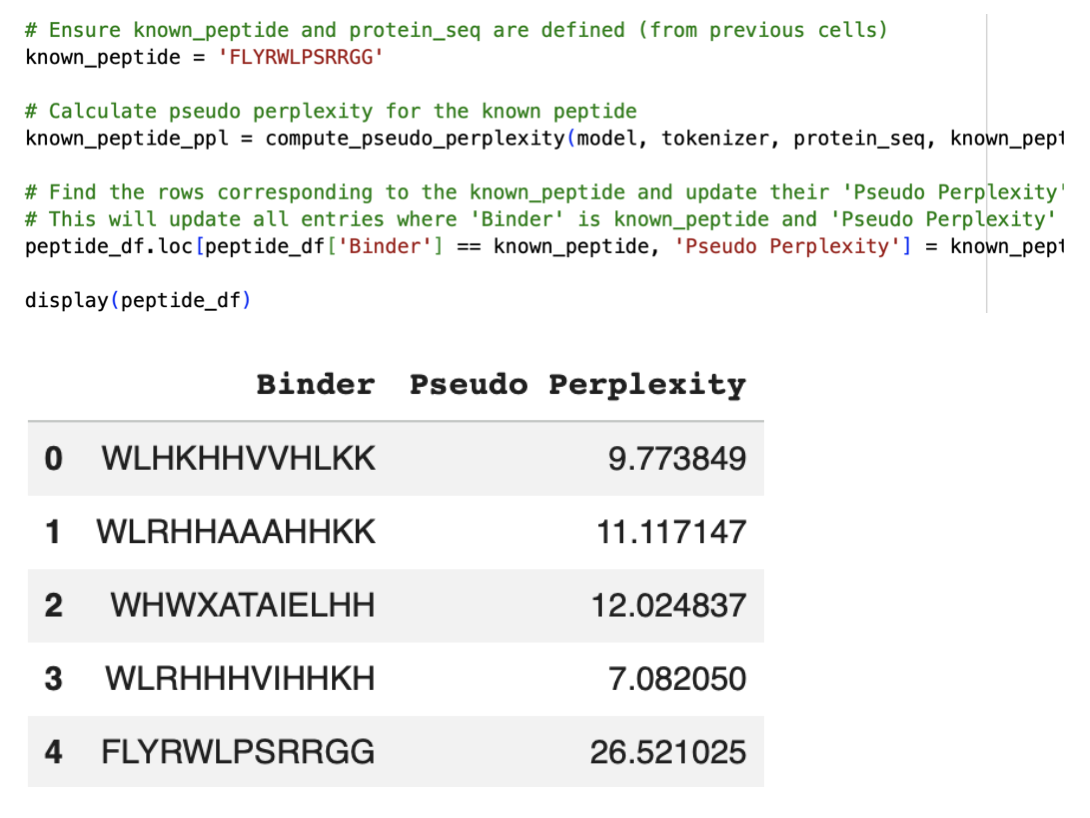
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
When entering the pepitdes, when adding sequence numbered 2 in the table above, the AlphaFold Server wasn’t letting me enter the ‘X’ Amino Acid as it isn’t an actual amino acid and will be used in a sequence for an unknown or undetermined AA. I therefore had to alter this sequence:
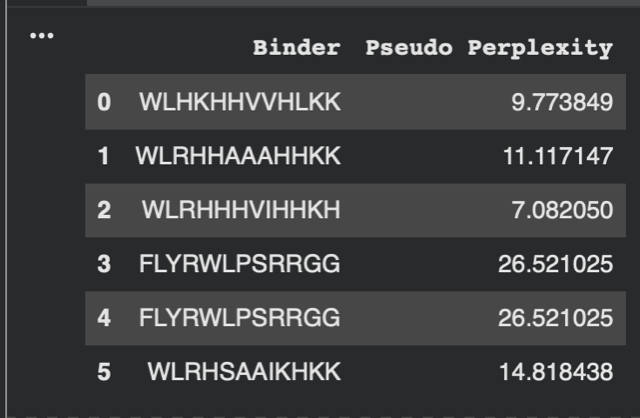
2. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localise near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
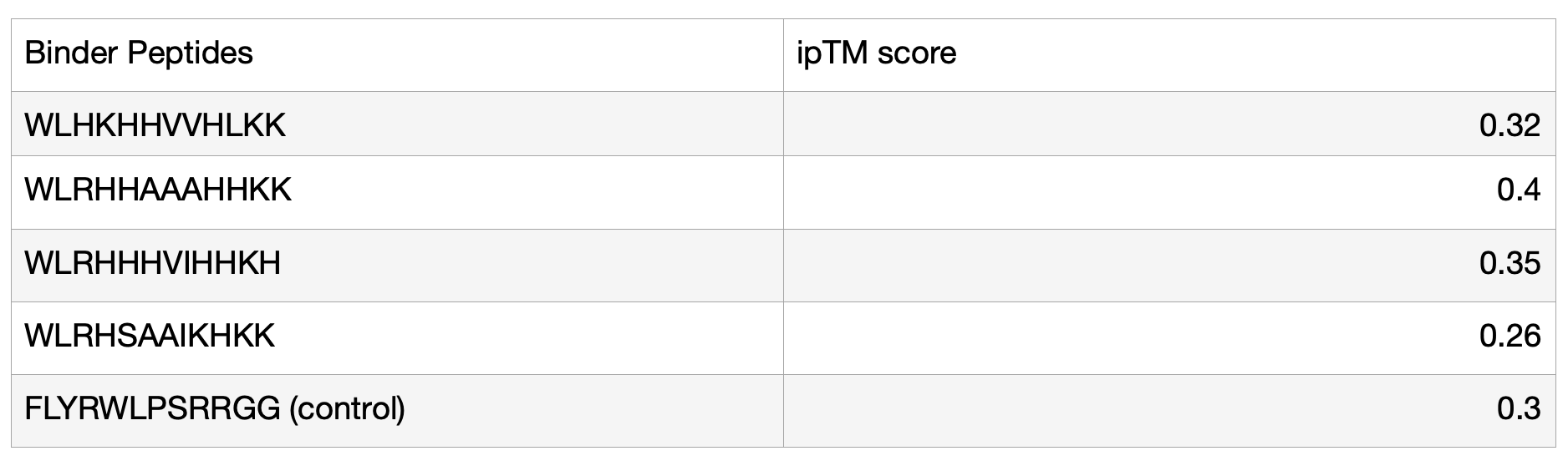
Binder 1: WLHKHHVVHLKK
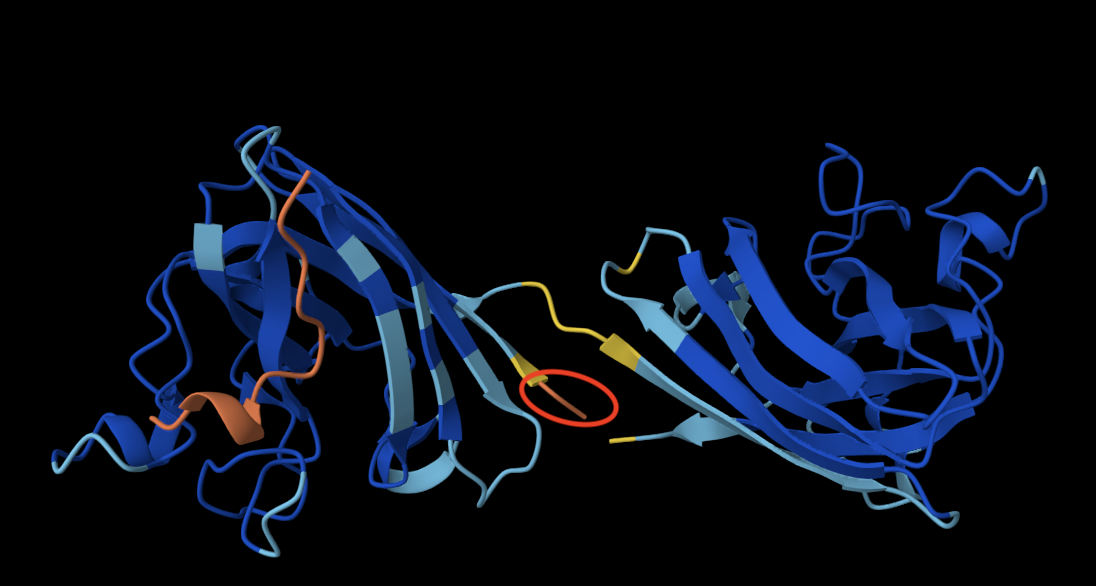
The peptide doesn’t bind directly to the protein and is not located close to the N terminus (circled in red in the image above, always the orange end). It engages with the Beta barrel of the lefmonomer. It also appears to be surface-bound, as it is essentially exposed to the solvent and not in contact with the protein. I was initially confused about why I had two identical monomers, but after some research, I learned that SOD1 is a homodimer composed of two identical subunits that mirror one another.
Binder 2: WLRHHAAAHHKK
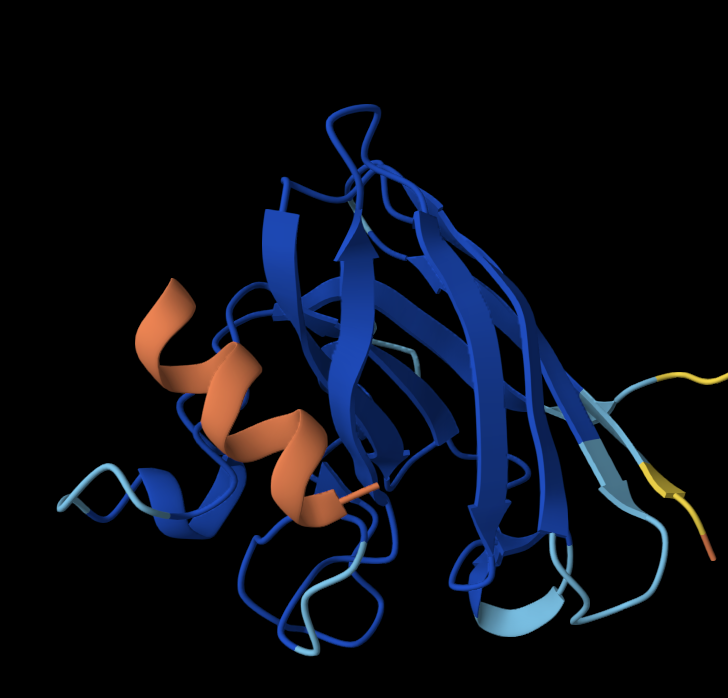
Once again, the peptide doesn’t localise close to the N terminus; it is close to the Beta barrel but doesn’t interact with it and is surface-bound.
Binder 3: WLRHHHVIHHKH
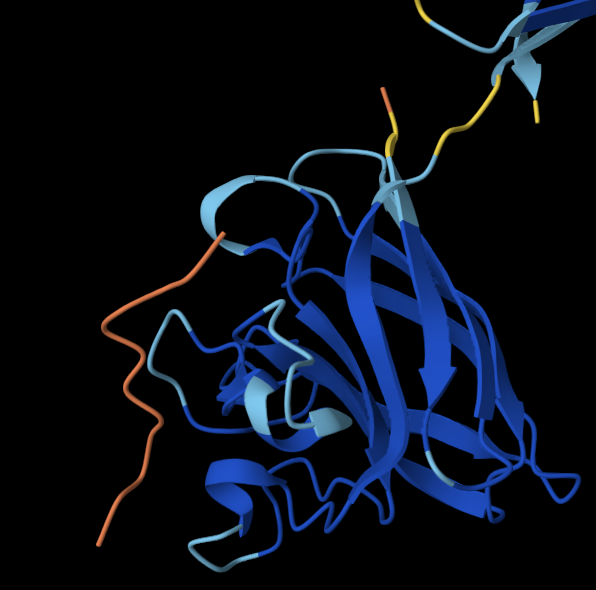
The peptide doesn’t localise close to the N terminus, and doesn’t engage with the
Beta barrel or the dimer interface and is surface-bound.
Binder 4: WLRHSAAIKHKK
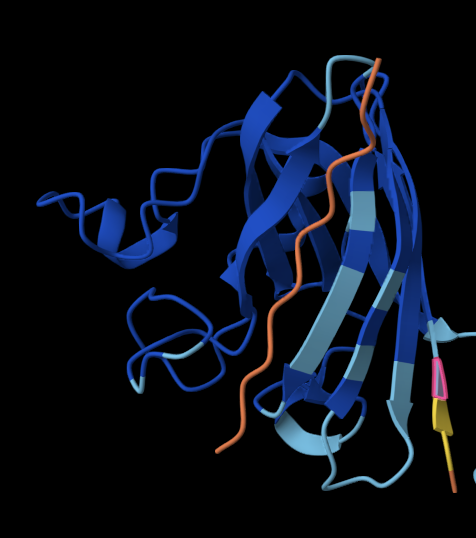
The peptide doesn’t localise close to the N terminus, and is the closest out of all the other peptides to the Beta barrel. It is also surface-bound.
Controlled Binder: FLYRWLPSRRGG
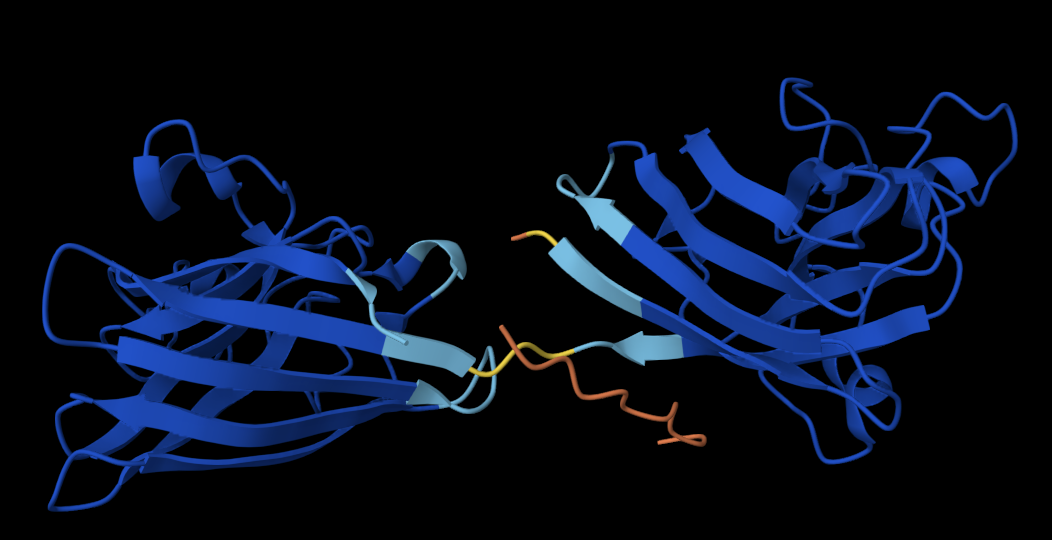
The peptide doesn’t localise close to the N terminus, is slightly partially buried and approaches the dimer surface, where the two monomers join.
3. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The ipTM values of my generated peptides are in a range of 0.26 to 0.4, with an average of 0.33. In comparison, the known (control) binder has a score of 0.3. Ultimately, they are relatively similar, but 3/4 of my generated peptides have a higher score than the known binder. Overall, all the binders have a relatively low confidence in the predicted interface, translating to weak interactions between the protein and the binder.
Evaluate Properties of Generated Peptides in the PeptiVerse
1. Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:Binder 1:
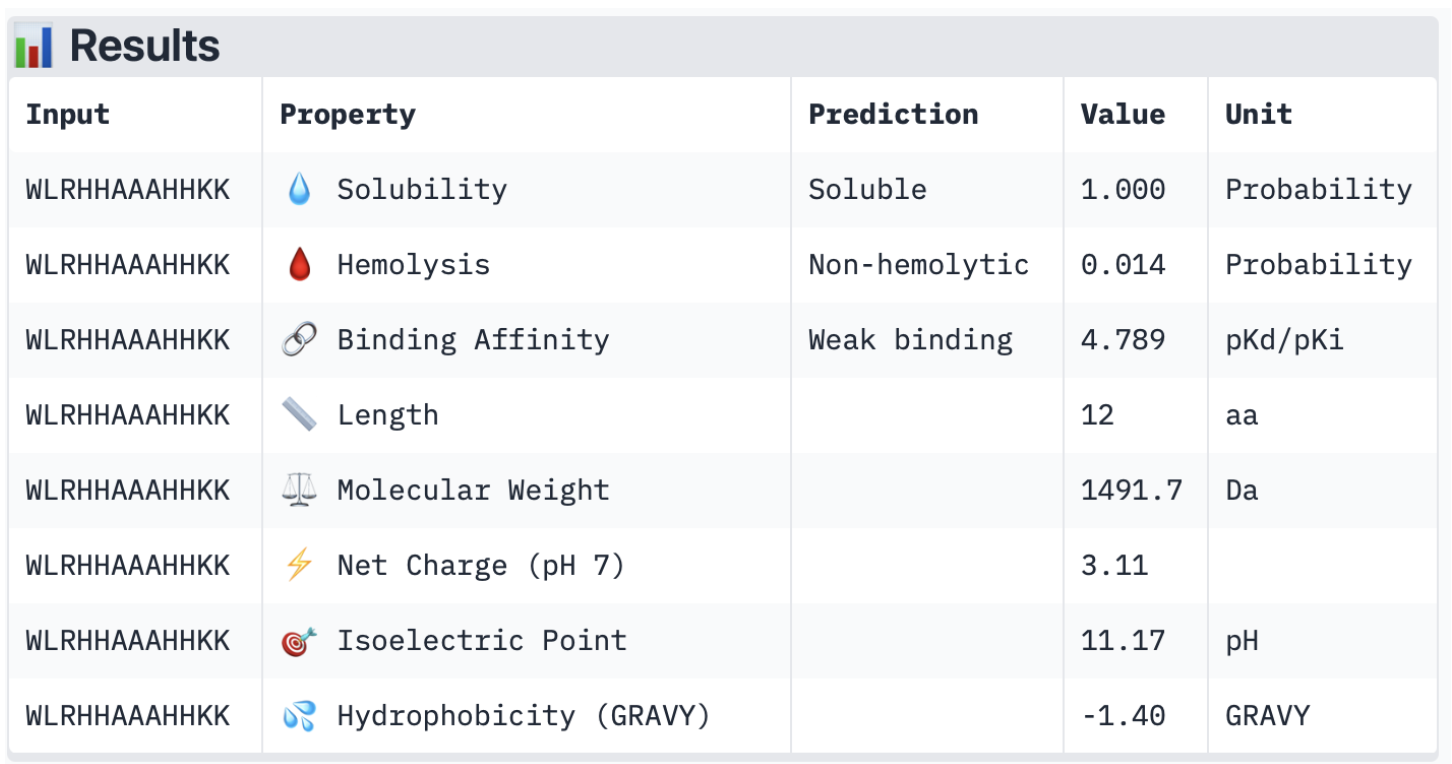 Binder 2:
Binder 2:
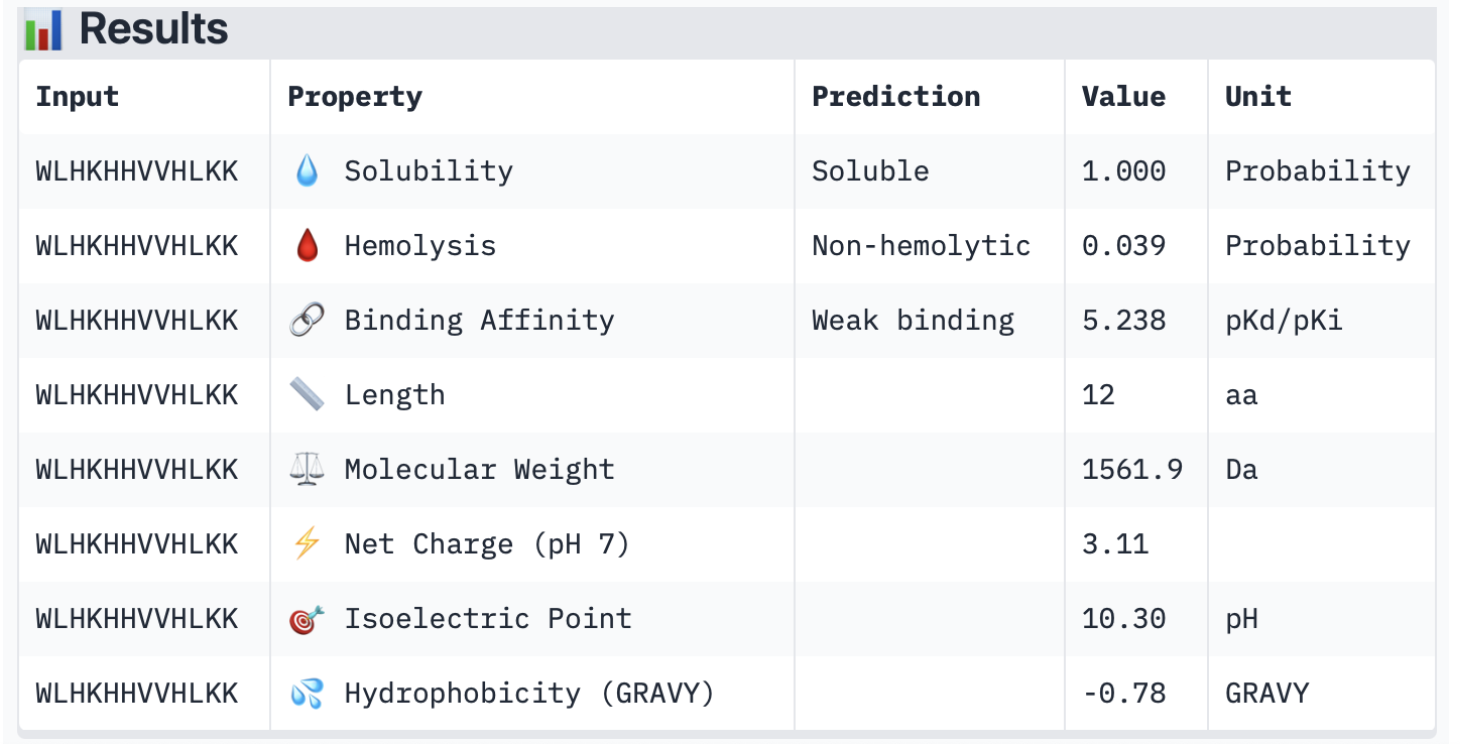 Binder 3:
Binder 3:
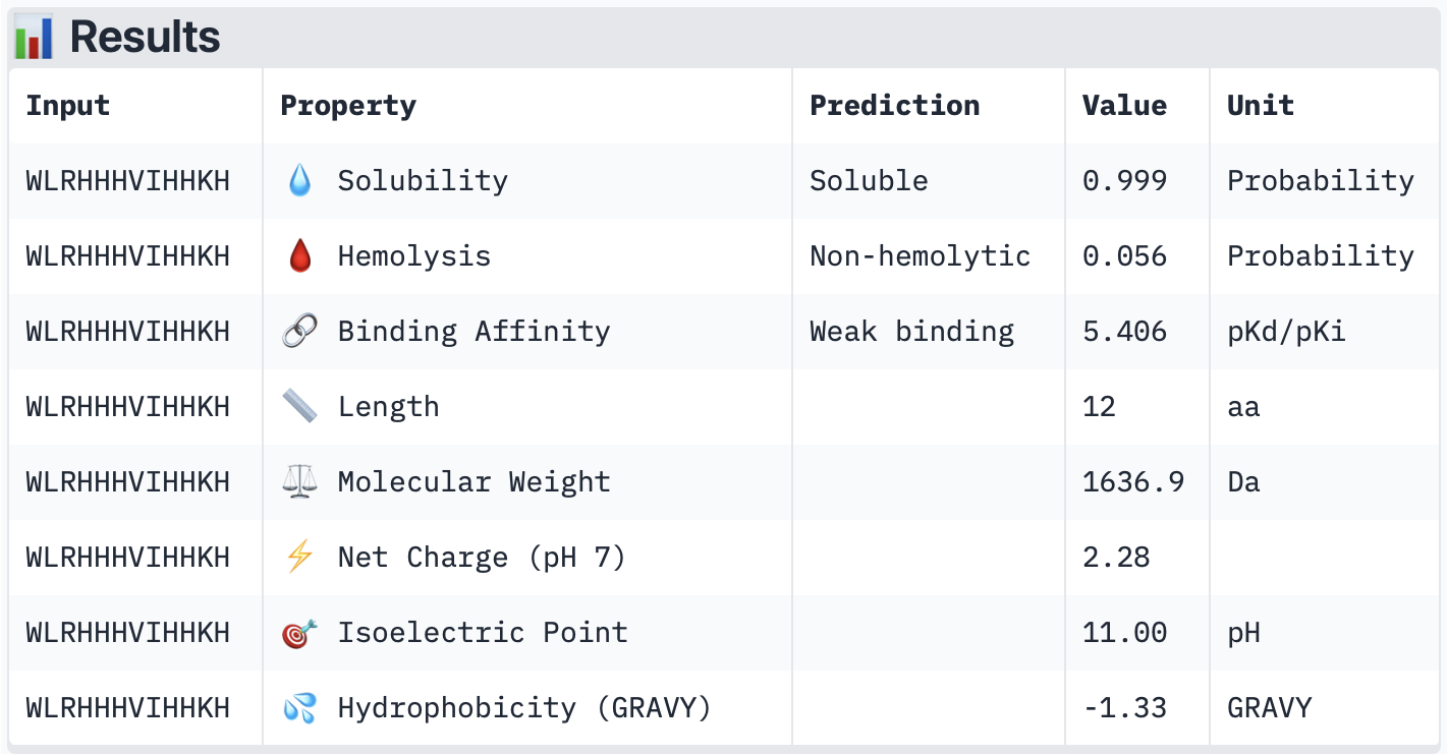 Binder 4:
Binder 4:
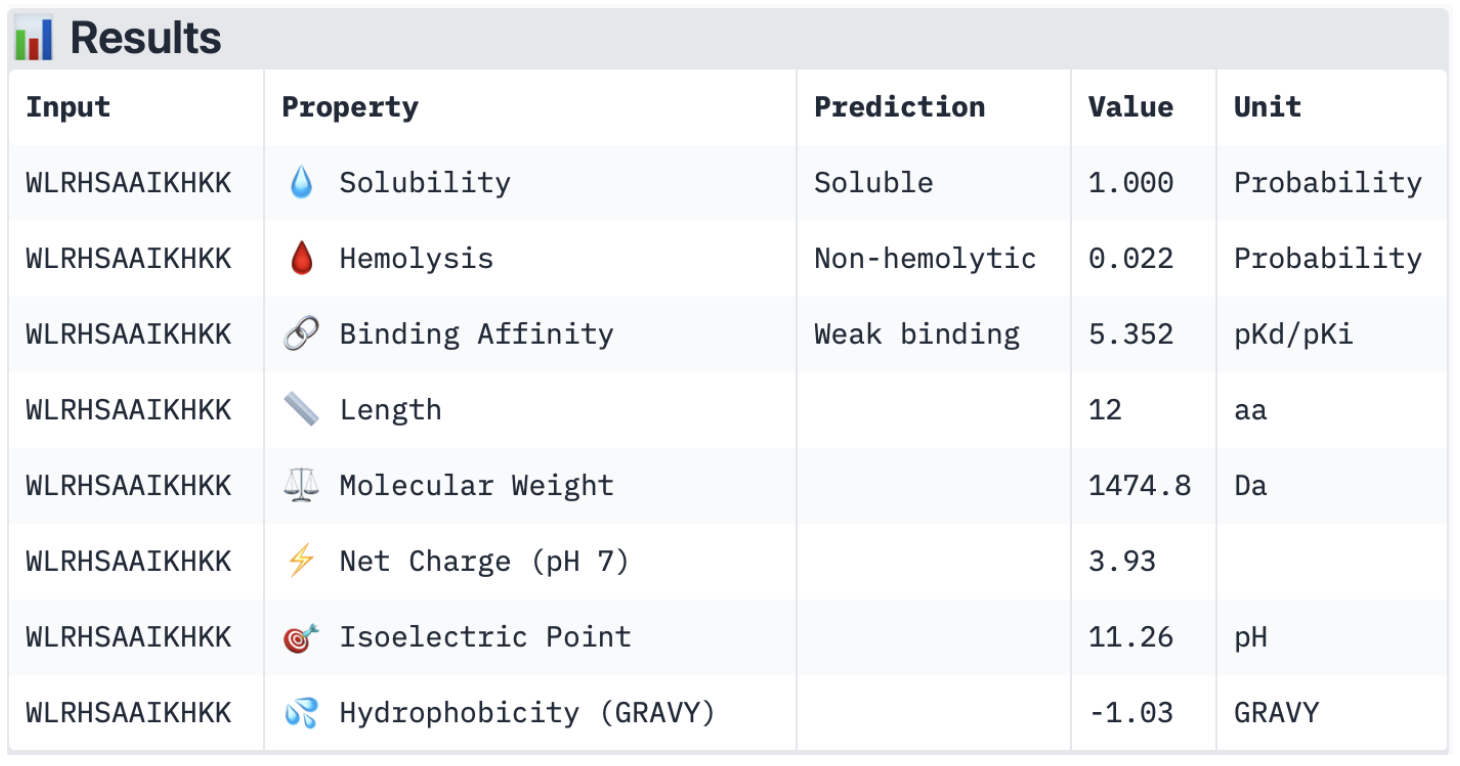
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Overall, all the binders have a weak binding affinity, which matches the ipTM scores described earlier. This also backs up the 3D structures where none of the peptides was closely interacting with the protein. In terms of solubility, all of them are soluble, which is therapeutically beneficial as it improves dissolution rates and increases absorption (Saeed et al., 2009). Furthermore, they are all non-hemolytic (low hemolysis probability), which is also therapeutically beneficial, as it ensures minimum red blood cell destruction (Cleveland Clinic, 2022). Looking at the net charge, SOD1 is negatively charged (-6 monomers) with the physiological pH of 7.4 (Shi et al., 2014), and all the binders are positively charged with a pH ranging from 2.28 to 3.93. This is generally advantageous as the positively charged binders will be attracted to the negatively charged SOD1 surface; however, when they are too positively charged, it may also cause the binders to bind to other negatively charged molecules. Lastly, in terms of molecular weight, all binders have a relatively low value, ranging from 1474.8 to 1636.9 Da, which minimises aggregation risks.
I believe the peptide which best balances predicted binding and therapeutic properties would be peptide 3 (WLRHHHVIHHKH), firstly due to having the highest ipTM score. Secondly, in terms of its therapeutic properties, it has the highest binding affinity out of all the peptides, as well as having high solubility and being non-hemolytic. Although all peptides had similar properties and scores, it appears to be the most appropriate option for further development.
References:
Cleveland Clinic (2022). Hemolysis: Types, causes & symptoms. [online] Cleveland Clinic. Available at: https://my.clevelandclinic.org/health/diseases/24108-hemolysis [Accessed 20 Apr. 2026].Saeed, M., Yang, Y., Deng, H-X., Hung, W-Y., Siddique, N., Dellefave, L., Gellera, C., Andersen, P.M. and Siddique, T. (2009). Age and founder effect of SOD1 A4V mutation causing ALS. Neurology, 72(19), pp.1634–1639. doi:https://doi.org/10.1212/01.wnl.0000343509.76828.2a.
with a value of around 1.0 and are non-hemolytic.Shi, Y., Abdolvahabi, A. and Shaw, B.F. (2014). Protein charge ladders reveal that the net charge of ALS‐linked superoxide dismutase can be different in sign and magnitude from predicted values. Protein Science, 23(10), pp.1417–1433. doi:https://doi.org/10.1002/pro.2526.Generate Optimised Peptides with moPPIt
Settings for moPPIt peptide generation towards specific residues and optimisation of the therapeutic properties: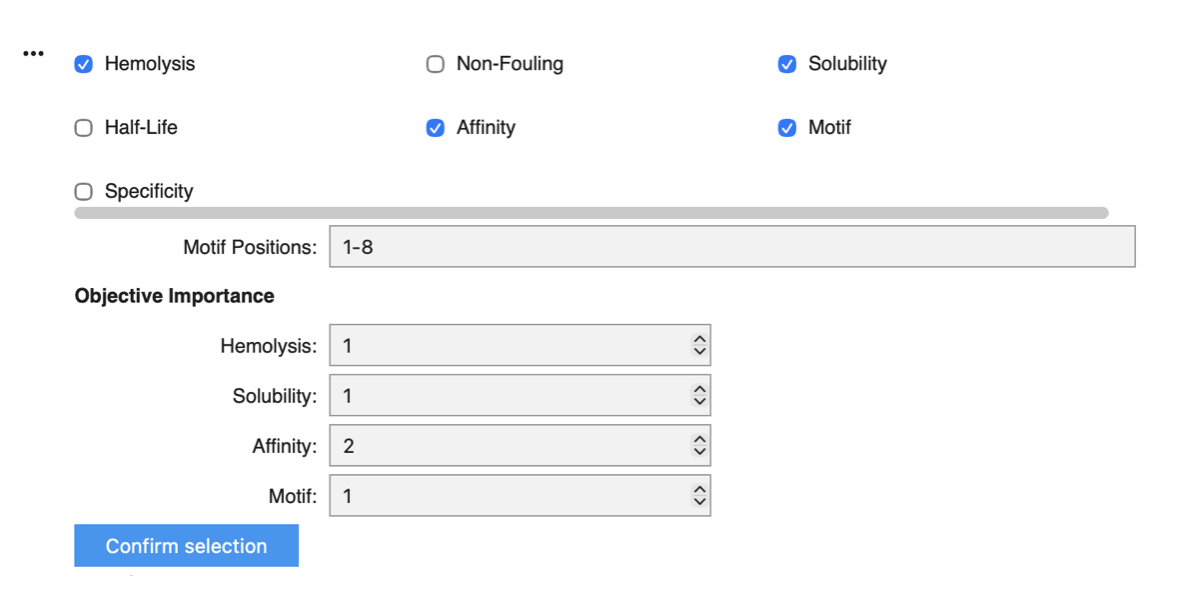
I focused on Motif positions 1–8 to keep the design space close to the mutation site at position 4. I increased the weight of binding affinity to 2, as this is ultimately the most critical objective, while assigning similar weights to hemolysis, solubility, and motif to preserve overall therapeutic properties.
Generated moPPit peptides (37-minute runtime):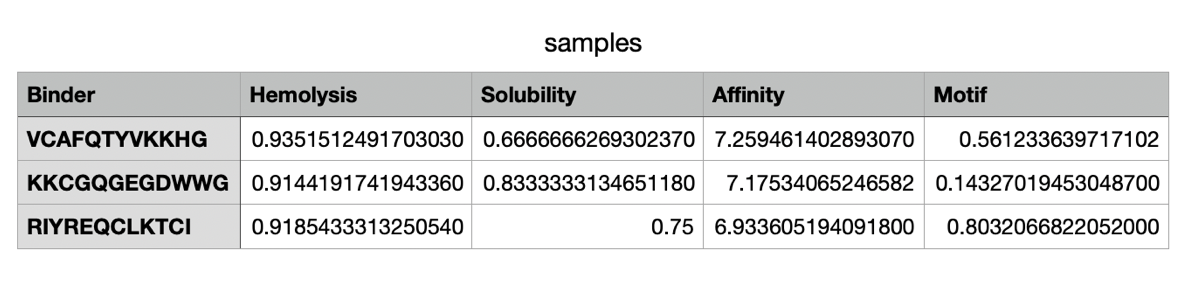 Briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?The moPPit peptides generated differ from the ones I have generated on PepMLM in their binding affinity, solubility and hemolysis risks. moPPit peptides have a significantly higher affinity score (6.93-7.26) in comparison to the PepMLM ones. This could essentially be due to the motif position, as well as increasing the weight of affinity in the settings. In terms of solubility, the moPPit peptides have a lower solubility value than the PepMLM peptides, which makes them slightly less advantageous in this specific therapeutic property. Additionally, they have a significantly higher hemolysis value, resulting in potentially higher risks of red blood cell destruction. Ultimately, the moPPit peptides appear to bind more strongly but appear less favourable for safety and developability. To evaluate these peptides before advancing them to clinical studies, I would begin by testing their predictions in an in-silico model through in vitro experiments before any animal or human trial. Furthermore, I would conduct a safety screening (Cyprotex in vitro hemolysis testing) to evaluate the level of toxic hemolysis (Bauch, 2026).
References:
Bauch, C. (2026). Hemolysis | Cyprotex ADME-Tox Solutions. [online] Evotec Website (English). Available at: https://www.evotec.com/solutions/drug-discovery-preclinical-development/cyprotex-adme-tox-solutions/toxicology/mechanistic-toxicity/hemolysis [Accessed 25 Mar. 2026].Final Project: L-Protein Mutants
Option 1: Mutagenesis
Step 1: Gathering information about Lysis proteins
DNA sequence of Lysis Protein Phage MS2 (E. coli) 225bp:
atggaaacccgctttccgcagcagagccagcagaccccggcgagcaccaaccgccgccgcccgtttaaacatgaagattatccgtgccgccgccagcagcgcagcagcaccctgtatgtgctgatttttctggcgatttttctgagcaaatttaccaaccagctgctgctgagcctgctggaagcggtgattcgcaccgtgaccaccctgcagcagctgctgacc
Reference: EMBL-EBI (2026). ENA Browser. [online] Ebi.ac.uk. Available at: https://www.ebi.ac.uk/ena/browser/view/V00642 [Accessed 26 Mar. 2026].
Chaperone DNAj sequence (E. coli, strain K12) 1358bp:
TCGACGCTGAATTTGAAGAAGTCAAAGACAAAAAATAATCGCCCTATAAACGGGTAATTA
TACTGACACGGGCGAAGGGGAATTTCCTCTCCGCCCGTGCATTCATCTAGGGGCAATTTA
AAAAAGATGGCTAAGCAAGATTATTACGAGATTTTAGGCGTTTCCAAAACAGCGGAAGAG
CGTGAAATCAGAAAGGCCTACAAACGCCTGGCCATGAAATACCACCCGGACCGTAACCAG
GGTGACAAAGAGGCCGAGGCGAAATTTAAAGAGATCAAGGAAGCTTATGAAGTTCTGACC
GACTCGCAAAAACGTGCGGCATACGATCAGTATGGTCATGCTGCGTTTGAGCAAGGTGGC
ATGGGCGGCGGCGGTTTTGGCGGCGGCGCAGACTTCAGCGATATTTTTGGTGACGTTTTC
GGCGATATTTTTGGCGGCGGACGTGGTCGTCAACGTGCGGCGCGCGGTGCTGATTTACGC
TATAACATGGAGCTCACCCTCGAAGAAGCTGTACGTGGCGTGACCAAAGAGATCCGCATT
CCGACTCTGGAAGAGTGTGACGTTTGCCACGGTAGCGGTGCAAAACCAGGTACACAGCCG
CAGACTTGTCCGACCTGTCATGGTTCTGGTCAGGTGCAGATGCGCCAGGGATTCTTCGCT
GTACAGCAGACCTGTCCACACTGTCAGGGCCGCGGTACGCTGATCAAAGATCCGTGCAAC
AAATGTCATGGTCATGGTCGTGTTGAGCGCAGCAAAACGCTGTCCGTTAAAATCCCGGCA
GGGGTGGACACTGGAGACCGCATCCGTCTTGCGGGCGAAGGTGAAGCGGGCGAGCATGGC
GCACCGGCAGGCGATCTGTACGTTCAGGTTCAGGTTAAACAGCACCCGATTTTCGAGCGT
GAAGGCAACAACCTGTATTGCGAAGTCCCGATCAACTTCGCTATGGCGGCGCTGGGTGGC
GAAATCGAAGTACCGACCCTTGATGGTCGCGTCAAACTGAAAGTGCCTGGCGAAACCCAG
ACCGGTAAGCTATTCCGTATGCGCGGTAAAGGCGTCAAGTCTGTCCGCGGTGGCGCACAG
GGTGATTTGCTGTGCCGCGTTGTCGTCGAAACACCGGTAGGCCTGAACGAAAGGCAGAAA
CAGCTGCTGCAAGAGCTGCAAGAAAGCTTCGGTGGCCCAACCGGCGAGCACAACAGCCCG
CGCTCAAAGAGCTTCTTTGATGGTGTGAAGAAGTTTTTTGACGACCTGACCCGCTAACCT
CCCCAAAAGCCTGCCCGTGGGCAGGCCTGGGTAAAAATAGGGTGCGTTGAAGATATGCGA
GCACCTGTAAAGTGGCGGGGATCACTCCCATAAGCGCT
Reference: EMBL-EBI (2026). ENA Browser. [online] Ebi.ac.uk. Available at: https://www.ebi.ac.uk/ena/browser/view/M12565
Conserved sites and known mutational effects:
Lysis proteins, and in particular single-gene lysis (Sgls) proteins, often feature conserved sequence sites despite their diversity. Analysis of L sequences from related F-specific MS2-like phages reveals four key elements in the L protein: a basic N-terminus, a hydrophobic region, a conserved Leu(48)-Ser(49) (LS) dipeptide essential for type II Sgl lytic function, and a variable C-terminus.(Antillon et al., 2024). Additionally, mutational studies of the MS2 lysis protein L demonstrate that several residues in the central domains are highly conserved across most homologs (Chamakura et al., 2017).
 Figure: Conserved LS and similarity of central domains
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
Figure: Conserved LS and similarity of central domains
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
The figure below analyses the mutational analysis of MS2 L. The sequence is separated into its four domains through the numbered boxes at the bottom of the figure. The Amino acids above the L sequence show single amino acid mutations (missense mutation) that have a lysis defect, but don’t cause protein accumulation. Below the sequence are single amino acid mutations that don’t affect lysis. The amino acids with green asterisks in the L sequence show where a single nucleotide of the sequence could be changed by a stop codon (nonsense mutation). (Chamakura et al., 2017)
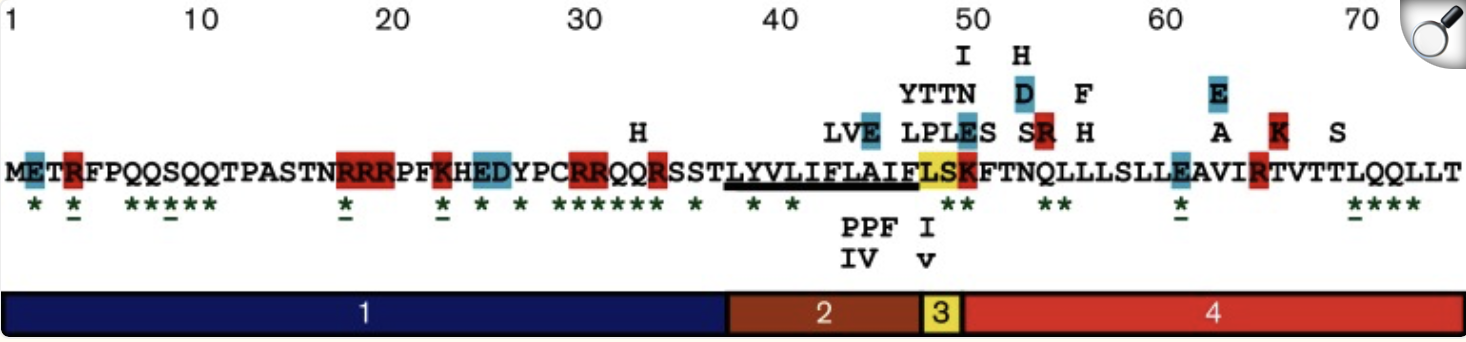 Figure: Mutational Analysis of MS2 L.
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
Figure: Mutational Analysis of MS2 L.
Reference: Mutational analysis of the MS2 lysis protein L. Microbiology
doi:https://doi.org/10.1099/mic.0.000485.
References:
Chamakura, K.R., Edwards, G.B. and Young, R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology, [online] 163(7), pp.961–969. doi:https://doi.org/10.1099/mic.0.000485.S. Francesca Antillon, Bernhardt, T.G., Karthik Chamakura and Young, R. (2024). Physiological characterisation of single-gene lysis proteins. Journal of bacteriology. doi:https://doi.org/10.1128/jb.00384-23.Step 2: Approach and Sequence variants
Below is the downloaded top 20 L-protein mutants dataset that I got from the ESM-2 Colab:
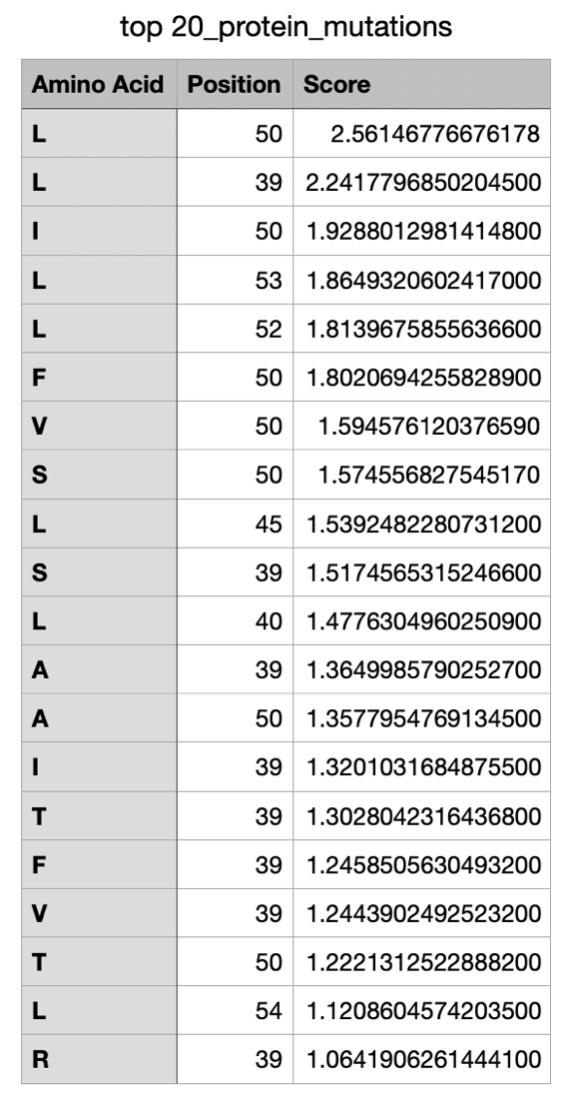
The LLR Scores: A higher log-likelihood ratio is considered to mean the protein mutation of that amino acid in the specific location of the sequence is more supported, whereas a lower LLR score shows a low tolerance for a mutation.
Now continuing through the Colab, I managed to get the top 10 of the mutations and added the experimental data as a cvs file to also gather the top 10 mutations from the data:
Top 10 from Colab:

Experimental data:
Top 10 of experimental Data:

Step 3: Filtering and Ranking sequences
To determine the location of the mutations, it is important to identify the soluble N-terminus (1–39 aa) and the transmembrane domain (40–75 aa) (Mezhyrova et al., 2023). As shown in the tables above, all experimental and generated mutations are located within these regions of the sequence.
By comparing the experimental data with the mutations generated in Colab, it is possible to identify the most likely mutations present in both datasets. The highest LLR-scored mutations are mainly located in the N-terminal region, particularly at position 29, with one mutation also found in the transmembrane domain at position 50. Since higher LLR scores indicate greater mutational tolerance, these mutations appear to be the most tolerated. The three mutations that appear in both datasets are:
K50LC29LY39LC29RC29SReferences:
Mezhyrova J., Martin, J., Börnsen, C., Volker Dötsch, Achilleas Stefanos Frangakis, Morgner, N. and Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4). doi:https://doi.org/10.20517/mrr.2023.28.p>Step 4: Submit 5 mutated sequences
*Note: in all the structure images below, the top protein is the original, and the bottom one is the mutated structure*
Mutation 1: K50L
Mutated sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
The mutated sequence seems slightly more elongated and compacted than the original sequence.
Mutation 2: C29L
Mutated sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPLRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
My observations reveal no noticeable structural differences between the original and mutated sequences.
Mutation 3: Y39L
Mutated sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The original sequence exhibits more elongated yet compacted alpha helices compared to the mutated sequence.
Mutation 4: C29R
Mutated sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
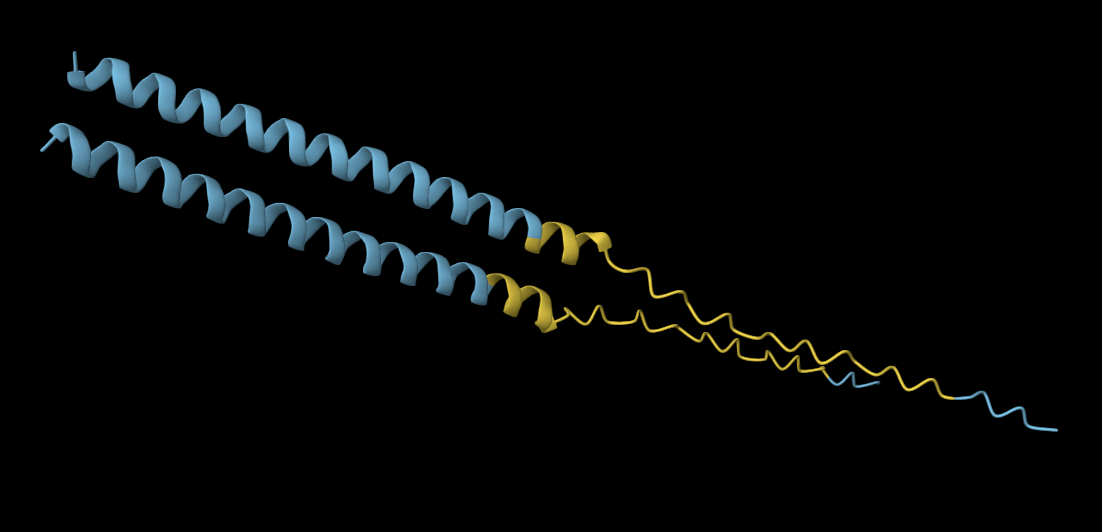
The mutated sequence produces a slightly more elongated structure compared to the more compact alpha-helical fold of the original protein. This suggests the mutation C29S in the N-terminus slightly impacts secondary structure without fully destabilising the fold.
Mutation 5: C29S
Mutated sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPSRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
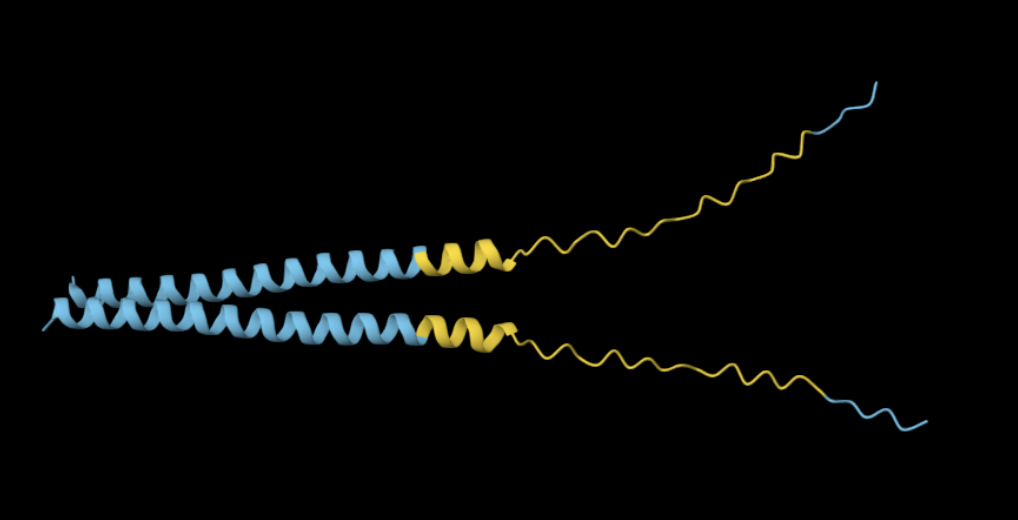
Similarly to the previous mutation in the same location, the mutated sequence produces a slightly more elongated structure compared to the more compact alpha-helical fold of the original protein.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix, and what is their purpose?
Phusion High-Fidelity PCR Master Mix offers high fidelity and performance for PCR. It consists of Phusion DNA polymerase (1), deoxynucleotides and reaction buffer that has been optimised and also includes MgCl2 (New England Biolabs, 2026). The DNA Polymerase allows for the rapid synthesis of a new DNA strand with high accuracy, as it generates long templates with a single enzyme (Thermo Scientific, 2018). The deoxynucleotides are the essential building blocks for the DNA polymerase to proceed with the synthesis of a new DNA strand, whilst the buffer is what provides the ideal environment for the DNA polymerase to function, resulting in high-yield and fidelity.
References:
New England Biolabs (2026).PhusionTM High-Fidelity PCR Master Mix with HF Buffer. [online] Neb.com. Available at: https://www.neb.com/en-gb/products/m0531-phusion-high-fidelity-pcr-master-mix-with-hf-buffer?srsltid=AfmBOooj2e0WKxJyWmTMyut6egCuogqvdwaFvEZFcWb2_X7GBZSD_15h [Accessed 20 Mar. 2026].Thermo Scientific (2018). Thermo Scientific Phusion High-Fidelity PCR Master Mix #_ Lot _ Expiry Date _ Store at -20 °C Ordering information. [online] Available at: https://documents.thermofisher.com/TFS-Assets/LSG/manuals/MAN0012771_Phusion_HiFi_PCR_MasterMix_100rxn_UG.pdf [Accessed 20 Mar. 2026].2. What are some factors that determine primer annealing temperature during PCR?
The main factor that determines primer annealing temperature during PCR is the calculation of the melting temperature of the primers selected for the amplification. The general rule is to start with an annealing temperature of 3-5°C lower than the lowest melting temperature of the primers (ThermoFisher Scientific, 2019). Another factor that will further determine the temperature is the length of the primers, as they have higher melting temperatures. If the annealing temperature exceeds a temperature of 65°C instead of an optimal temperature of approximately 54°C, there are risks of secondary annealing (Andreas Ebertz, 2022). Furthermore, salt concentration (NA+) further impacts primer annealing as higher levels allow for higher annealing temperature (ThermoFisher Scientific, 2019).
References:
ThermoFisher Scientific (2019). PCR Cycling Parameters—Six Key Considerations for Success | Thermo Fisher Scientific - UK. [online] Thermofisher.com. Available at: https://www.thermofisher.com/uk/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html [Accessed 20 Mar. 2026].Ebertz, A. (2022). Primer Design Guide. [online] The DNA Universe BLOG. Available at: https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/ [Accessed 20 Mar. 2026].3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Polymerase Chain Reaction (PCR) is a laboratory nucleic acid amplification technique used to generate multiple copies of a specific target DNA sequence in vitro. It relies on repeated cycles of thermal denaturation, primer annealing, and extension by a DNA polymerase, leading to exponential amplification of the target DNA section and enabling its detection and analysis. (Khehra et al., 2023).
Protocol(Khehra et al., 2023):
- PCR begins with the extraction of a small nucleic acid sample of DNA or RNA into a reaction tube.
- Denaturation: the DNA is heated to 95°C to break the hydrogen bonds between its complementary base pairs of the double-stranded DNA, separating them into two seperate strands.
- Annealing: the denatured DNA is then cooled down to a temperature of 55°C to 72°C, which allows the binders to bind to each complementary sequence on single-stranded DNA through pairing their 3’ ends to the template strand, providing a start point for DNA synthesis.
- Extension: during the final stage, the temperature is raised to 75°C to 80°C to optimise the DNA polymerase, which promotes strand elongation of the new strand.
DNA polymerase synthesises in a 3’ to 5’ direction, generating new sequences complementary to the template strand. The full process is regulated by the use of a thermal cycler that regulates the time and temperature of each step mentioned above. Conducting multiple cycles results in the amplification of multiple copies of the target DNA within the tube.
Restriction enzyme digestion is a process that involves the cutting of DNA into fragments by restriction enzymes (endonucleases) at specific recognition sequences. This results in DNA fragments that vary in size depending on their location in these recognition sequences. It is usually performed in a microcentrifuge tube with the necessary components: the template DNA, restriction enzymes and Mg2+ under the right conditions (Shen, 2019). There are two possible products depending on the used restriction enzymes; the first one creates staggered cuts (majority of cuts), leaving single-stranded overhangs that will easily anneal to complementary DNA strands (sticky) and the second results in cuts in the middle of the recognition sequence, leaving no overhangs (blunt-end) causing them to ligate less and are therefore harder to clone (QIAGEN, 2013).
Protocol (QIAGEN, 2013):
- Pipette reaction components into a tube (water, DNA, buffer, enzyme)
- Centrifuge the tube.
- Incubate the content in a water bath (usually around 37°C for 1-4 hours).
- In certain applications, it is needed to heat-inactivate the enzyme after digestion ( around 65°C for 20 minutes).
Ultimately, PCR is the amplification of specific DNA sequences to create large amounts of copies, whilst restriction enzyme digestion uses endonucleases to cut DNA in specific sites, meaning they don’t serve the same purpose. PCR is quantity-based, whilst restriction enzyme digestion is used to create smaller DNA fragments. Furthermore, PCR synthesises new DNA strands whilst PCR cuts already existing DNA strands. Lastly, PCR relies on the binding of primers to the DNA strands, whilst restriction enzyme digestion relies on the recognition site of the enzyme.
References:
Khehra, N., Padda, I.S. and Swift, C.J. (2023). Polymerase Chain Reaction (PCR). [online] PubMed. Available at: https://www.ncbi.nlm.nih.gov/books/NBK589663/ [Accessed 20 Mar. 2026].Shen, C.-H. (2019). Restriction Digest - an overview | ScienceDirect Topics. [online] www.sciencedirect.com. Available at: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/restriction-digest [Accessed 20 Mar. 2026].QIAGEN (2013). Restriction endonuclease digestion of DNA. [online] www.qiagen.com. Available at: https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/dna/handling-dna/restriction-endonuclease-digestion-of-dna [Accessed 20 Mar. 2026].4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that the DNA sequences will be appropriate, several factors need to be considered. Firstly, Gibson Assembly are designed to be 20 to 40 nucleotides long (Yu, 2021); it is essential for successful annealing that the primers are designed within that range of base pairs. In addition to this, Gibson Assembly’s optimal temperature melting point typically ranges between 60°C and 70°C, so making sure the primers from PCR also have a similar temperature melting point will ensure a more successful assembly. Additionally, using high-fidelity PCR instead of regular PCR will reduce the risks of mutations during amplification (Eggert et al., 2005). Furthermore, purifying your DNA post-PCR to eliminate any unnecessary byproducts will increase the accuracy of Gibson cloning.
References:
Yu, C. (2021). What is the optimal Tm temperature for overlaps in Gibson Assembly? | ResearchGate. [online] ResearchGate. Available at: https://www.researchgate.net/post/What_is_the_optimal_Tm_temperature_for_overlaps_in_Gibson_Assembly [Accessed 20 Mar. 2026].Eggert, T., Funke, S.A., Rao, N.M., Acharya, P., Krumm, H., Reetz, M.T. and Jaeger, K.-E. (2005). Multiplex-PCR-Based Recombination as a Novel High-Fidelity Method for Directed Evolution. ChemBioChem, 6(6), pp.1062–1067. doi:https://doi.org/10.1002/cbic.200400417.5. How does the plasmid DNA enter the E. coli cells during transformation?
There are two seperate methods of how the plasmid DAN enters the E. Coli cells during transformation:
Chemical Competence method: This method consists of growing E. coli to log phase, which is then chilled on ice and further treated with ice-cold CaCl₂. This binds cell membrane lipids and DNA phosphates, causing the neutralisation of charges, making the negatively charged plasmid DNA less repelled by the membrane. The mix is then heat-shocked at 42°C for 30-90 seconds to temporarily open membrane pores for DNA entry. Lastly, the cell is iced again shortly to reseal the membrane (Froger and Hall, 2007)
Electroporation: The cells ells are mixed with DNA and zapped with a high-voltage pulse (e.g., 2.5 kV). This creates transient membrane holes through dielectric breakdown. Ultimately, the electric field drives charged DNA into the cell before the pores close (Biology LibreTexts, 2021)
References:
Froger, A., & Hall, J. E. (2007). Transformation of plasmid DNA into E. coli using the heat shock method. Journal of visualized experiments : JoVE, (6), 253. https://doi.org/10.3791/253Biology LibreTexts (2021) Transforming E. coli. Available at: https://bio.libretexts.org/Bookshelves/Biotechnology/Lab_Manual:_Introduction_to_Biotechnology/01:_Techniques/1.13:_Transformation6. Describe another assembly method in detail (such as Golden Gate Assembly)
Part 1: Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a DNA cloning technique that allows multiple fragments to be combined in a defined order without leaving extra sequences at the junctions. It uses Type IIS restriction enzymes, such as BsaI, which cut DNA outside of their recognition sites to create specific 4-base overhangs. These overhangs are designed so that each fragment connects only to its correct neighbours.
The process takes place in a single reaction mixture that alternates between temperatures, favouring enzyme cutting (37°C) and ligation by T4 DNA ligase (16°C), which aids in correcting incorrect assemblies automatically. To avoid unintended cleavage, DNA sequences are “domesticated” by removing internal enzyme recognition sites. Overall, this method is more efficient than traditional cloning techniques and is especially useful for building complex genetic systems (Bird et al., 2022).
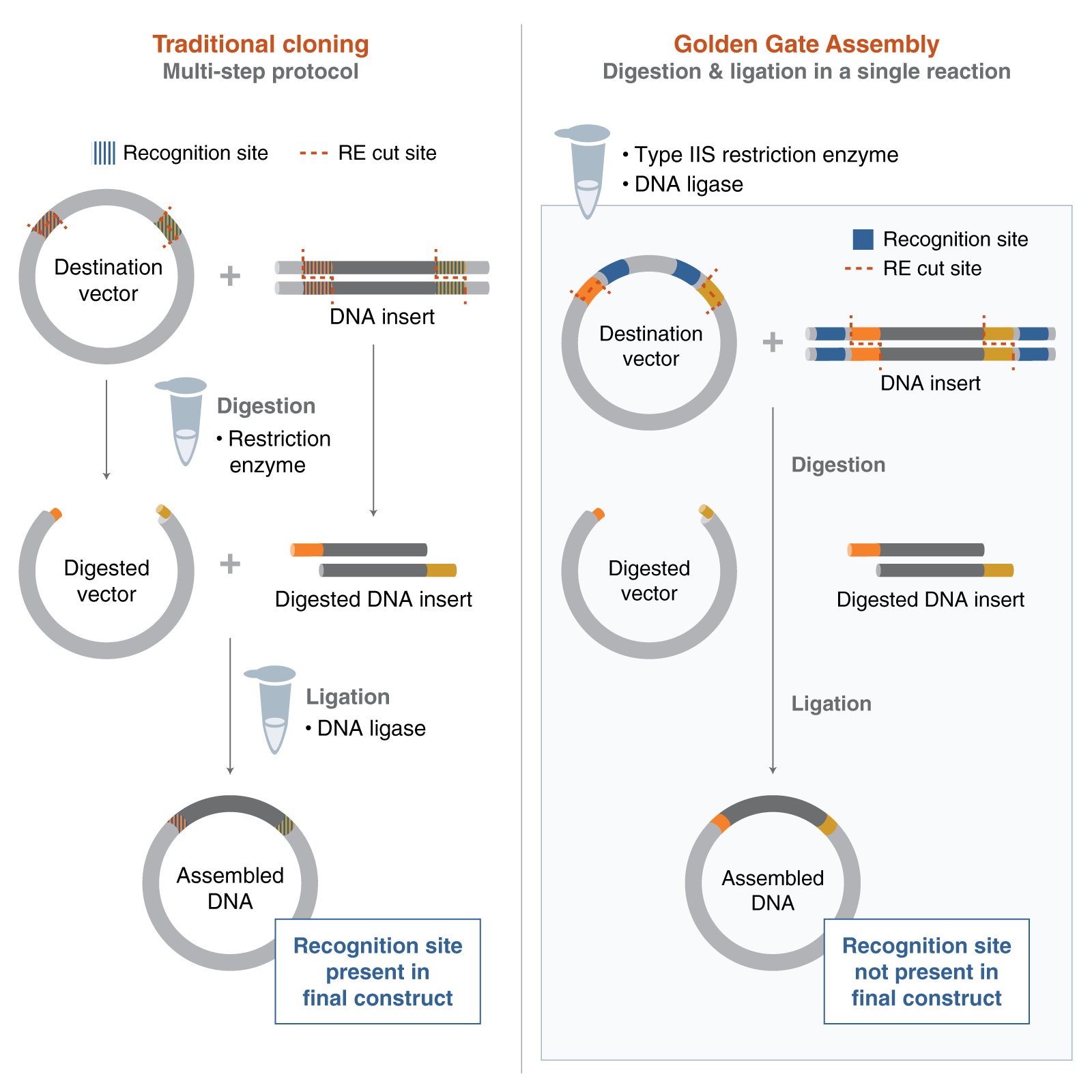 Figure 1: Golden Gate Assembly vs. Traditional Cloning
Image reference: New England Biolabs (2024). Getting Started with Golden Gate Assembly. [online] Neb.com. Available at: https://www.neb.com/en-gb/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoo7ZCRSqsQ1DI7fCFa1awuGBgToH2L_sRbw7czYQBuj4tPjf8lh [Accessed 20 Mar. 2026].
Figure 1: Golden Gate Assembly vs. Traditional Cloning
Image reference: New England Biolabs (2024). Getting Started with Golden Gate Assembly. [online] Neb.com. Available at: https://www.neb.com/en-gb/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoo7ZCRSqsQ1DI7fCFa1awuGBgToH2L_sRbw7czYQBuj4tPjf8lh [Accessed 20 Mar. 2026].
References:
Bird, J.E., Marles-Wright, J. and Giachino, A. (2022). A User’s Guide to Golden Gate Cloning Methods and Standards. ACS Synthetic Biology, 11(11). doi:https://doi.org/10.1021/acssynbio.2c00355.Part 2: Model this assembly method with Benchling or Asimov Kernel!
To start, I selected the pGGA backbone (a common domesticated vector used for Golden Gate assembly) to use in my construct. After obtaining the sequence and adding it to Benchling as a circular plasmid map, I inserted the PsiH codon-optimised enzyme sequence from a previous assignment. This enzyme, involved in psilocybin biosynthesis, aligns with the concept for my Project 1 final design.
PGGAselect: 2020bp DNA
Full sequence and details of the cloning vector are available at:
https://www.neb.com/en/-/media/nebus/page-images/tools-and-resources/interactive-tools/dna-sequences-and-maps/text-documents/pggaselectgbk.txt?rev=5882d0f571c34713a43c67e6d64c25ea&hash=5C1F89A2A1E3E98F94CA667DE322DDF0

Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviours are Boolean functions?
The main limitation of traditional genetic circuits is their restricted control over the strength, timing, and cellular context of therapeutic effects, as their input–output behaviour is usually constrained to simple Boolean logic. In contrast, IANNs can provide finer control of gene expression and cellular behaviour by tuning promoters, repressors, and other genetic components, and they can also sum and weight multiple inputs within a single network, rather than relying on many individually wired logic gates, which become complex and error‑prone.
References:
Carneiro, D.C., Rocha, C., Patrícia K. F. Damasceno, Barbosa, V. and Soares, P. (2024). Therapeutic applications of synthetic gene/genetic circuits: a patent review. Frontiers in Bioengineering and Biotechnology, 12. doi:https://doi.org/10.3389/fbioe.2024.1425529.Seak, L.C.U., Lo, O.L.I., Suen, W.C.-W. and Wu, M.-T. (2021). Next-generation biocomputing: mimicking artificial neural network with genetic circuits. bioRxiv. doi:https://doi.org/10.1101/2021.03.12.435120.2) Describe a useful application for an IANN; include a detailed description of input/output behaviour, as well as any limitations an IANN might face to achieve your goal.
Exploring my Final Project 2 idea (Bacillus subtilis biofilm paint), creating IANN genetic circuits in this spore-forming bacterium could transform the living paint into a smarter system for VOC pollution mapping. Rather than only showing simple surface darkening, the paint could form responsive biofilm patterns that record not just the presence of pollution, but also the environmental context in which exposure occurred. This would create a richer “pollution memory map,” making the project more useful for long-term environmental monitoring, scientific analysis, and the development of an educational and artistic installation.
I was inspired by the paper “A single-layer artificial neural network with engineered bacteria” by Sarkar et al. (2020). In this work, the authors designed an IANN in which extracellular chemical signals acted as inputs. These signals were linearly combined and processed through a nonlinear log-sigmoid activation function generated by synthetic genetic circuits. Each artificial neuron was encoded in bacteria, with weight and bias values tuned by engineering molecular interactions within the cell. This allowed bacterial “neurons” to perform specific logical functions, effectively creating an ANN using living cells.
For this living paint system, the IANN would integrate several environmental inputs. Different VOCs could be detected through separate promoter pathways, where each promoter contributes a weighted signal to the final pigmentation response. For example, VOC1 and VOC2 could each activate distinct sensory promoters whose outputs are combined inside the circuit. Additional contextual inputs could include airflow-sensitive signalling, which would influence spore germination and help distinguish stagnant pollution from outdoor exposure, and UV exposure, which would act as an indicator of spore viability and environmental stress. The output would be a nonlinear pigmentation or biofilm pattern response, where the intensity, shape, or layering of darkening reflects the combined history of the environment’s pollution.
A major limitation of this system is the dormant nature of Bacillus subtilis spores. Because IANN function depends on active transcription and translation, the circuit can only “compute” during short wet periods when spores germinate. During prolonged dry weather or strong UV exposure, the spores remain dormant, meaning the paint cannot continuously process inputs. This could lead to inconsistent activation and patchy or faded biofilm patterns over time. Another important challenge is mutation and genetic instability. A multi-gene IANN containing several nodes alongside VOC sensing pathways would be at high metabolic risk of creating mutations, especially over months of outdoor exposure. There are also regulatory and public acceptance limitations. Engineered spore-forming bacteria with multi-input decision-making circuits would likely face greater biosafety scrutiny than simpler VOC-degrading systems. Finally, the nonlinear nature of the IANN output may make interpretation difficult. Because the visible darkening is based on multiple weighted inputs, complex shade gradients may be hard for scientists to decode into a clear pollution history. While this complexity improves sensing, it may reduce interpretability unless paired with computational decoding tools.
References:
Sarkar, K., Bonnerjee, D. and Bagh, S. (2020). A single-layer artificial neural network with engineered bacteria. doi:https://doi.org/10.48550/arXiv.2001.00792.3) Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
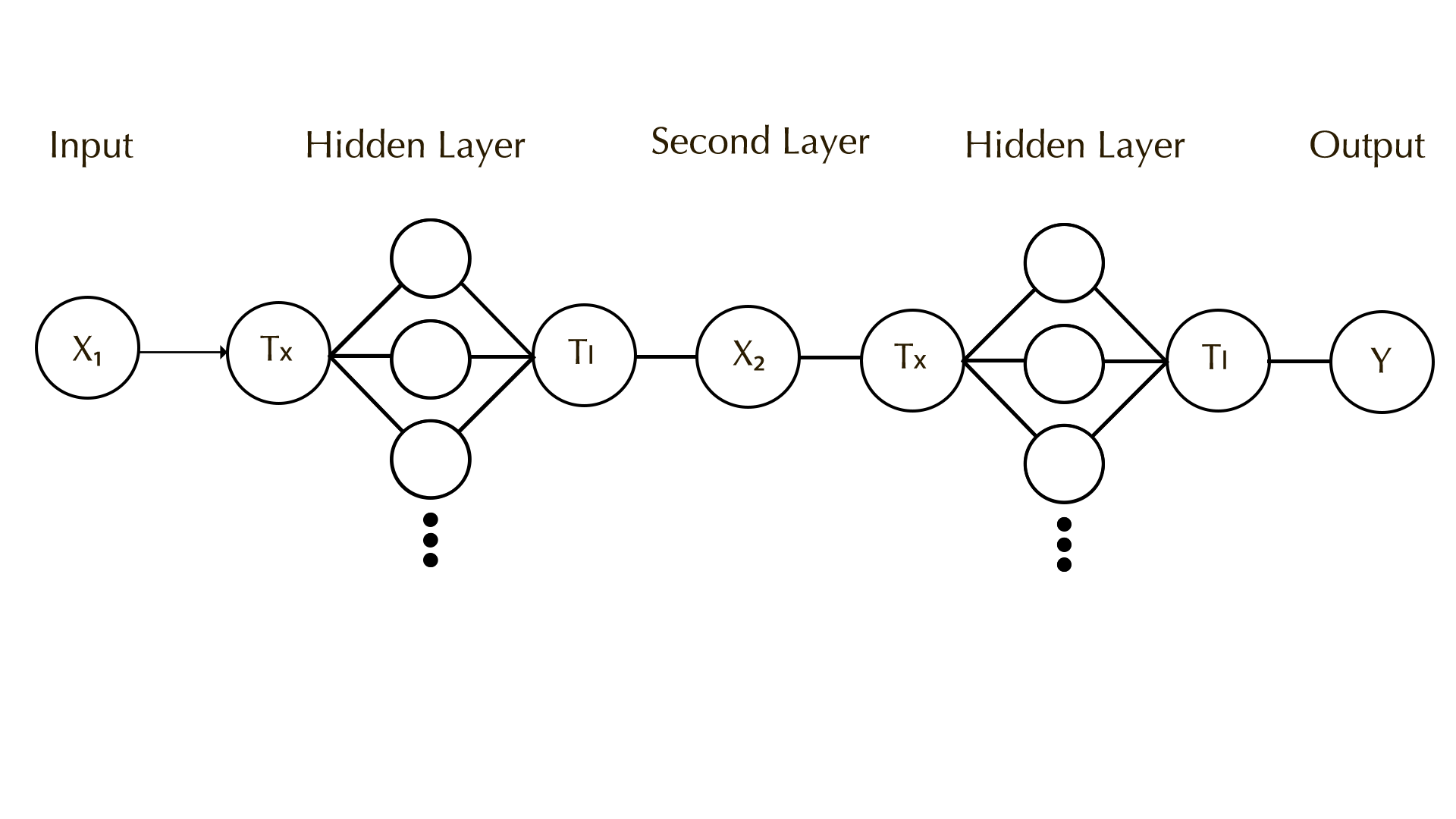
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials, and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials have been an increasing area of development for the production of sustainable alternative materials, in particular, mycelium, the underground root network of fungi. There are two primary ways of processing mycelium into a material.
Composite mycelium materials (CMM): An organic growing material (medium) like woodchips or agricultural waste is pressed inside a mould and is then inoculated with mycelium, allowing for the network to grow and fill in the cracks of the medium, creating a solid material. In order to stop the mycelium from growing, the material is dehydrated through heat or compression. The outcome is a solid foam-like material whose characteristics can be manipulated depending on the selected growing medium. All the parts of this processing technique are composed of biological elements, resulting in its biodegradability over a short period of time.
Pure mycelium materials (PMM): The mycelium is grown without a medium, but still has a nutrient source to allow its growth. Once grown. It can be combined with additional processing techniques, allowing greater control over the final product. For example, the material can be coated with a chemical to increase its durability, but this can make it non-biodegradable. In comparison to CMM, it allows the production of flexible materials and its characteristics can be altered depending on the strain of fungi used.
Examples of mycelium materials:
Mycelium has been expanding in the world of textiles thanks to a 2018 patent that enabled the use of fungi in textile production. Mycelium leather has been tested and proven to, in some cases, have a higher tensile strength and resistance to tearing than synthetic leather (Gandia et al., 2021). However, it may degrade more quickly unless treated with a plastic coating. The company MycoWorks has been exploring how to replicate woven fabric with mycelium by directing the orientation of its growth. In theory, this would allow to create a stronger woven material. Another example of mycelial materials includes packaging such as CMM styrofoam (hemp and mycelium), developed by a company called Ecoactive (Ecoactive LLC, 2026). Furthermore, it is being explored as a paper-like material produced by growing pellets of fungi that are agitated into a pulp. This is a fast process, but it is prone to contamination. In construction, mycelium has a lot of advantageous properties, such as high thermal insulation and fire resistance, whilst still allowing for good airflow. Currently, it is being used for carbon-neutral architecture (Myceen, 2024) in a variety of materials (bricks, insulation, pastes…).
Lastly, I discovered that mushroom material can be grown to carry out an electrical wiring pattern. In Ecovative’s patent, a fungus is grown on a nutrient medium (like potato dextrose agar or broth) that contains metal salts such as copper sulfate or copper chloride. As the mycelium spreads, it absorbs these metals and forms thin sheets that grow in the shape of the desired circuit pattern, creating conductive “wires” within the fungal material (Cerimi et al., 2019). In comparison to standard circuits, it would essentially be compostable as well as having a lower carbon footprint in production. In terms of functionality, being a ‘living electronic’ could, in theory, be combined with self-regenerative repair and sensing (Adamatzky, 2022).
Advantages
They are biodegradable, so they can break down over a relatively short time.Their processing generally involves lower toxicity and energy use than that produced from petrochemicals.By changing the fungal strain, substrate and post-processing, the same system can be adapted for many different applications.They can have a lower overall carbon footprint than conventional materials, especially when they replace high‑impact products.In some cases, they can be designed as “living” or responsive materials (e.g. self‑healing structures or conductive), which is not possible with inert synthetic materials.Disadvantages
When kept fully bio-based and uncoated, they are often less durable and more sensitive to biological decay than traditional materials.Their mechanical properties can be more variable and sometimes lower than standardised industrial materials, which makes engineering and certification more challenging.They usually take longer to produce because the mycelium needs time to grow and colonise the substrate, slowing down manufacturing cycles.At the moment, they are often more expensive than mass‑produced plastics, foams or leathers.They can be sensitive to environmental conditions such as humidity and contamination, which can limit certain outdoor or high‑load uses unless additional protection is added.Being a relatively new class of materials, they come with more uncertainty around regulation, long‑term performance and risk assessment compared to well‑known conventional materials.References:
Adamatzky, A., Ayres, P., Beasley, A.E., Chiolerio, A., Dehshibi, M.M., Gandia, A., Albergati, E., Mayne, R., Nikolaidou, A., Roberts, N., Tegelaar, M., Tsompanas, M.-A., Phillips, N. and Wösten, H.A.B. (2022). Fungal electronics. Biosystems, 212, p.104588. doi:https://doi.org/10.1016/j.biosystems.2021.104588.Ecoactive (n.d.). Ecovative. [online] Ecovative. Available at: https://ecovative.com [Accessed 26 Mar. 2026].Cerimi, K., Akkaya, K.C., Pohl, C., Schmidt, B. and Neubauer, P. (2019). Fungi as source for new bio-based materials: a patent review. Fungal Biology and Biotechnology, 6(1). doi:https://doi.org/10.1186/s40694-019-0080-y.Gandia, A., van den Brandhof, J.G., Appels, F.V.W. and Jones, M.P. (2021). Flexible Fungal Materials: Shaping the Future. Trends in Biotechnology, 39(12). doi:https://doi.org/10.1016/j.tibtech.2021.03.002.Myceen (2024). Materials for carbon-neutral architecture. [online] Myceen.com. Available at: https://myceen.com/about [Accessed 26 Mar. 2026].Pale Blue dots (2026). Fungi-based materials. [online] Notion. Available at: https://pbdvc-research.notion.site/Fungi-based-materials-3b088667784f416e90169be831fb6105 [Accessed 26 Mar. 2026].1. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For my project idea of developing an IBD treatment via psilocybin's effects on gut microbiota and serotonin signalling, a key challenge is confirming whether microbial reshaping is a direct effect of psilocybin (Fung et al., 2019),(Robinson, et al., 2023). I would therefore propose to start with the engineering of one psilocybin-producing fungal strain (Psilocybe cubensis) and genetically engineer variants that differ by only one feature, for example: one with active PsiH enzyme (IPsiH catalyses the 4-hydroxylation of tryptamine to 4-hydroxytryptamine, which is an essential and unique part of psilocybin biosynthesis that allows for the production of psilocin) (Huang et al., 2025), one with PsiH knocked out, one with tuned production levels (via promoter strength), and one producing an inactive analogue. These variants could be applied to ex vivo gut models to compare microbiome composition and immune markers, isolating whether psilocybin itself and not other mushroom components drives changes. Additionally, a reporter circuit with fluorescence under a gut condition-responsive promoter would track fungal activity timing and location. This genetic engineering approach would provide a controlled platform to test the hypothesis of psilocybin's gut therapeutic potential.
References:
Dragos Ciocan and Eran Elinav (2023). Engineering bacteria to modulate host metabolism. Acta physiologica, 238(3). doi:https://doi.org/10.1111/apha.14001.Fung, T.C., Vuong, H.E., Luna, C.D.G., Pronovost, G.N., Aleksandrova, A.A., Riley, N.G., Vavilina, A., McGinn, J., Rendon, T., Forrest, L.R. and Hsiao, E.Y. (2019). Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nature Microbiology. doi:https://doi.org/10.1038/s41564-019-0540-4.Gregory Ian Robinson, Li, D., Wang, B., Rahman, T., Gerasymchuk, M., Hudson, D., Kovalchuk, O. and Kovalchuk, I. (2023). Psilocybin and Eugenol Reduce Inflammation in Human 3D EpiIntestinal Tissue. Life, 13(12), pp.2345–2345. doi:https://doi.org/10.3390/life13122345.Huang, Z., Yao, Y., Di, R., Zhang, J., Pan, Y. and Liu, G. (2025). De Novo Biosynthesis of Antidepressant Psilocybin in Escherichia coli. Microbial biotechnology, [online] 18(4), p.e70135. doi:https://doi.org/10.1111/1751-7915.70135.Week 9 HW: Cell - free systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions/Lab Questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis provides greater flexibility and control compared to traditional in vivo expression, as the reaction occurs outside living cells, allowing factors such as DNA concentration and energy components to be adjusted without affecting cell viability. In in vivo expression methods, amplifying the bacterial plasmid, sequencing it, expressing the strain, and purifying the protein must be done separately for every single protein in the pathway. In contrast, in cell-free protein expression using a synthetic cell, proteins can be expressed directly from linear PCR fragments without the need for plasmid construction. This makes the process much more time‑efficient and allows results to be obtained within hours. Furthermore, all proteins can be expressed in one pot by adjusting the concentrations of the DNA, unlike in vivo systems that require different promoters for each protein and expression level.
Answer based on Kate Adamala, 2/03/2026 lecture
Cases where cell-free expression is more beneficial than cell production:1. In the production of toxic or membrane proteins, cell-free systems are advantageous because the reaction occurs outside a living cell. Internal metabolism does not need to be maintained, and there are no cellular barriers limiting translation control (Zemella et al., 2015). Traditionally, genetic circuit prototyping using plasmid DNA is time‑consuming, taking several days per validation cycle. In contrast, linear DNA can complete the same cycles within 4–8 hours, making it possible to validate large circuits rapidly and to study molecules that were previously considered too toxic for in vivo work (Brookwell et al, 2021).
2. Cell-free systems also benefit other prototyping efforts, such as drug discovery, by shortening the time from compound identification to validation. Moreover, reactions can be monitored in real time, providing insights into the underlying mechanisms and improving the design and optimisation of experimental pathways.
References
Brookwell, A., Oza, J.P. and Caschera, F. (2021). Biotechnology Applications of Cell-Free Expression Systems.Life , 11(12), p.1367. doi:https://doi.org/10.3390/life11121367.Zemella, A., Thoring, L., Hoffmeister, C. and Kubick, S. (2015). Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem, [online] 16(17), pp.2420–2431. doi:https://doi.org/10.1002/cbic.201500340.2.Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system contains all the molecular machinery needed for transcription and translation without using living cells. Its main components include:
Lipid membrane: Composed of phospholipids for structural stability and cholesterol additives that increase membrane fluidity and durability.Cell extract: Provides ribosomes, enzymes, and other necessary cofactors for transcription and translation.Cytoplasmic components: Small molecules such as salts, metabolites, and energy substrates that support biochemical reactions.tRNAs and nucleotides: Supply the genetic code and building blocks for protein synthesis.Membrane channels: Allow controlled exchange of small molecules, enabling communication and regulation.Genetic template: A minimal genome in the form of a plasmid or linear DNA containing the genes to be expressed.Answer based on Kate Adamala, 2/03/2026 lecture
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because protein synthesis requires a continuous supply of ATP. Translation is highly energy‑intensive, using roughly 4–5 ATP equivalents per peptide bond. As the reaction proceeds, phosphate by‑products accumulate and deplete the energy pool, limiting protein yield. To maintain ATP levels, energy regeneration schemes can be introduced.
One effective example is the Protein Synthesis Using Recombinant Elements (PURE) system, a minimal biochemical setup capable of carrying out cell‑free protein synthesis using defined enzymatic components. In this system, pyruvate oxidase is added to catalyse the conversion of pyruvate and inorganic phosphate into acetyl phosphate. This intermediate can then regenerate ATP through its conversion to acetate, catalysed by acetate kinase naturally present in the extract(Yadav et al., 2025).
references
Yadav, S., Perkins, A.J.P., Liyanagedera, S.B.W., Bougas, A. and Laohakunakorn, N. (2025). ATP Regeneration from Pyruvate in the PURE System. ACS Synthetic Biology, 14(1), pp.247–256. doi:https://doi.org/10.1021/acssynbio.4c00697.4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems typically use E. coli extracts and provide high protein yields at low cost. They are rapid to prepare and are easily genetically engineered, which makes them suitable for large-scale production and the incorporation of non-canonical amino acids. However, they have limited capacity for complex protein folding and lack most post-translational modifications. In contrast, eukaryotic cell-free systems use extracts from sources such as wheat germ, insect cells, or protozoa. They are better suited for proteins that require proper folding and post-translational modifications, allowing the production of more complex and functional proteins. However, they are generally slower, more expensive, and often yield less protein than prokaryotic systems (Zemella et al., 2015).
For eukaryotic systems: Monoclonal Antibodies (mAbs)
These are complex, large glycoproteins that require proper folding and post-translational modifications, which eukaryotic systems handle better. Heavily used in drug manufacturing, their higher solubility in eukaryotic extracts makes the final drug more effective (Ding and Huang, 2024).
For prokaryotic systems: Human Serum Albumin (HSA)
This small protein needs no complex glycosylation. Commonly used as a drug carrier where production time matters, prokaryotic systems provide the high yields needed for scalable manufacturing (Raoufinia et al., 2016).
references
Creative Biostructure (2025). Protein Synthesis in Prokaryotes vs. Eukaryotes: What’s the Difference? [online] Creative-biostructure.com. Available at: https://www.creative-biostructure.com/protein-synthesis-in-prokaryotes-vs-eukaryotes.htm [Accessed 4 Apr. 2026].Ding, Z. and Huang, Y. (2024). Production of Monoclonal Antibodies for Therapeutic Purposes: Applications, Techniques, and Improvement. doi:https://doi.org/10.20944/preprints202405.1854.v1.Zemella, A., Thoring, L., Hoffmeister, C. and Kubick, S. (2015). Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem, [online] 16(17), pp.2420–2431. doi:https://doi.org/10.1002/cbic.201500340.Raoufinia, R., Mota, A., Keyhanvar, N., Safari, F., Shamekhi, S. and Abdolalizadeh, J. (2016). Overview of Albumin and Its Purification Methods. Advanced Pharmaceutical Bulletin, 6(4), pp.495–507. doi:https://doi.org/10.15171/apb.2016.063.5. How would you design a cell-free experiment to optimise the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
What needs to be added:
Insoluble expression:If lipids or detergents are not included, membrane proteins tend to aggregate due to their hydrophobic regions. Although aggregation can sometimes allow refolding into functional proteins, it more often results in misfolded or inactive proteins.Detergents: These are commonly added during expression to increase solubility by surrounding hydrophobic regions of the protein. However, they can sometimes destabilise the protein or interfere with downstream applications.MSP-Nanodiscs:These provide a stabilising lipid environment for membrane proteins. They enable proteins to remain in a near-native state and can be used in a wide range of assays without requiring detergents.Liposomes and lipids:These create a membrane-like environment that supports proper folding and insertion of membrane proteins, particularly when used with insect cell lysates.There are two main methods of cell-free membrane protein expression: batch and Continuous exchange cell-free (CECF) / dialysis systems. I would choose CECF/dialysis methods over batch systems because they allow longer reaction times and generally result in higher protein yields.(Cube Biotech, 2014)
Steps:
Follow a medium-scale protein production approach, which results in a high protein yield whilst optimising reaction conditions for maximum purity:
Preparation of plasmid template: Use circular DNA templates, as they are more stable and optimised for high-yield protein expression.Reaction setup: Use a large reaction volume within a continuous exchange system to support sustained protein synthesis.Reaction conditions optimisation: Maximise yield by adjusting key variables such as the composition of the feeding solution.Protein analysis: Assess the yield and purity of the expressed protein and prepare it for downstream applications.(Synthego, 2026)
Challenges and how to address them:
Membrane proteins are difficult to study because their hydrophobic surfaces require detergents or lipid environments for extraction and stabilisation. This creates challenges not only during expression, but also throughout purification and downstream analysis.
In addition, membrane proteins are often flexible and structurally unstable, which reduces the likelihood of obtaining a functional or well-behaved protein. This issue can be addressed by providing a more native-like environment during expression, such as incorporating lipids or nanodiscs to improve stability and folding. These inherent properties impact multiple stages of research, including expression, solubilisation, purification, crystallisation, and structure determination. As a result, it is often necessary to adopt broader screening strategies.
One way to overcome these challenges is to test a range of targets or homologues, increasing the chances of identifying a protein that behaves well under experimental conditions. This approach has been particularly successful in the study of G-protein-coupled receptors, where reducing protein flexibility has been essential for structural studies. For example, stabilising flexible regions through engineered modifications or binding partners has enabled successful structure determination.
(Carpenter et al., 2008).references
Carpenter, E.P., Beis, K., Cameron, A.D. and Iwata, S. (2008). Overcoming the challenges of membrane protein crystallography. Current Opinion in Structural Biology, 18(5), pp.581–586. doi:https://doi.org/10.1016/j.sbi.2008.07.001.Cube Biotech (2014). Why use cell-free protein expression? [online] Cube Biotech. Available at: https://cube-biotech.com/our-science/cell-free-lysates/cell-free-expression/ [Accessed 4 Apr. 2026].Synthego (2026). Cell-Free Protein Expression: A Guide - Synthego. [online] Synthego. Available at: https://www.synthego.com/cell-free-protein-expression-guide/ [Accessed 4 Apr. 2026].6.Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reasons for low yield of target protein:
1)Low ATP Level: Use Protein Synthesis Using Recombinant Elements (PURE) for ATP regeneration. (Yadav et al., 2025)
Poor DNA template: Use circular DNA to increase stability. (Synthego, 2026)
Poor protein folding: Add protein folding factors such as Chaperones to the in vitro extract. (Anantha, 2025)
references
Anantha, S. (2025). Solved: Low Yields in Cell-Free Protein Synthesis. [online] Bitesize Bio. Available at: https://bitesizebio.com/10234/solvedlow-yields-in-cell-free-protein-synthesis/ [Accessed 4 Apr. 2026].Synthego (2026). Cell-Free Protein Expression: A Guide - Synthego. [online] Synthego. Available at: https://www.synthego.com/cell-free-protein-expression-guide/ [Accessed 4 Apr. 2026].Yadav, S., Perkins, A.J.P., Liyanagedera, S.B.W., Bougas, A. and Laohakunakorn, N. (2025). ATP Regeneration from Pyruvate in the PURE System. ACS Synthetic Biology, 14(1), pp.247–256. doi:https://doi.org/10.1021/acssynbio.4c00697.Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a) What would your synthetic cell do? What is the input, and what is the output?
Based on my Final Project proposal: A synthetic minimal cell for PAH paint bioremediation.
Function: Continuously degrades PAH carcinogenic pollutants produced by incomplete combustion (eg. traffic emissions and industrial processes) when deposited on the paint surfaces.Input: Gaseous PAHs from air pollutionOutput: Carbon dioxide and water —> PAH-free surfacesb) Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, this function could not be realised by cell-free Tx/Tl alone and would require encapsulation. Encapsulation would protect the synthetic minimal cells within the paint from harsh outdoor environmental conditions, allowing the PAH-degrading enzymes to function effectively.
c) Could this function be realized by genetically modified natural cell?
A genetically natural cell could realise PAH degradation, but would face serious challenges. PAHs like benzo[a]pyrene (BaP) form toxic reactive diol epoxides (BPDE) that bind to DNA adducts that halt replication and enzyme production (Wang et al., 2023). Additionally, living cells also need complex nutrients for long-term survival, plus growth control to prevent overgrowth.
However, in my project proposal examining B. subtilis's PAH-degrading properties, its spore-forming ability and environmental robustness. In relation to food source, a study conducted showed strain BMT4i (MTCC 9447) utilising BaP as a sole carbon and energy source with 84.66% degradation efficiency (Lily et al, 2010). Additionally, the degradation capacity of microbes may be induced by exposing them to higher PAH concentrations, resulting in genetic adaptation or changes responsible for high-efficiency removal/degradation. Overall, this keeps it as a strong candidate for the development of a PAH-degrading paint (Sakshi and Haritash, 2020).
references
Hazan, A., Lee, H.Y., Tiong, V. and AbuBakar, S. (2025). Bacillus subtilis Spores as a Vaccine Delivery Platform: A Tool for Resilient Health Defense in Low- and Middle-Income Countries. Vaccines, [online] 13(10), pp.995–995. doi:https://doi.org/10.3390/vaccines13100995.Lily, M.K., Ashutosh Bahuguna, Koushalya Dangwal and Garg, V. (2010). Optimization of an inducible, chromosomally encoded benzo [a] pyrene (BaP) degradation pathway in Bacillus subtilis BMT4i (MTCC 9447). Annals of Microbiology, 60(1), pp.51–58. doi:https://doi.org/10.1007/s13213-009-0010-y.Sakshi and Haritash, A.K. (2020). A comprehensive review of metabolic and genomic aspects of PAH-degradation. Archives of Microbiology, 202(8), pp.2033–2058. doi:https://doi.org/10.1007/s00203-020-01929-5.Wang, H., Liu, B., Cui, H., Xu, P., Xue, H. and Yuan, J. (2023). Dynamic changes of DNA methylation induced by benzo(a)pyrene in cancer. Genes and Environment, 45(1). doi:https://doi.org/10.1186/s41021-023-00278-1.d) Describe the desired outcome of your synthetic cell operation.
PAH-contaminated paint surfaces become PAH-free, preventing re-volatilisation and runoffs.
2. Design all components that would need to be part of your synthetic cell.
a) What would be the membrane made of?
Phospholipid bilayer with additive cholesterol (increases stability and fluidity) for paint surface adhesion and environmental stability. Additionally, PEG-lipids (polyethene glycol-lipids) as it acts as an interface between hydrophobic polymer cores and aqueous environments (Esteve, 2025).
References:
Esteve, M.Á. (2025). Beyond Lipids and Polymers: Understanding Hybrid Nanoparticles. [online] Curapath.com. Available at: https://blog.curapath.com/beyond-lipids-and-polymers-understanding-hybrid-nanoparticles [Accessed 4 Apr. 2026].b) What would you encapsulate inside? Enzymes, small molecules.
Catabolic enzymes such as dehydrogenase, monooxygenase, catechol dioxygenase, and aromatic-ring-hydroxylating dioxygenase (found in bacterias gene that degrade PAHs, key components of the pathway). (Tesfaye et al., 2025)Linear DNA encoding for the catabolic enzymes.E. Coli cell extract containing tRNAs, ribosomes and polymerases.Cholesterol and phospholipids.references
Tesfaye, E.L., Bogale, F.M. and Aragaw, T.A. (2025). Biodegradation of polycyclic aromatic hydrocarbons: The role of ligninolytic enzymes and advances of biosensors for in-situ monitoring. Emerging Contaminants, [online] 11(1), p.100424. doi:https://doi.org/10.1016/j.emcon.2024.100424.c) Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promoters, like Tet-ON, you need mammalian)
I would use E. Coli as my Tx/TI bacterial system, as PAH degradation is present in prokaryotic enzymes and pathways; therefore, no mammalian systems would be needed. Additionally, they also give a higher yield production and have a lower cost compared to eukaryotic and mammalian systems.
d) How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The catabolic PAH-degrading enzymes are hydrophilic proteins that remain inside the cell extract. The PAHs (small, hydrophobic) are what will passively diffuse through the phospholipid membrane and due to their high permeability, no specific channels need to be expressed (Cao et al., 2021).
references
Cao, Y., Zhang, L., Geng, Y., Li, Y., Zhao, Q., Huang, J., Ning, P. and Tian, S. (2021). Evaluation of the permeability and potential toxicity of polycyclic aromatic hydrocarbons to pulmonary surfactant membrane by the parallel artificial membrane permeability assay model. Chemosphere, p.132485. doi:https://doi.org/10.1016/j.chemosphere.2021.132485.3. Experimental details
a) List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids:
CholesterolPolymer coatingSpecific genes:
For ATP regeneration: pyruvate oxidase and acetate kinaseCatabolitic PAH-degrading enzymes: dehydrogenase, monooxygenase, catechol dioxygenase, and aromatic-ring-hydroxylating dioxygenaseb) How will you measure the function of your system?
Use Mass spectrometry coupled to gas chromatography (GC-MS) quantification to determine the exact levels of volatile compounds (VOCs), specifically the PAHs (Dimitrios,2024), present before and after application of paint.
referencesDimitrios Tsikas (2024). Perspectives of Quantitative GC-MS, LC-MS, and ICP-MS in the Clinical Medicine Science—The Role of Analytical Chemistry. Journal of Clinical Medicine, [online] 13(23), pp.7276–7276. doi:https://doi.org/10.3390/jcm13237276.Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
I will continue to apply my final project proposal from above to explore my development options.
Self-monitoring, self-remediating anti-pollution paint that continuously degrades volatile Polycyclic Aromatic Hydrocarbons (PAH) using freeze-dried Bacillus subtilis BMT4i expressing PAH-degrading enzymes.
How will the idea work, in more detail? Write 3-4 sentences or more.
The paint contains microencapsulated, freeze-dried cell-free extracts from Bacillus subtilis BMT4i, containing their PAH-degrading enzymes such as laccases and dioxygenases. When exposed to environmental moisture, including rain, dew, or humidity, the microcapsules rehydrate and reactivate the enzymatic systems within localised regions of the paint matrix. These enzymes then catalyse the oxidation and breakdown of PAHs deposited on the coated surface into less toxic and more water-soluble intermediates. To maintain long-term functionality across repeated wet–dry cycles, the formulation incorporates stabilising agents such as protective polymer matrices, along with slow-release cofactors necessary for enzymatic activity.
What societal challenge or market need will this address?
PAHs are associated with urban pollution sources such as vehicle exhaust and industrial emissions, which persist in the air and soil and pose significant health risks via inhalation, ingestion, and dermal contact (Wu et al., 2025). This freeze-dried cell-free system addresses these issues and could be a promising tool for environmental and human health.
references
Wu, Y., Meng, Y., Zhang, H., Hao, L., Zeng, T., Shi, Y., Chen, Y., Qiao, N. and Ren, Y. (2025). Ecological and Health Risks of Polycyclic Aromatic Hydrocarbons in Particulate Matter in Chinese Cities. GeoHealth, [online] 9(6). doi:https://doi.org/10.1029/2024gh001126.How do you envision addressing the limitations of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation: Uses outdoor rainwater and humidity to hydrate the freeze-dried system, so no manual activation is needed.One-time use: Bacillus subtilis can use PAHs as a carbon source, so the system is designed to keep breaking down fresh pollutant deposits over repeated exposure.Stability: The encapsulation layer in the paint matrix helps protect the enzymes from environmental exposures.Homework question from Ally Huang
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Galactic cosmic radiation poses a major health risk to astronauts, causing DNA damage, cancer risk, neurocognitive decline, and cardiovascular disease (Chancellor et al., 2014). This challenge is critical as long-duration missions to Mars exceed current radiation safety limits. Addressing it is vital for enabling deep-space exploration and protecting crew health. Scientifically, it is compelling to investigate whether BioBits cell-free systems can both detect DNA-damaged biomarkers and respond by producing DNA repair enzymes on demand, offering a real-time response.
references
Chancellor, J., Scott, G. and Sutton, J. (2014). Space Radiation: The Number One Risk to Astronaut Health beyond Low Earth Orbit. Life, [online] 4(3), pp.491–510. doi:https://doi.org/10.3390/life4030491.2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
γ-H2AX (phosphorylated histone H2AX) as a biomarker for cosmic radiation (Sethu et al., 2017) and the PARP-1 gene for DNA repair enzyme production (Ko and Ren, 2012).references
Ko, H.L. and Ren, E.C. (2012). Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules, 2(4), pp.524–548. doi:https://doi.org/10.3390/biom2040524.Hande, Mp., Zeegers, D., Venkatesan, S., Koh, S., Low, G.M., Srivastava, P., Sundaram, N., Sethu, S., Banerjee, B., Jayapal, M., Belyakov, O., Baskar, R. and Balajee, A. (2017). Biomarkers of ionizing radiation exposure: A multiparametric approach. Genome Integrity, 8(1), p.6. doi:https://doi.org/10.4103/2041-9414.198911.3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Galactic cosmic rays can cause severe DNA damage in astronauts’ immune cells, creating clustered double-strand breaks. Within about 30 minutes, this damage leads to the formation of γ-H2AX foci, which act as an early signal that DNA repair may be needed. By detecting γ-H2AX, a BioBits® cell-free system can be triggered to produce the repair enzyme PARP-1. PARP-1 helps coordinate the repair process, allowing the cell to fix DNA breaks before mutations start to build up. This kind of self-activating biosensor-and-response system is especially useful in deep space, where long resupply delays make it difficult to rely on traditional medicines. Instead, it provides on-demand DNA repair exactly when astronauts need it.
references
Redon, C.E., Dickey, J.S., Bonner, W.M. and Sedelnikova, O.A. (2009). γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Advances in Space Research, 43(8), pp.1171–1178. doi:https://doi.org/10.1016/j.asr.2008.10.011.Wang, Y., Luo, W. and Wang, Y. (2019). PARP-1 and its associated nucleases in DNA damage response. YDNA Repair, 81, p.102651. doi:https://doi.org/10.1016/j.dnarep.2019.102651.4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Goal:
Create a toehold switch that senses γ-H2AX and turns on PARP-1 production in a BioBits® reaction. Then test whether the PARP-1 made this way can actually repair damaged DNA, as a proof-of-concept for a wearable radiation-response patch for astronauts.
Reasoning:
γ-H2AX appears quickly at sites where radiation causes DNA double-strand breaks, making it a reliable early warning signal. PARP-1 is one of the first proteins to respond to this damage, helping recruit the cell’s repair machinery. BioBits® cell-free systems have already been shown to work on the ISS, so combining them with toehold switches could create a self-contained system that detects DNA damage and immediately produces a repair protein without needing outside intervention. If small fragments of γ-H2AX can activate the toehold switch, this would demonstrate a detect and respond system for space radiation damage.
references
Podhorecka, M., Skladanowski, A. and Bozko, P. (2010). H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. Journal of Nucleic Acids, 2010, pp.1–9. doi:https://doi.org/10.4061/2010/920161.Kinner, A., Wu, W., Staudt, C. and Iliakis, G. (2008). -H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Research, 36(17), pp.5678–5694. doi:https://doi.org/10.1093/nar/gkn550.Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Samples: Human lymphocytes exposed to simulated cosmic radiation and non-exposed controls.Methods: Use miniPCR to amplify PARP-1 DNA. Measure protein production and γ-H2AX signals with a P51 Fluorescence Viewer.Data: Track fluorescence (protein expression), percentage of repaired DNA, and number of γ-H2AX foci before and after treatment.Controls: Non-irradiated cells and a scrambled toehold switch.Week 10 HW: Advanced Imaging & Measurment Technology
Homework: Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
For my project, I want to measure the activity of the CotA laccase from Bacillus subtilis (whose gene is inserted into my pETite‑based plasmid) against PAH compounds using ABTS as a colour‑indicating substrate. I will measure the initial reaction rate of CotA, expressed as the change in absorbance over time, under different conditions, such as pH and temperature, to determine the enzyme’s optimal working range.
I would also like to measure the half‑life and stability of CotA in a paint‑like formula by embedding cultured CotA‑expressing cells in a water‑based gel or coating to then sample at regular intervals and quantify any residual activity through ABTS assays. Finally, I will record colour‑change metrics of the ABTS PAH‑surrogate solution over time, using percentage colour loss as a quantitative readout of degradative activity. This will help characterise the functional performance of the system in conditions relevant to a PAH‑degrading paint.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Considering PAH are not LBS1 safe, I will be using ABTS, a synthetic water‑soluble compound that laccases (like CotA) oxidise to a coloured radical as a PAH substitute. After mixing my pETite‑CotA plasmid with BL21(DE3) competent cells, performing heat shock and plating on LB kanamycin to grow a colony. I can then proceed to scaling up the culture to have enough E.coli to proceed with the following tests:
Reaction Rate of CotA Laccase - Change in absorbance over time:
1) Centrifuge the culture to pellet the cells and pipette away the surplus (LB and kanamycin) to obtain only the bacterial cells.2) Prepare the solution: dissolve ABTS in 100 mM potassium phosphate buffer (achieved by mixing Potassium phosphate monobasic and dibasic in water).
3) Centrifuge the culture to pellet the cells and pipette away the surplus (LB and kanamycin) to obtain only the bacterial cells.
4) From the obtained cells, I will then resuspend the pellet in a smaller volume of buffer to create a cell suspension.
5) I will then measure the ABTS assay of the cell suspensions of my prepared bacterial samples at different temperatures and pHs by using a 96-well plate reader or UV-Vis spectrophotometer, depending on lab availability.
6) I will measure the change in absorbance at 420nm over time during the ABTS oxidation assay.
7) I will calculate the initial rate as ∆A₄₂₀·min⁻¹, which serves as a quantitative measure of CotA activity under each condition.
8) Additionally, adding copper to the reaction will enhance the activity of CotA Laccase.
Colour‑change metrics of the ABTS PAH‑surrogate solution over time:At each time point t, take or prepare a small sample and measure Aλ at 420 nm (Aₜ)
Keep the unreacted ABTS at time 0 as your reference (A₀).For each timepoint, I will calculate the % colour loss: % colour loss t = 100 x A₀ - Aₜ / A₀If Aₜ is significantly smaller than A₀, there is a high % colour loss.Half‑life and stability of CotA‑expressing cells embedded in a paint‑like gel
Use some of the suspended cells and mix them with a water‑based gel or coating (e.g., agar hydrogel) as a conceptual paint.Spread a layer of the mixture on tiles or other surfaces and let it solidify.At different selected times (over a couple of days at least), scrape off a defined amount of the surface.Resuspend the sample in the buffer, centrifuge to get a CotA sample.Rerun an ABTS assay at 420nm on each sample, calculate the absorbance at the start and the initial rate: ∆A₄₂₀/minI can then express the activity and determine the time it takes for the enzyme’s activity to drop below 50% of its initial value: activity remaining at t= 100 x rateₜ/rate₀References
Ardila-Leal, L.D., Monterey-Gutiérrez, P.A., Poutou-Piñales, R.A., Quevedo-Hidalgo, B.E., Galindo, J.F. and Pedroza-Rodríguez, A.M. (2021). Recombinant laccase rPOXA 1B real-time, accelerated and molecular dynamics stability study. BMC Biotechnology, 21(1). doi:https://doi.org/10.1186/s12896-021-00698-3.BenchChem Technical Support Team (2026). Application Notes: ABTS Assay for Laccase Activity. [online] Available at: https://pdf.benchchem.com/7949/Application_Notes_ABTS_Assay_for_Laccase_Activity.pdf [Accessed 12 Apr. 2026].Dias, A.A., António J.S. Matos, Fraga, I., Sampaio, A. and Rui (2017). An Easy Method for Screening and Detection of Laccase Activity. The Open Biotechnology Journal, 11(1), pp.89–93. doi:https://doi.org/10.2174/1874070701711010089.JenaBios (n.d.). 4 benzenediol + O 2 4 benzosemiquinone + 2 H 2 O Activity. [online] Available at: https://www.jenabios.de/wp-content/uploads/2016/03/Datenblatt_Laccase_Cu.pdf [Accessed 13 Apr. 2026].Margot, J., Bennati-Granier, C., Maillard, J., Blánquez, P., Barry and Holliger, C. (2013). Supporting Information: Bacterial versus fungal laccase: Potential for micropollutant degradation. AMB Express.MartinsL.O., Soares, C.M., Pereira, M.M., Teixeira, M., Costa, T., Jones, G.H. and Henriques, A.O. (2002). Molecular and Biochemical Characterization of a Highly Stable Bacterial Laccase That Occurs as a Structural Component of the Bacillus subtilis Endospore Coat. Journal of Biological Chemistry, 277(21), pp.18849–18859. doi:https://doi.org/10.1074/jbc.m200827200.Shin-ichi Sakasegawa, Ishikawa, H., Imamura, S., Haruhiko Sakuraba, Goda, S. and Toshihisa Ohshima (2006). Bilirubin Oxidase Activity of Bacillus subtilis CotA. Applied and Environmental Microbiology, 72(1), pp.972–975. doi:https://doi.org/10.1128/aem.72.1.972-975.2006.JenaBios (n.d.). 4 benzenediol + O 2 4 benzosemiquinone + 2 H 2 O Activity. [online] Available at: https://www.jenabios.de/wp-content/uploads/2016/03/Datenblatt_Laccase_Cu.pdf [Accessed 13 Apr. 2026].What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
For all three protocols, the core instrument is a UV‑Vis spectrophotometer (or a microplate reader with absorbance capabilities) to measure A₄₂₀ during ABTS‑based laccase assays. For the settings, I will set the wavelength at 420 nm (ABTS has the highest absorbance point). I will then create a ‘blank’ cuvette to calibrate it (buffer + ABTS with no CotA cells) and place it first in the spectrometer. Place each sample in a cuvette and place those in the spectrophotometer one by one. Calculation details are explained in the bullet point above.
Additionally, to relate to the lecture, my aim 3 for my project aims to develop a field‑ready bioremediation paint by evaluating CotA performance in a real‑world‑like coating matrix and validating its safety profile. Toxicity validation would focus on verifying that degradation yields safe byproducts rather than more hazardous compounds, a concern highlighted by recent studies showing that PAH transformation products could be toxic (Huizenga et al., 2025). In this context, mass spectrometry (e.g., LC‑MS/MS) would be used to identify and monitor the formation of degradation products (Nakken et al., 2025).
References
Agilent Technologies (2025). The Basics of UV-Vis Spectrophotometry. [online] Agilent. Available at: https://www.agilent.com/cs/library/primers/public/primer-uv-vis-basics-5980-1397en-agilent.pdf?srsltid=AfmBOopupFXxpTMDA1XLxTrhBD3EBGXNJ9ukjBHqXIJSZoU1jsVGuLr2 [Accessed 12 Apr. 2026].Huizenga, J.M., Semprini, L. and Garcia-Jaramillo, M. (2025). Identification of Potentially Toxic Transformation Products Produced in Polycyclic Aromatic Hydrocarbon Bioremediation Using Suspect and Non-Target Screening Approaches. Environmental Science & Technology, 59(15), pp.7561–7573. doi:https://doi.org/10.1021/acs.est.4c13093.Nakken, C.L., Sørhus, E., Holmelid, B., Meier, S., Mjøs, S.A. and Donald, C.E. (2025). Transformative knowledge of polar polycyclic aromatic hydrocarbons via high-resolution mass spectrometry. Science of The Total Environment, 960, p.178349. doi:https://doi.org/10.1016/j.scitotenv.2024.178349.Homework: Waters Part I - Molecular Weight
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Calculated molecular weight: 27,988.97 Da
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1)
- Determine for each adjacent pair of peaks (n, n +1) using:
I selected:
Peak (m/zn): 875.4421Peak (m/znn+1): 848.9758Z = 848.9758 / 875.4421 - 848.9758 = 848.9758/26.4663 = 32.077615 (+32)
Peak (m/zn) charge state: +32Peak (m/znn+1) charge state: +33- Determine the MW of protein using the relationship between m/Zⁿ, MW and z
Only one of the peaks is needed to calculate the molecular weight. Using the charge peak (m/zn), here is how I calculated the molecular weight:
Formula: z (m/z - 1.0073)
Valu 1.0073 is the Proton mass.
Peak (m/zn): 32 x (875.4421- 1.0073) = 27.998 Da
- Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein:
Accuracy = ( MWexperiment - MWtheory / MWtheory ) x 106Accuracy = (27.98190 - 27.988.97 / 27.988.97) x 106 = 217 ppm- Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, the charge state of the zoomed-in peak cannot be observed directly because the protein is denatured and highly charged, so the isotopic peaks are too closely spaced to be resolved at the instrument’s resolution.
Homework: Waters Part II — Secondary/Tertiary structure
1. Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
When a protein is denatured, it unfolds and exposes many basic amino acids. These exposed sites can easily gain protons, so the protein ends up with a high number of charges. This leads to many peaks at lower m/z values. In contrast, a protein in its native (folded) state hides many of these protonatable sites inside its structure. As a result, it gains fewer protons, has lower charge states, and produces peaks at higher m/z values. In Figure 2, the unfolded protein shows a wider distribution of peaks, while the folded protein shows fewer, more clustered peaks, reflecting its lower charge.
2. Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800? What is the charge state? How can you tell?
Yes, the charge state is 10+.
By zooming in on Figure 3, we can see the individual isotopic peaks are baseline-resolved, and the distance between adjacent isotopes is 0.1 m/z. Because the isotope spacing in a mass spectrum is given by 1/z, a spacing of 0.1 m/z corresponds to a charge of 10+
Homework: Waters Part III — Peptide Mapping - primary structure
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
 Lysine (K): 19Arginines (R): 6
Lysine (K): 19Arginines (R): 62. How many peptides will be generated from tryptic digestion of eGFP?
- Navigate to https://web.expasy.org/peptide_mass/
19 peptides were generated from the tryptic digestion of eGFP.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
There are 15 chromatographic peaks that meet the >10% relative abundance.
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
I observed fewer peaks (4 less) than the number of peptides predicted.
5. Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ( [M + H] + ) based on tissue m/z and z.
The most abundant charge state peptide is (m/z)526.25918 - 525.76712 = 0.49206Z = 2Formula: ([M + H]+ ) = (m/Z x Z) - (Z - 1) x 1.0073( [M + H] + ) = (525.76712 x 2) - (2-1) x 1.0073( [M + H] + ) = 1,050.527 Da6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
(Recall that MWexperiment - MWtheory/ MWexperiment
The Theoatritical MH+ = 1049.52 DaAccuracy= (1,050.527 - 1,049.52)/ 1,050.527 x 106Error Accuracy = 5.7ppmThis is highly accurate and does confirm the peptide identity.
7. What is the percentage of the sequence that is confirmed by peptide mapping?
In reference to Figure 6., 88% of the sequence is confirmed by the peptide mapping.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU = 340 kDa8FU = 400 kDa.Note: 1 MDa corresponds to 1000 kDa.ANSWERS:
7FU Decamer: 10 x 340 = 3,400 kDa —> 3.4 MDa8FU Didecamer: 20 × 400 = 8,000 kDa —> 8.0 MDa8FU 3-Decamer: 12.67 MDa: 30 × 400 = 12,000 kDa —> 12 MDa8FU 4-Decamer: 16 MDa: 40 × 400 = 16,000 kDa —> 16MDaHomework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
This table is based on MIT Homework Review Session:
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
Well: Q2L5Placement number: 1297The pixel I contributed I circled in yellow:
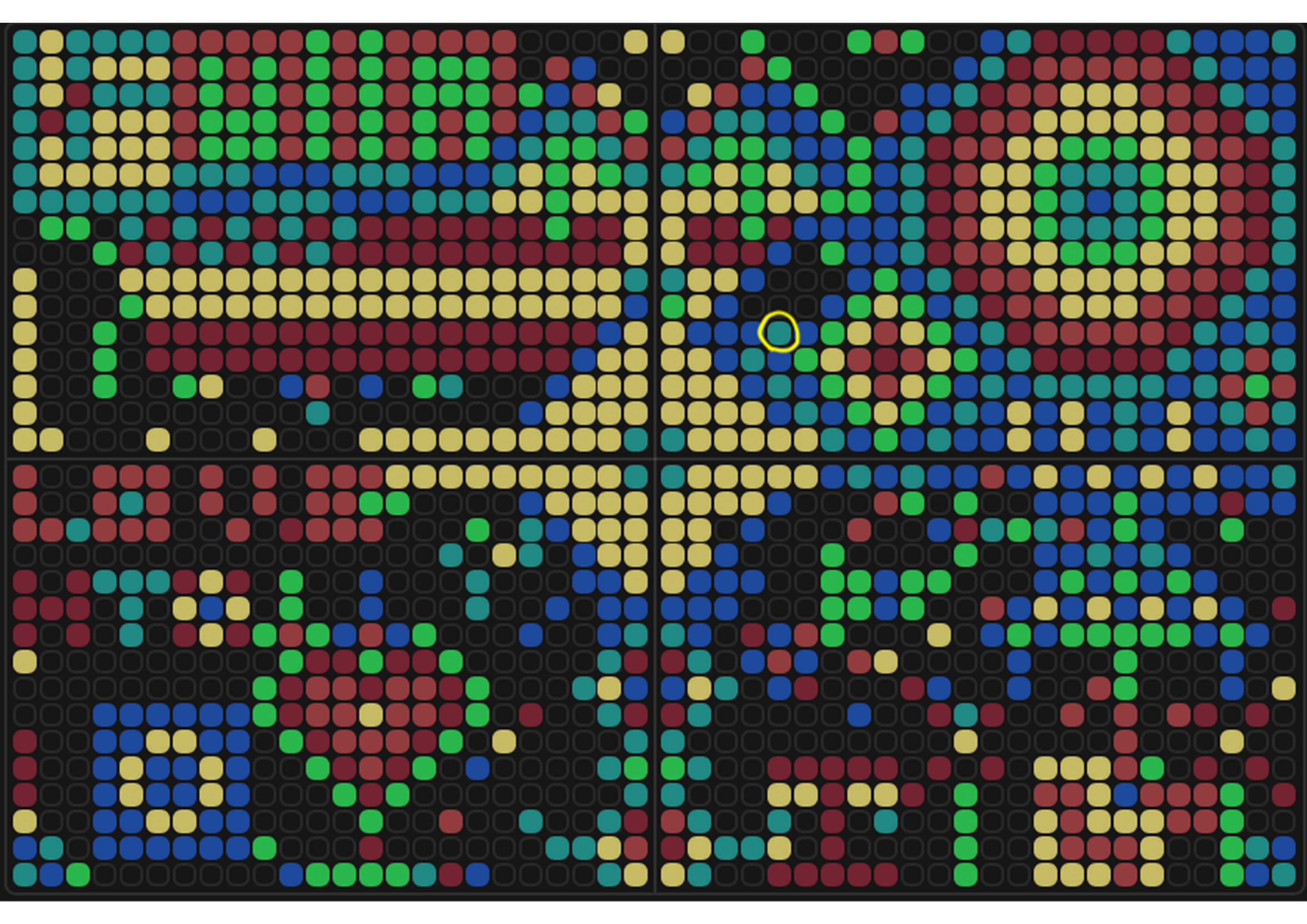
2. Make a note on your HTGAA webpages, including:What you liked about the projectWhat about this collaborative art experiment could be made better for next year?
What I liked about this project was visualising the change through time, although my final contribution didn’t make it to the final drawing, it was lovely to see it actively changing form, shape and colour. My suggestion would be to have more colours for next year.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): The core element for transcription and translation for the reaction to synthesise protein outside of the cell.Salts & Buffers
Potassium Glutamate: Maintains ionic strength and osmotic balance to enhance enzyme activity and translation.HEPES-KOH pH 7.5: Acts as the buffering system for the reaction at a physiological pH where ribosomes and enzymes are most stable and active.Magnesium Glutamate: A magnesium supplier that is essential for many enzymes involved in transcription and translation.Potassium phosphate monobasic: Aids the phosphate buffer system, which aids in controlling pH and supplying phosphate to nucleotides.Potassium phosphate dibasic: Works with phosphate monobasic (they work as a buffer pair).Energy/Nucleotide System
Ribose: A sugar precursor that supports nucleotide pools and the energy metabolism of the lysate.Glucose: The main carbon and energy source. Endogenous enzymes of the lysate use it to regenerate ATP and sustain protein synthesis.AMP: Provides adenine nucleotides that are phosphorylated up to ATP as well as building RNA.CMP: Provides cytidine nucleotides that are converted to higher-phosphates and incorporated into RNA during transcription.GMP: Provides guanosine nucleotides that can be used to form GTP during translation.UMP: Provides uridine nucleotides, which can be phosphorylated to UTP and used in RNA synthesis.Guanine: Acts as a nucleobase precursor to maintin guanine containing nucleotide pools.Translation Mix (Amino Acids)
17 Amino Acid Mix): Supplies most of the amino acids required as building blocks for polypeptide synthesis (translation stage).Tyrosine: Essential amino acid, but usually added individually to control its stability.Cysteine: Essential amino acid, but usually added individually due to its high reactivity.Additives
Nicotinamide: A precursor for NADH to support metabolic reactions and regenerate energy.Backfill
Nuclease-Free Water: To achieve the desired final volume for the reaction, whilst avoiding the use of nucleases that may degrade the DNA or RNA.References:
Gregorio, N.E., Levine, M.Z. and Oza, J.P. (2019). A User’s Guide to Cell-Free Protein Synthesis. Methods and Protocols, 2(1). doi:https://doi.org/10.3390/mps2010024.Miguez, A.M., McNerney, M.P. and Styczynski, M.P. (2019). Metabolic Profiling of Escherichia coli-Based Cell-Free Expression Systems for Process Optimization. Industrial & Engineering Chemistry Research, 58(50), pp.22472–22482. doi:https://doi.org/10.1021/acs.iecr.9b03565.Tuckey, C., Asahara, H., Zhou, Y. and Chong, S. (2014). Protein Synthesis Using a Reconstituted Cell‐Free System. Current Protocols in Molecular Biology, 108(1). doi:https://doi.org/10.1002/0471142727.mb1631s108.2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP master mix already contains the NTPs (ATP, GTP, CTP, and UTP) along with PEP, which acts as a quick energy source. This lets the system start making proteins fast, but the reaction doesn’t last very long. On the other hand, the 20-hour NMP-Ribose-Glucose mix starts with simpler building blocks like ribose, glucose, and nucleoside monophosphates (AMP, CMP, UMP, etc.). The lysate gradually converts these into NTPs over time, so protein production starts more slowly but can continue for many hours.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Superfolder GFP is an engineered green fluorescent protein for robust folding even under stressful conditions. It is also reported to be a very rapidly maturing weak dimer.mRFP1: A basic red fluorescent protein known as a slow-maturing monomer with low acid sensitivity.mKO2: A basic orange fluorescent protein that has a moderate acid sensitivity and has a long maturation time.mTurquoise2: A basic turquoise fluorescent protein which is rapidly matures and has a very low acid sensitivity.mScarlet_I: A basic red fluorescent protein that has a moderate acid sensitivity and a slow maturation.Electra2: A basic blue fluorescent protein, published in 2022, it overcomes generally lower sensitivity in the blue channel, therefore making it easier to detect in cell-free systems in comparison to previous blue fluorescent proteins.References
Lambert, T. (n.d.). FPbase: The Fluorescent Protein Database. [online] FPbase. Available at: https://www.fpbase.org.2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mScarlet_I, which matures pretty slowly and is somewhat sensitive to acidic conditions, I hypothesise that increasing the amount of HEPES-KOH buffer (pH 7.5) and adding a potassium phosphate buffer (monobasic and dibasic) to the mastermix could help keep the pH more stable. This could reduce acid-related loss of the red fluorescence signal and help maintain brightness over time. I also hypothesise that slightly increasing the ribose and glucose levels could improve ATP/NTP regeneration. This may give more of the newly made mScarlet_I enough time and energy to fully mature its chromophore, potentially leading to stronger fluorescence at later time points
References
Calhoun, K.A. and Swartz, J.R. (2005). Energizing cell-free protein synthesis with glucose metabolism. Biotechnology and Bioengineering, 90(5), pp.606–613. doi:https://doi.org/10.1002/bit.20449.Lambert, T. (n.d.). mScarlet at FPbase. [online] FPbase. Available at: https://www.fpbase.org/protein/mscarlet/.Lazzari-Dean, J.R., Maria Clara Ingaramo, John C.K. Wang, Yong, J. and Ingaramo, M. (2022). mScarlet fluorescence lifetime reports lysosomal pH quantitatively. [online] Github.io. Available at: https://andrewgyork.github.io/mScarlet_lifetime_reports_pH/.3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
I chose the mScarlet_I protein and decided to base my experiment on my hypothesis from question 2 (above). Below are the alterations I made for my master-mix experimentation:
HEPES-KOH pH 7.5: increased from 45mM to 55mM (+ 10.000 mM).Potassium phosphate mono/dibasic: increased from 5.625mM to 7.500mM each (+ 1.875 mM).Ribose: increased from 11.625g/L to 15 g/L (+3.375).Glucose: increased from 1.25g/L to 2g/L (+0.750).
References
Angelo Cardoso Batista. Enabling and optimizing cell-free systems for synthetic biology applications. Molecular biology. Université Paris-Saclay, 2022. English. ⟨NNT : 2022UPASL030⟩. ⟨tel-04147740⟩Moon, B.-J., Lee, K.-H. and Kim, D.-M. (2018). Effects of ATP regeneration systems on the yields and solubilities of cell-free synthesized proteins. Journal of Industrial and Engineering Chemistry, 70, pp.276–280. doi:https://doi.org/10.1016/j.jiec.2018.10.027.www.aatbio.com. (n.d.).Potassium Phosphate (pH 5.8 to 8.0) Preparation and Recipe | AAT Bioquest. [online] Available at: https://www.aatbio.com/resources/buffer-preparations-and-recipes/potassium-phosphate-ph-5-8-to-8-0 [Accessed 22 May 2026].4. The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:6 μL of Lysate10 μL of 2X Optimized Master Mix from above2 μL of the assigned fluorescent protein DNA template2 μL of your custom reagent supplementsTotal: 20 μL reaction
This question isn’t applicable, but if I were to receive the fluorescence data, I would compare both the fluorescence time courses and endpoint intensities of mScarlet_I across wells with different buffer and energy compositions. I hypothesise that my modified mix will produce higher fluorescence at later time points compared with the course default formulation.
Subsections of Projects
FINAL PROJECT
B. subtilis as a PAH Degrading
Bioremediative Paint

Abstract
Polycyclic aromatic hydrocarbons (PAHs) are organic compounds characterised by two or more fused aromatic rings composed of carbon and hydrogen. These environmental contaminants are widely found in air, soil, water, sediments, and food, originating from both natural sources (such as wildfires and volcanic activity) and anthropogenic sources (including fossil fuel combustion, industrial processes, and incomplete biomass burning). Due to their environmental persistence and well-documented toxicity, PAHs pose significant risks to both ecosystems and human health, making their detection and remediation an ongoing challenge.
The overall goal of this project is to explore the potential of the bacterium Bacillus subtilis in degrading PAHs through the expression of its laccase enzyme, CotA, within a bioremediative paint system. This system is designed to incorporate a colourimetric output, enabling visualisation of pollutant presence while actively contributing to PAH degradation. When applied to urban surfaces, this living paint aims to function as a self-monitoring and self-remediating interface.
The hypothesis of this project is that fusion of the CotA enzyme with a His-tagged SUMO protein will enhance its stability and activity when expressed in Escherichia coli, improving its performance for potential environmental applications. While CotA has previously been expressed in E. coli and explored in indoor bioremediation contexts, this project investigates whether this modification can further optimise its function for outdoor use.
The aims of this project are divided into three key milestones. The first focuses on the expression and activity testing of the SUMO-tagged CotA enzyme in E. coli BL21, using a colourimetric ABTS assay to confirm functionality. Enzyme activity will also be assessed across different temperature conditions to determine optimal operating ranges. The second aim explores the development of a bio-based paint formulation incorporating the engineered bacteria, including further condition testing (pH and humidity), alongside testing cell viability and enzyme activity on surface applications. The third, more exploratory aim, involves evaluating PAH degradation potential and assessing the safety of byproducts, supporting the future development of a deployable and commercially relevant bioremediative paint.
The experimental approach begins with the transformation of E. coli BL21 competent cells using a pET-based plasmid (kanamycin resistance, T7 promoter) encoding the SUMO-tagged CotA enzyme. Following heat shock transformation and selection, protein expression will be induced, and the enzyme will be obtained using a SUMO protease-based purification system. As PAHs are not suitable for initial laboratory assays, ABTS will be used as a synthetic substrate, as it is oxidised by laccases to produce a measurable colour change. Enzyme activity will be quantified using a 96-well absorbance plate reader across varying temperatures, providing a proxy for its potential PAH-degrading capability.
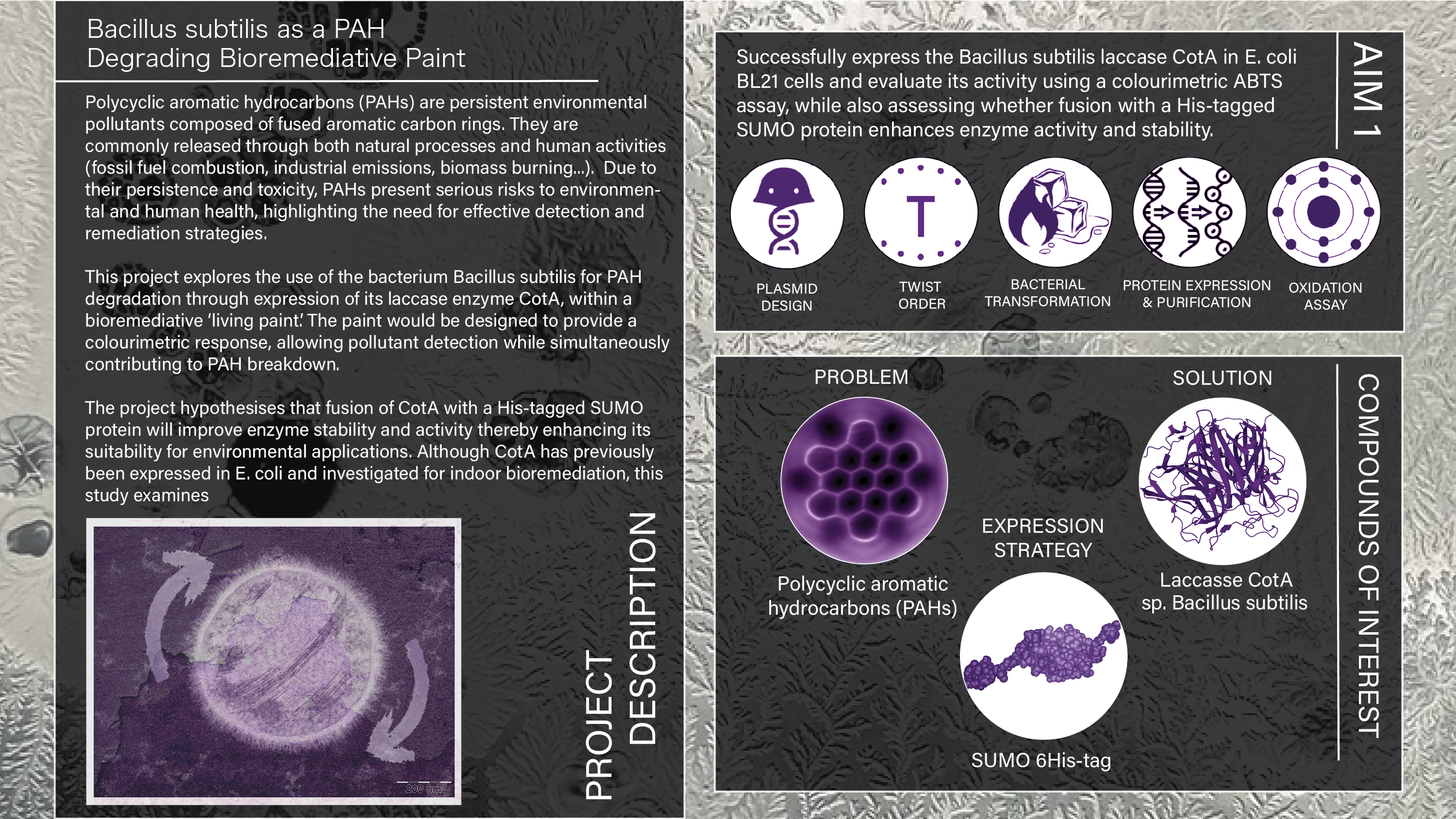

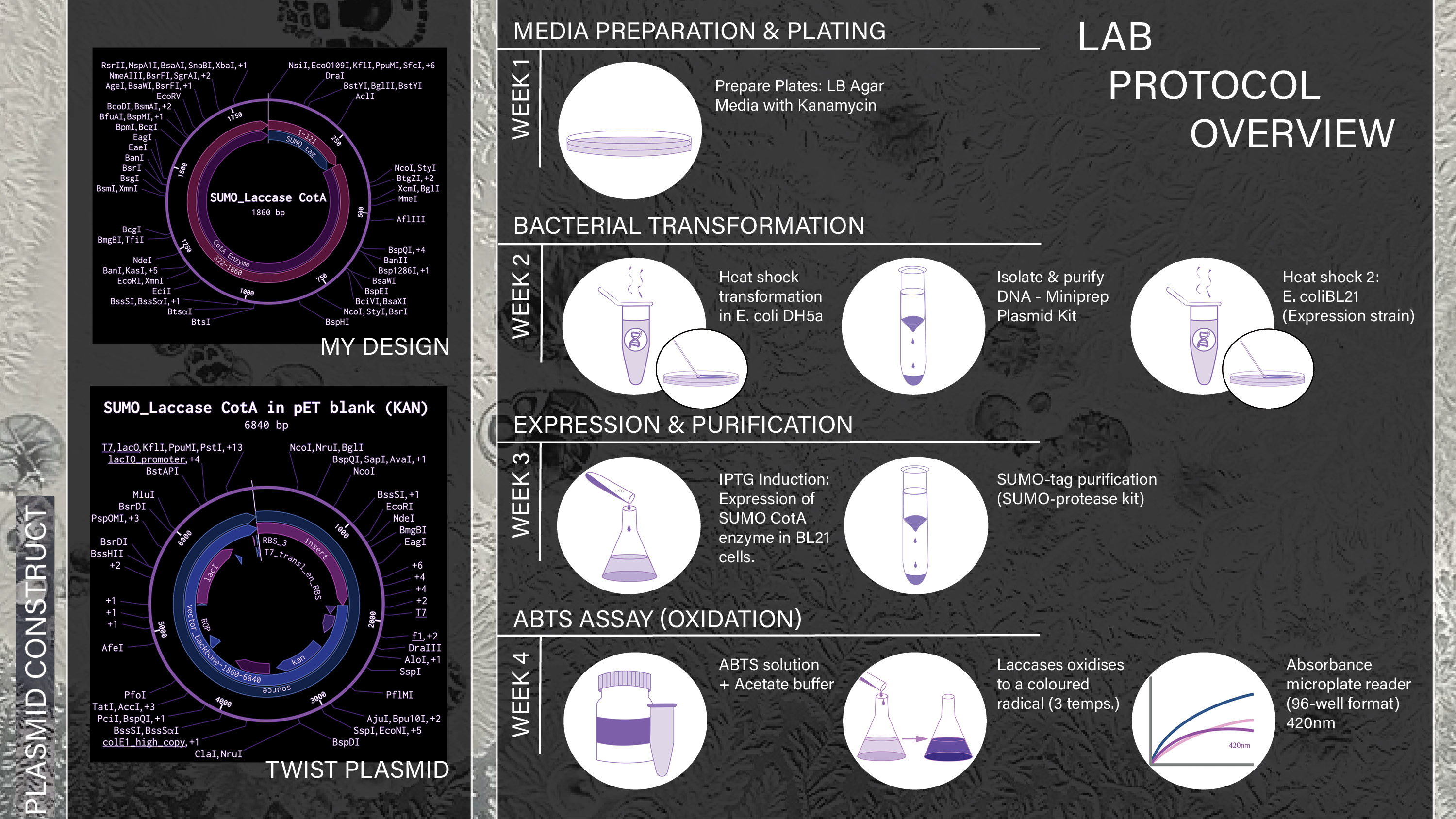
Project Aims
Aim 1: Experimental Aim
The first aim of my final project is to successfully express the Bacillus subtilis laccase CotA in E. coli BL21 cells and evaluate its activity using a colourimetric ABTS assay, while also assessing whether fusion with a His-tagged SUMO protein enhances enzyme activity and stability. To achieve this, a plasmid construct encoding the SUMO-tagged CotA enzyme was designed in Benchling and synthesised via Twist Bioscience using a pET-based expression backbone (kanamycin resistance, T7 promoter). The construct will be transformed into E. coli BL21 competent cells via heat shock, followed by antibiotic selection. Protein expression will be induced, and enzyme activity will be characterised using ABTS oxidation assays measured by a 96-well absorbance plate reader. Enzyme performance will be further evaluated across different temperature conditions to determine optimal activity and stability profiles.
Aim 2: Development Aim
The second aim of this project is to develop a bioremediative paint formulation containing the engineered E. coli cells expressing CotA, and to assess whether the system remains viable and functional when applied to a surface. Following successful confirmation of enzyme expression and activity in Aim 1, this step would focus on adapting the biological system into a usable paint-based format and testing its stability under conditions relevant to outdoor deployment. The aim is to determine whether the living paint can maintain bacterial viability as well as retain enzyme activity, and function as a self-remediating system in outdoor environments.
Aim 3: Visionary Aim
The third aim of this project is to explore the long-term potential of the living paint system as a safe scalable environmental technology for the detection and degradation of PAHs on urban surfaces. If fully realised, this approach could provide a new form of self-reporting and self-remediating material that not only indicates pollution hotspots through a colourimetric response but also actively contributes to contaminant breakdown. This would address a major barrier in environmental bioremediation by combining pollutant sensing and remediation within a single surface-applied platform. In the long term, the project could support the development of responsive building materials and offer a novel experimental framework for integrating synthetic biology with sustainable urban design.
Initial Interest
My project grew out of a particular interest in Bacillus subtilis, which I first explored during a previous biodesign group project. In that work, we investigated soil-dwelling bacteria and their benefits for both soil health and human wellbeing, and used this research to develop an educational game kit designed to teach children about the importance of caring for soil.

Initial group project with fellow biodesign students: Luiggi Marresse, Qian Qian Yutoung Hou, Christine Xiaoyan Weng, sparked my interest in the potential health benefits of bacteria, more specifically, Bacillus subtilis.
Background
1.Coming across using Bacillus Subtilis as a paint for bioremediation
While exploring ideas for my HTGAA final project, I took a closer interest in these bacteria and came across the paper “The Utilization of Bacillus subtilis to Design Environmentally Friendly Living Paints with Anti-Mold Properties” (Yuval et al., 2024). Because the study focused on exterior applications, I began to question how such a paint would perform in outdoor conditions and what additional capabilities this bacterium might offer as a bioremediative agent. This led me to a broader body of research examining its potential to degrade volatile organic compounds (VOCs), particularly polycyclic aromatic hydrocarbons (PAHs).
Reference: Yuval Dorfan, Avichay Nahami, Morris, Y., Shohat, B. and Kolodkin-Gal, I. (2024). The Utilization of Bacillus subtilis to Design Environmentally Friendly Living Paints with Anti-Mold Properties. Microorganisms, 12(6), pp.1226–1226.
The following are some of my key findings whilst extending my research:
Paper: Bacillus subtilis as a powerful weapon in the removal of environmental pollutants
Baciullus subtilitis: non-pathogenic Gram-positive bacteriumRenowned as the second most studied microorganism after Escherichia coliContributes significantly to the biodegradation of organic pollutants.According to the United Nations' 2030 Agenda for Sustainable Development, beneficial microbes are key tools for removing environmental contaminants and controlling plant diseases under the "One Health" framework (Sarrocco, 2023; Lee et al., 2023).Low molecular weight aromatic compounds (LMW - subset of PAH’s, 2-3 aromatic rings vs HMW PAHS have more than 4)Among polycyclic aromatic hydrocarbons, benzo[a]pyrene (BaP) and phenanthrene (PHE) have attracted significant attention due to their notable carcinogenicity, and have been classified by the International Agency for Research on Cancer (IARC) as Group 1 and Group 3 carcinogens.Both BaP and PHE, with their fused aromatic rings, are highly stable and resistant to biodegradation. However, B. subtilis has shown impressive abilities in its degradation.Reference: Liu, M., Chen, W.-J., Si, G., Yan, C., Song, H., Mishra, S., Ghorab, M.A. and Chen, S. (2025). Bacillus subtilis as a powerful weapon in the removal of environmental pollutants. Journal of Environmental Management, 396, p.127894. doi:https://doi.org/10.1016/j.jenvman.2025.127894.
Paper: Enzymatic Pathways and Mechanisms of Polycyclic Aromatic Hydrocarbon (PAH) Degradation by Bacillus subtilis
PAHs: Organic pollutants made of fused aromatic rings, formed mainly by incomplete combustion (coal, oil, gas, biomass) and industrial activity.Structural diversity: Range from simple 2-ring compounds (e.g., naphthalene) to complex 3+ ring PAHs (e.g., anthracene, phenanthrene).Biodegradability: Low-molecular-weight (LMW) PAHs are more biodegradable; high-molecular-weight (HMW) PAHs are more persistent → initial focus on LMW, extend to HMW in later work.Key challenge: PAH degradation can produce toxic intermediates → toxicity assays are needed to ensure safe and complete bioremediation (biosafety consideration).Health risks: PAHs are toxic, mutagenic, and carcinogenic (e.g., benzo[a]pyrene); exposure occurs via inhalation, ingestion, or skin contact and is linked to respiratory disease and cancer.Why Bacillus subtilis: Non-pathogenic and safe, capable of degrading multiple PAHs (including some HMW), genetically tractable for enhancing degradation efficiency.Pseudomonas spp.: Highly adaptable degraders, carry PAH-degrading genes on plasmids (enabling horizontal transfer), produce enzymes like dioxygenases to break down a wide range of PAHs.B. subtilis BMT4i: Degrades benzo[a]pyrene via chromosomally encoded genes, meaning the genes responsible for this function are integrated into the bacterium’s chromosome rather than carried on plasmids. This was confirmed through experiments demonstrating that removing plasmids had no impact on BaP degradation.Enzymatic Pathways and Mechanisms of Polycyclic Aromatic Hydrocarbon (PAH) Degradation by Bacillus subtilis
Enzymatic capability: Bacillus subtilis can degrade PAHs using multiple enzymes, though full pathways are not yet fully understood.Initial oxidation: Dioxygenases and monooxygenases add hydroxyl groups to PAH rings, starting the breakdown process.Intermediate conversion: Dehydrogenases convert dihydrodiols into diols, helping stabilise and further process intermediates.Ring cleavage: Ring-cleaving dioxygenases break aromatic rings via ortho- or meta-cleavage pathways.Catechol breakdown: Catechol-degrading enzymes process intermediates formed after ring cleavage.Additional enzymes: Likely includes lignin peroxidase and cytochrome P450 monooxygenases, similar to other PAH-degrading microbes.Knowledge gap: Exact degradation pathways in B. subtilis remain incompletely characterised and require further research.Reference:Nayra Niño (2024). Review Paper: Bacillus subtilis BMT4i a Bioremediation Agent for High-Molecular-Weight PAH…. [online] Medium. Available at: https://medium.com/insights-of-nature/review-paper-bacillus-subtilis-bmt4i-a-bioremediation-agent-for-high-molecular-weight-pah-f62438f1c574 [Accessed 2 Mar. 2026].
Paper:Degradation of Benzo [a] Pyrene by a novel strain, Bacillus subtilis BMT4i (MTCC 9447)
Unlike prior studies, where BaP degradation occurred only co-metabolically in the presence of additional carbon sources, BMT4i efficiently utilised BaP as its sole carbon source.Highlights the adaptability of BMT4i, which can also degrade other polycyclic aromatic hydrocarbons (PAHs) such as naphthalene, anthracene, and dibenzothiophene.Reference:Lily, M.K., Bahuguna, A., Dangwal, K. and Garg, V. (2009). Degradation of Benzo [a] Pyrene by a novel strain Bacillus subtilis BMT4i (MTCC 9447). Brazilian Journal of Microbiology, 40(4), pp.884–892. doi:https://doi.org/10.1590/s1517-83822009000400020.
Future Applications / Research Gap based on literature review:
- Toxicity & safety assessment
Analyse the toxicity of metabolites formed during PAH degradation.Ensure breakdown products are less harmful than parent compounds.Incorporate toxicity assays as part of biosafety evaluation.- Mineralisation studies
Determine whether B. subtilis can fully mineralise PAHs (e.g., pyrene, benzo[a]pyrene).Assess conversion into harmless end products (CO₂ and H₂O).- Metabolic pathway elucidation
Identify enzymes and pathways involved in PAH degradation.Address the current lack of fully characterised pathways.Improve understanding of degradation mechanisms.- Environmental stability
Evaluate long-term performance of B. subtilis in contaminated environments.Assess reliability and consistency under real-world conditions.- Genetic optimisation
Address gap: Most studies focus on natural degradation capacity.Use genetic engineering to enhance PAH degradation efficiency.Upregulate or introduce key degradation genes.Modify metabolic pathways to improve enzyme activity.Enable breakdown of more complex (HMW) PAHs.Project Proposal: 1st Draft
Project Proposal Slide: Second Draft
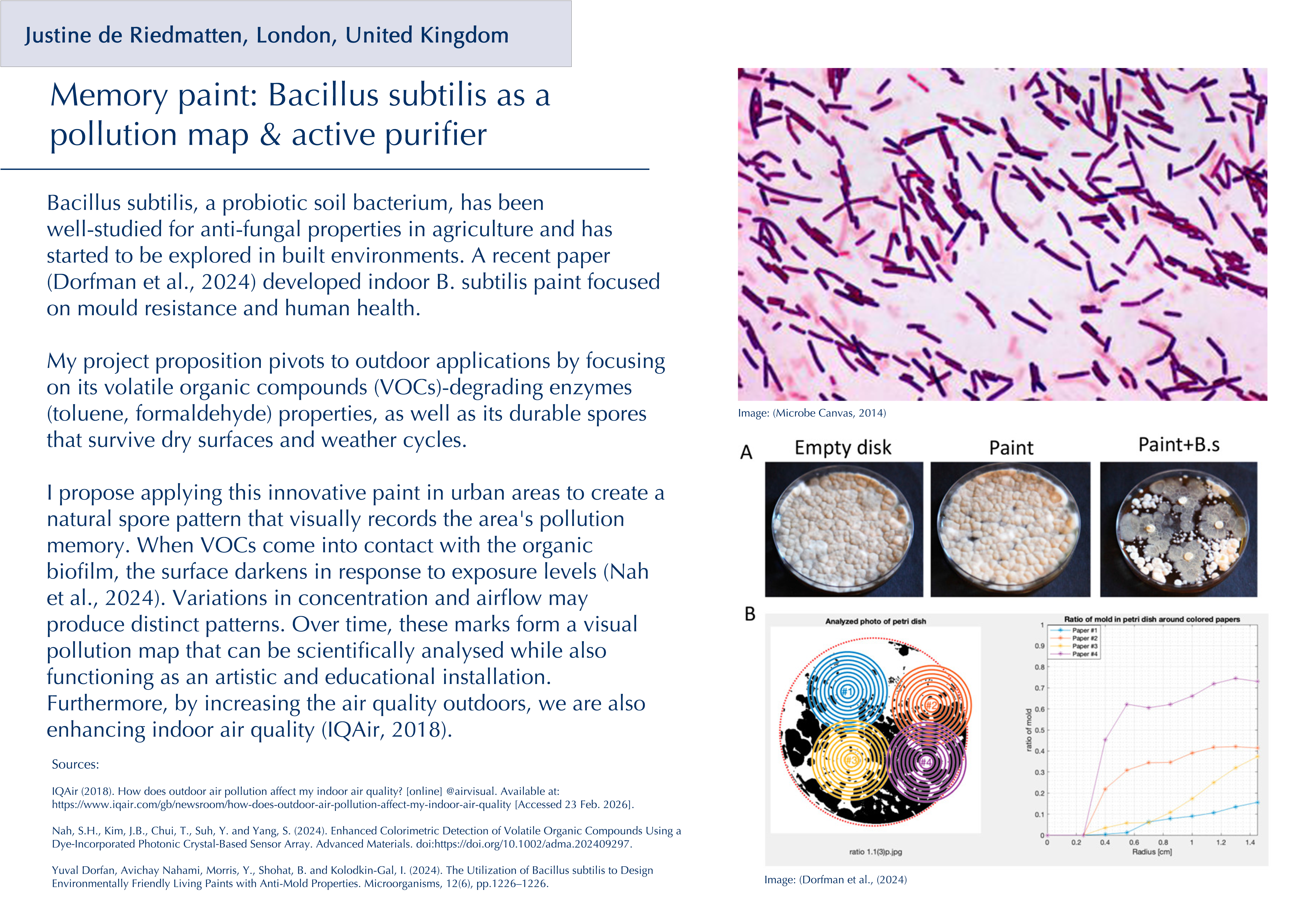
After more research and getting feedback from my node leader, I chose to develop this idea (out of 3) further:

Specific Enzyme Research
I furthered my research to identify the specific enzymes that Bacillus subtilis has that have been studied for their ability to degrade PAH. Through this research, I found two key enzymes:
Enzyme 1: polycyclic aromatic hydrocarbon ring-hydroxylating dioxygenase alpha subunit, partial [Bacillus sp. SB26]
Paper reference: Bhatt, K.K., Lily, M.K., Joshi, G. and Dangwal, K. (2018a). Benzo(a)pyrene degradation pathway in Bacillus subtilis BMT4i (MTCC 9447). Turkish Journal of Biochemistry, 43(6), pp.693–701. doi:https://doi.org/10.1515/tjb-2017-0334.
Enzyme 2: Laccase CotA
Paper reference: NPU CHINA (2022). | NPU-CHINA - iGEM 2022. [online] Igem.wiki. Available at: https://2022.igem.wiki/npu-china/design#gene [Accessed 2 Mar. 2026].
quote from reference: "One study found that laccase CotA from Bacillus subtilis exhibit a higher laccase-specific activity than laccase CueO from Escherichia coli, indicating that CotA is a better candidate for the remediation of PAHs than CueO. This is why we chose laccase cotA from Bacillus subtilis as a member of the ligninolytic enzymes system in our project."
After further research into the individual enzymes, I chose to focus on the CotA laccase due to the stronger body of literature and the number of projects developing plasmid constructs incorporating this enzyme for the bioremediation of PAHs. In addition, E. coli is widely used as a model organism in laboratory settings, and as I was the only in-person student at Lifefabs aiming to explore expression directly in Bacillus subtilis, this provided a practical and well-supported alternative. Previous studies have successfully expressed CotA laccase in E. coli strains, reinforcing its suitability for this approach.
Sequence of Bacillus subtilis strain WD23 laccase (cotA) gene, complete cds (1542bp):
atgacacttg aaaaatttgt ggatgctctc ccaatcccag atacactaaa gccagtacag caatcaaaag aaaaaacata ctacgaagtc accatggaag aatgcactca tcagctccat cgcgatctcc ctccaacccg cctgtgggga tacaacggct tatttccggg gccgaccatt gaggttaaaa gaaatgaaaa cgtatatgta aaatggatga ataaccttcc ttccacacat ttccttccga ttgatcacac cattcatcac agtgacagcc agcatgaaga gcccgaggta aagactgttg ttcatttaca cggcggcgtc acgccagatg acagtgacgg gtatccggag gcttggtttt ccaaagactt tgaacaaaca ggaccttatt tcaaaagaga ggtttatcat tatccaaacc agcagcgcgg ggctatattg tggtatcacg atcacgccat ggcgctcacc aggctaaatg tctatgccgg acttgtcggt gcttatatca ttcatgaccc aaaggaaaaa cgcttaaaac tgccttcaga cgaatacgat gtgccgcttc ttatcacaga ccgcacgatc aatgaggacg gttctttgtt ttatccaagc gcaccggaaa acccttctcc gtcactgcct aatccttcaa tcgttccggc tttttgcgga gaaaccatac tcgtcaacgg gaaggtatgg ccatacttgg aagtcgagcc aaggaaatac cgattccgtg tcatcaacgc ctccaataca agaacctata acctgtcact cgataatggc ggagagttta ttcagattgg ttcagatgga gggctcctgc cgcgatctgt taaactgaat tctttcagcc ttgcgcctgc tgaacgttat gatatcatca ttgacttcac agcatatgaa ggagaatcga tcattttggc aaacagcgcg ggctgcggcg gtgacgtcaa tcctgaaaca gatgcgaata tcatgcaatt cagagtcaca aaaccattgg cacaaaaaga cgaaagcaga aagccgaagt acctcgcctc atacccttcg gtacagcatg aaagaataca aaacatcaga acgttaaaac tggcaggcac ccaggacgaatacggcagac ccgtccttct gcttaataac aaacgctggc acgatcccgt cacagaagca ccaaaagtcg gcacaactga aatatggtcc attatcaacc ccacacgcgg aacacatccgatccacctgc atctagtctc cttccgtgta ttagaccggc ggccgtttga tatcgcccgttatcaagaaa gcggggaatt gtcatatacc ggtccggctg tcccgccgcc gccaagtgaaaaaggctgga aagacaccat tcaagcgcat gcaggtgaag tcctgagaat cgcggcgacattcggtccgt acagcggacg atacgtatgg cattgccata ttctagagca tgaagactatgacatgatga gaccgatgga tataactgat ccccataaataa
Uniprot page: https://www.uniprot.org/uniprotkb/H8WGE7/entryUniprot page: https://www.uniprot.org/uniprotkb/H8WGE7/entryBACKGROUND
1. Briefly summarize two peer-reviewed research citations relevant to your research
Paper 1: Butt, T.R., Edavettal, S.C., Hall, J.P. and Mattern, M.R. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification, 43(1), pp.1–9. doi:https://doi.org/10.1016/j.pep.2005.03.016.
This paper reviews how fusing target proteins to the small ubiquitin‑like modifier (SUMO) can dramatically improve their expression, solubility, and stability in both prokaryotic and eukaryotic systems. It explains that SUMO behaves as a well‑folded N‑terminal fusion partner that promotes proper folding and protects proteins from degradation, and that the SUMO tag can be removed very cleanly using a highly specific SUMO protease to regenerate the native N‑terminus. The authors compare SUMO to traditional fusion tags and show that SUMO often outperforms them for “difficult” proteins, making it a powerful general strategy for functional recombinant protein production.
Paper 2:Chemosphere, 148, pp.1–7. doi:https://doi.org/10.1016/j.chemosphere.2016.01.019.
This paper investigates whether recombinant Bacillus subtilis CotA laccase can efficiently oxidise polycyclic aromatic hydrocarbons (PAHs) and how it compares to other bacterial and fungal laccases. The authors express CotA, characterise its laccase activity, and show that it can oxidise a range of EPA‑priority PAHs with relatively high reaction rates and with less dependence on copper supplements than other bacterial laccases. They demonstrate that CotA retains activity over a broad range of pH and temperature, suggesting it is a robust biocatalyst for PAH degradation in environmental conditions. Overall, the study concludes that CotA is a particularly promising bacterial laccase candidate for bioremediation of PAH‑contaminated environments due to its high activity and practical independence from copper under the tested conditions.
2. Explain how your project is novel or innovative.
The novelty of this project lies in applying an N‑terminal SUMO-His fusion tag to CotA laccase from Bacillus subtilis for recombinant expression in Escherichia coli. CotA is generally considered difficult to express and fold correctly in standard bacterial hosts. Whilst His‑tagged CotA and various other fusion strategies have been reported, I found no studies that use a SUMO (or His‑SUMO) tag for CotA in E. coli. By combining the established solubility and folding‑enhancing properties of SUMO with CotA, this project explores a previously unreported expression strategy that could improve yields of active CotA and provide a useful platform for future application studies.3. Explain why your project matters and what impact it could have.
i. What pressing real-world problem does your project attempt to solve?
The project tackles the problem of polycyclic aromatic hydrocarbons (PAHs), which are persistent environmental pollutants composed of fused aromatic carbon rings. They are commonly released through both natural processes (e.g., volcanic eruptions) and human activities (e.g., fossil fuel combustion, industrial emissions, biomass burning). Due to their persistence and toxicity, PAHs present serious risks to environmental and human health, highlighting the need for effective detection and remediation strategies.
ii. Importance of the problem: Why is this problem significant, or what critical barrier to progress in the field does it represent?
The problem is significant due to its effect on planetary health and its presence in water, soil and air. In humans, many PAHs are classified as carcinogens and mutagens. Once inside the human body, metabolic enzymes transform PAHs into reactive epoxides and diol epoxides, which bind to DNA and form DNA adducts, which may trigger mutations and lead to cancer. Additionally, their consumption includes a variety of routes such as inhalation, dermal touch and ingestion.
Their persistence and high accumulation rates due to their lipophilic nature make them a major threat to terrestrial and aquatic life, as high concentrations will disrupt microbial communities, inhibit plant growth and lead to an overall disruption of biodiversity and the metabolic function of local ecosystems.
iii. Broader societal contribution: How could the outcomes of your project benefit society beyond the immediate research context?
Beyond the research context, this project envisions a bioremediative “living paint” that not only contributes to the breakdown of PAHs but also provides a clear colourimetric readout of pollution levels. By targeting PAHs in the air, it addresses a key pathway through which these pollutants enter and circulate within ecosystems. Designed for use in urban environments, such a system could both reduce human exposure to harmful contaminants while also acting as an accessible early‑warning indicator, empowering communities and policymakers to respond more quickly to deteriorating air quality.
iv. Advancement of knowledge or capability: How might the project improve scientific understanding, technical capability, or clinical practice within one or more fields?
Aim 1 of the project may improve scientific understanding and technical knowledge by validating whether a N-terminal SUMO-His tag protein may facilitate expression, stability and activity of the expression of CotA Lacasse for bioremediative applications due to its moderately difficult expression and low yield production. Ultimately, it would generate new data on SUMO-based fusion strategies intended for bioremediation strategies.
The future aims of the project (2 and 3) would extend that molecular knowledge into the technical capability of a living paint through testing it in realistic environmental conditions and the analysis and identification of the by-products. In turn, these outcomes could inform upstream enzyme engineering as well as downstream deployment of bioremediation technologies in outdoor urban environments.
v. Field-level change: If your aims are achieved, how could the concepts, methods, technologies, treatments, services, or preventative approaches used in this field of research change?
The use of Bacillus subtilis in a paint matrix has been explored in indoor settings for the effective treatment of black mould. In contrast, an outdoor living paint system to degrade harmful compounds/pollutants hasn’t been documented, which could change the field in the potential use of CotA Laccases for such an application. Additionally, chemical treatments for certain treatments of PAHs often transfer those pollutants from air to water sources, whilst if this system works, bacteria break down the stable ring structures of PAH down into natural byproducts, resulting in more eco-friendly and effective degradation.
4.Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility).
a.First paragraph: Include what ethical implications are involved in your project. Try to suggest ethical the principle(s) you may apply (e.g. non-maleficence, justice)?
This project raises clear ethical questions about safety, responsibility, and fairness in how engineered microbes are used in an outside lab setting. Unintended release into the environment could have potential unknown impacts on human and ecosystem health. This calls for strict non-maleficence and responsibility in both the lab development and testing, as well as the final users and application into the world. Furthermore, the work must prioritise strict risk assessment, biosafety measures, and transparent reporting of limitations as well as benefits.
Additionally, by cross-collaboration of disciplines (microbiologists, chemists, environmental scientists, product designers, policy makers), an improved system can be developed. Lastly, rigorous testing of degradation byproducts for toxicity, careful consideration of where and how such a paint would be deployed, and engagement with stakeholders who might be most affected, such as affected communities of polluted urban areas, are essential for the development of such a project.
b. Describe the measures that should be taken to ensure that your project is ethical (both in how the research is conducted and in its broader implications for society). You may wish to answer the following questions:
i.What action(s) do you propose?
1. Safety and non‑maleficence
Develop and follow a detailed biosafety protocol for all lab work, including waste disposal, and deactivation of engineered strains before leaving the lab.Build in genetic safeguards so cells cannot persist or spread if accidentally released.Run toxicity tests on all PAH degradation byproducts before proposing any real‑world deployment.2. Risk assessment and responsibility
Perform a structured risk assessment covering environmental release, human exposure, and misuse, which will be updated at each stage of the project.Propose clear usage guidelines for any future “living paint” product, such as where it can be used, how it should be applied, how long it can remain active, and how to deactivate or remove it.3. Stakeholder involvement
Explicitly propose a multidisciplinary advisory group (microbiologists, chemists, environmental scientists, designers, ethicists, and policy makers) to review design and deployment scenarios.Commit to depositing key data (e.g. degradation rates, byproduct profiles, safety tests) in an open database to allow full transparency.4. Cross‑disciplinary collaboration and transparency
Explicitly propose a multidisciplinary advisory group (microbiologists, chemists, environmental scientists, designers, ethicists, and policy makers) to review design and deployment scenarios.Commit to depositing key data (e.g. degradation rates, byproduct profiles, safety tests) in an open database to allow full transparency.ii. What are the potential unintended consequences of your proposed actions?
1. Biosafety protocols and genetic safeguards
Even with strict lab protocols and kill switches, containment systems might fail in unexpected conditions (e.g. unexpected weather changes), allowing engineered bacteria to survive or spread beyond intended areas. If safeguards malfunction, the project could still cause ecological disruption.2. Byproduct toxicity testing and risk assessment
Toxicity tests are always limited, so some harmful byproducts might remain undetected until after deployment.3. Stakeholder engagement and public consultations
Public consultations might attract only a narrow group of participants, so some affected communities could remain unheard or unconvinced, creating a false impression. If their feedback is not meaningfully incorporated, this could lead to mistrust or feelings of exploitation.4. Cross‑disciplinary collaboration and open data
While improving transparency, it also increases the risk that others might deploy the technology inappropriately or without the same safety standards.iii. What could you have been wrong (e.g., incorrect assumptions and uncertainties)?
There are several ways my assumptions in this project could be wrong. The SUMO–His–CotA construct may not improve expression, folding, or activity as much as anticipated. Additionally, it could potentially even impair CotA’s suitability for PAH degradation. Furthermore, the assumption that bacterial degradation will oxidise into safe and natural byproducts may be too optimistic if some intermediates turn out to be toxic or persistent.
I also assume at this stage of the project that bacteria will remain contained and functional within the paint matrix, but in practice, they may either die rapidly (reducing effectiveness) or spread beyond intended areas. Similarly, the colour change may not provide a clear, reliable signal of PAH levels, leading to misinterpretation or false reassurance. Finally, I may be underestimating social and regulatory resistance to releasing engineered microbes in public spaces, making the acceptance and deployment more challenging.
iv. What are alternatives to your proposed actions?
If my proposed actions fail, I would like to work more closely with community organisations or NGOs to redesign engagement so that affected groups genuinely shape decisions. If support remained low, I would redirect the concept toward more contained industrial facilities instead of public urban spaces. Lastly, in communicating results, I would publish only safety‑relevant information and laboratory performance data. This would avoid the share of detailed protocols that would enable easy reproduction, leading to unsupervised deployment of the living paint system.
v. Ethics statement:
The project aims to design a living‑paint system that safely reduces PAH air pollution through the rigorous testing of byproducts and biosafety risks, the involvement of affected communities in deployment decisions, and the reporting of methods and limitations transparently.
Experimental Design, techniques, tools, and technology
Bacterial Transformation: Week 1-2
I will start by transforming the Twist-synthesised CotA-SUMO-His plasmid into E. coli DH5α competent cells using a standard heat shock protocol. Transformed cells will be plated on LB agar containing kanamycin to select for colonies carrying the plasmid.
I will then pick several DH5α colonies and grow them in LB + kanamycin to amplify the plasmid for expression. A miniprep kit will be used to isolate plasmid DNA from these cultures, providing clean DNA for transformation into the expression strain (E. coli BL21).
I will transform the verified plasmid into E. coli BL21 competent cells, again using heat shock followed by selection on LB + kanamycin plates. BL21 is chosen because it is a standard host for T7 promoter protein expression.
Expression and Purification: Week 2
I will pick BL21 colonies and grow them in LB + kanamycin, then induce expression of the CotA-SUMO-His fusion using IPTG induction. This step is aimed at producing the SUMO-tagged CotA enzyme inside the BL21 cells.
I will harvest the induced BL21 cultures and use a SUMO-tag purification (His-tag/SUMO protease kit) to yield a purified enzyme preparation suitable for activity measurements.
ABTS Assay under different conditions: Week 3
Once I have the purified CotA‑SUMO enzyme, I will set up a colourimetric ABTS assay to test laccase activity. ABTS will be used as a synthetic, water‑soluble substrate that laccases oxidise to a coloured radical. The resulting colour change will be measured by absorbance using an absorbance microplate reader (96‑well format) at 420 nm in a sodium acetate buffer.
For Aim 1, the assay will be performed at a single, literature‑supported sodium acetate buffer pH, selected based on reported optima for CotA with ABTS. Keeping pH constant in this way will allow me to focus specifically on how temperature affects CotA‑SUMO activity under realistic application conditions.
I will test enzyme activity at three temperatures, 20 °C, 25 °C, and 35 °C, to represent a realistic range for an outdoor paint rather than extreme laboratory optima. This will help indicate whether the SUMO‑tagged enzyme can remain active across temperatures that a bioremediative paint might actually experience, while systematic variation of pH and humidity will be explored in Aim 2.
NOTES - Experimental Plan
For each temperature condition, I will record the change in absorbance over time as ABTS is oxidised, using technical replicates where possible to make the readout more reliable. These measurements will give me a simple activity profile for the CotA-SUMO enzyme across the tested condition, with the main focus on proof‑of‑concept activity rather than full kinetic characterisation. If the SUMO‑tagged CotA consistently produces a clear colour change with ABTS, this will support my hypothesis that the construct is functional and worth developing further and will pay particular attention to conditions that make sense for a future paint application. Overall, the expected outcome is to show that the CotA-SUMO-His enzyme can be expressed in E. coli BL21, enriched using a SUMO‑based purification step, and that it shows measurable laccase‑like activity in a colourimetric ABTS assay under at least some of the pH and temperature conditions I test. Even though I won’t fully optimise the system in this short amount of time, the results should give me a first idea of which conditions support activity and whether they overlap with realistic ranges for a bioremediative paint, and help define clear next steps, such as deeper optimisation.
1. We discussed and practised various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.

2. Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project.
To validate this aspect of my project, I first used DNA construct design tools in Benchling to build a pET‑based plasmid encoding a His‑tagged SUMO–CotA fusion. In Benchling, I designed the coding sequence, promoter and resistance cassette, checked reading frames, and confirmed the presence of key features such as the T7 promoter, SUMO tag, CotA coding region, and His tag.
I also used online sequence databases ( NCBI/GenBank and UniProt) to retrieve and cross‑check the Bacillus subtilis CotA sequence, confirm annotations, and ensure that my design was compatible with expression in E. coli. Based on these designs, I then performed chassis selection by assigning E. coli DH5α for plasmid amplification and E. coli BL21(DE3) as the expression host.
I initially created two plasmids with the backbone on Benchling, one containing the SUMOtag and the other only having CotA Lacasse as a gene of interest:
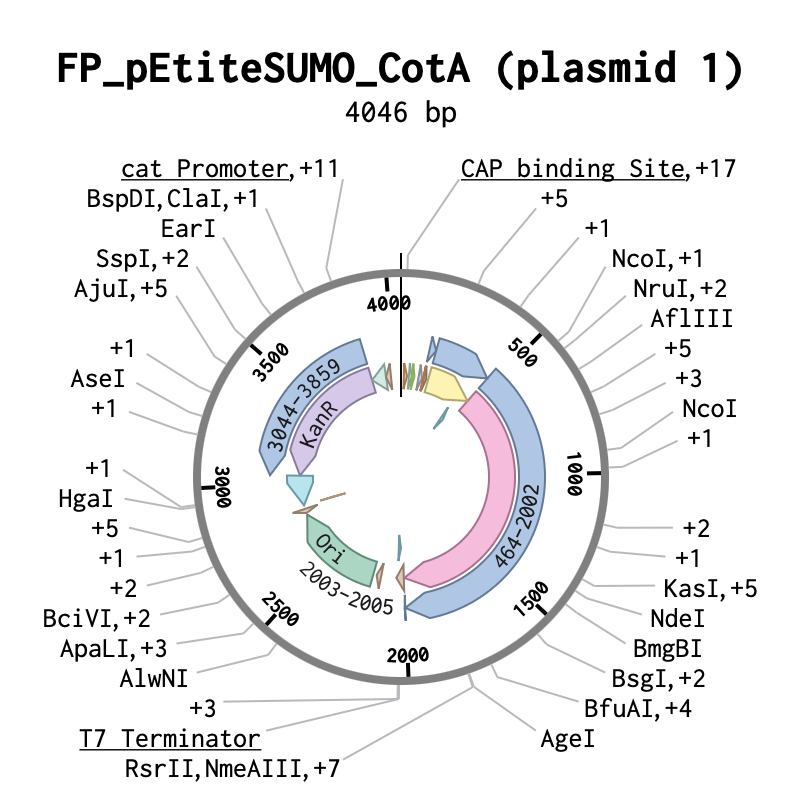

I then decided to only focus on my desired genes of interest:
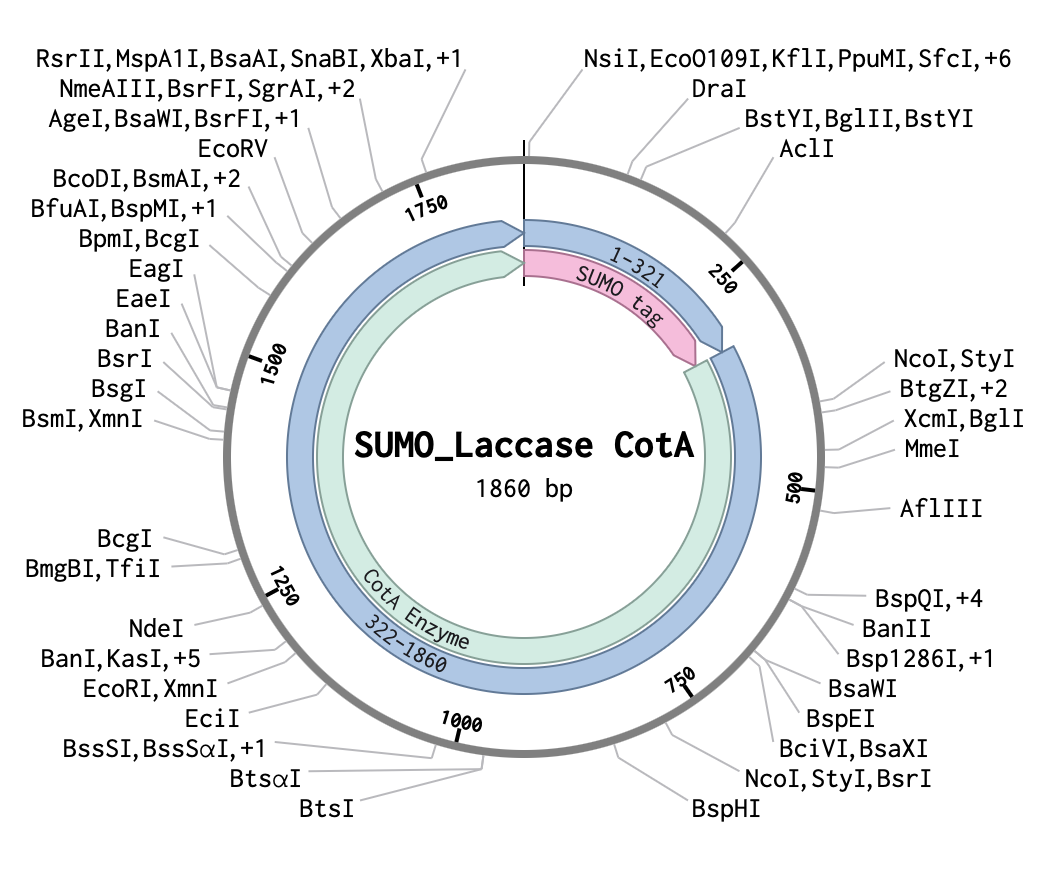
For accuracy, I selected a similar PET-compatible backbone on Twist:
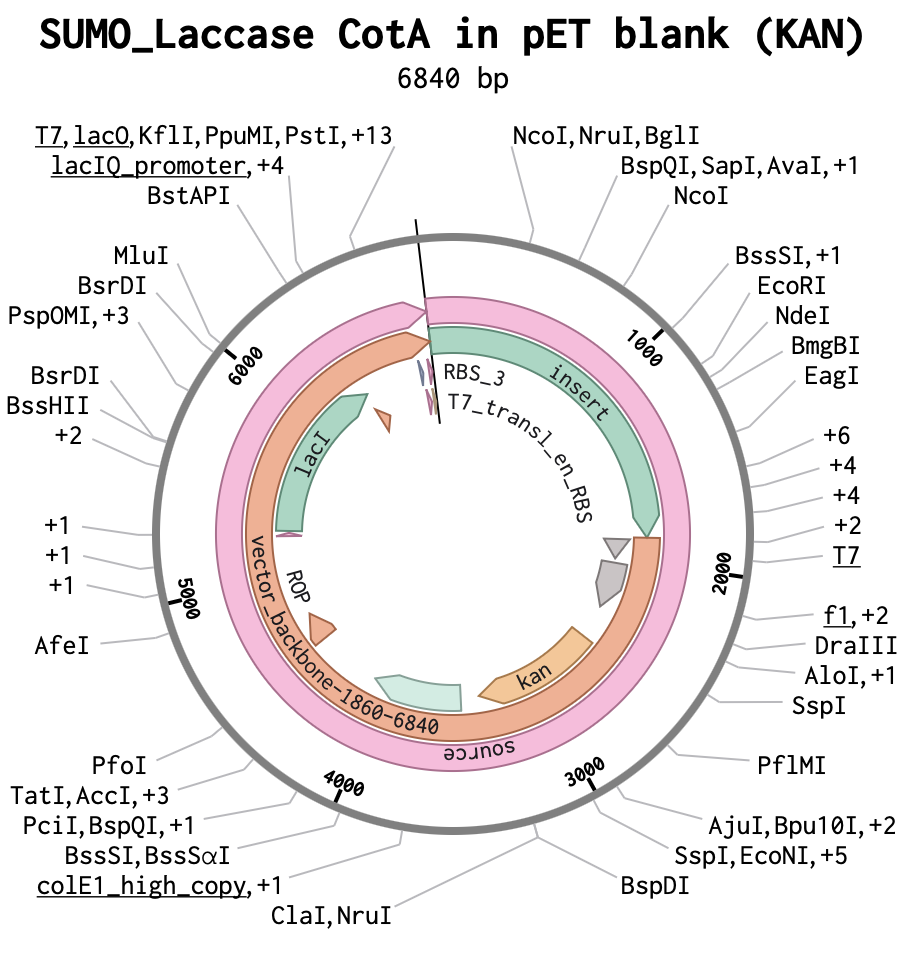
Benchling link to Project development: https://benchling.com/justine_biodesign/f_/Q6rz7UZkjP-laccase-cota-bacillus-s-_-petite-n-his-sumo-kan/
Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project

Results & Quantitative Expectations
You are required to validate at least one aspect of your final project aims. This is to ensure that you are able to successfully apply a relevant synthetic biology technique to your project. Include figures if you have them—accuracy is critical in figures, tables, and graphs
Here is a non-exhaustive list of acceptable validations:

Highlighted in green is what I have been able to validate, whilst yellow is still in progress and will be able to continue developing after Twist order delivery.
What aspect of your final project did you choose to validate?
I chose to validate the expression of the Bacillus subtilis laccase CotA in E. coli BL21 cells and to evaluate its enzymatic activity using a colourimetric ABTS assay. Additionally, validate whether fusion with a His-tagged SUMO protein can enhance the enzyme’s stability and overall activity.
Write down a detailed protocol of how you validated this aspect of your final project.
1. Plasmid Amplification and Verification in DH5α
The competent Escherichia coli DH5α cells will be thawed on ice.Twist-synthesised CotA-SUMO-His plasmid DNA (1–5 µL) will be added to 50 µL competent cells, gently mixed by flicking, and incubated on ice for 30 minutes.Cells will be heat shocked at 42 °C for 30 seconds and immediately returned to ice for 2 min.LB medium without antibiotic (950 µL) will then be added, and cells will be recovered at 37 °C with shaking (250 rpm) for 45 minutes.Recovered cells will be plated onto LB agar supplemented with kanamycin (50 µg/mL) and incubated overnight at 37 °C.Three to five kanamycin-resistant colonies will be selected and inoculated into 5 mL LB medium containing kanamycin, followed by overnight incubation at 37 °C with shaking.Plasmid DNA will be isolated using the GeneJET Miniprep Kit according to the manufacturer’s instructions.2. Transformation into BL21(DE3)
Competent E. coli BL21(DE3) cells will be thawed on ice.Verified CotA-SUMO-His plasmid DNA (1–5 µL) will be added to 50 µL competent cells and incubated on ice for 30 minutes.Cells will be heat shocked at 42 °C for 30 seconds, chilled on ice for 2 min, and recovered in 950 µL LB medium at 37 °C with shaking (250 rpm) for 45 minutes.Transformants will be plated onto LB agar containing kanamycin (50 µg/mL) and incubated overnight at 37 °C.Single colonies will be retained on plates for subsequent protein expression experiments.3. IPTG-Induced Expression of CotA-SUMO-His
A single BL21(DE3)/CotA-SUMO-His colony will be inoculated into 5–10 mL LB medium supplemented with kanamycin and cultured overnight at 37 °C with shaking (250 rpm).The overnight culture will be diluted 1:100 into 50–200 mL fresh LB medium containing kanamycin in a baffled flask and incubated at 37 °C with shaking until the culture reaches mid-log phase (OD₆₀₀ = 0.5–0.8), usually around 2.5-3 hours for BL21.Protein expression will be induced by the addition of IPTG to a final concentration of 0.5–1 mM.The cultures will then be shifted to 20–25 °C and incubated for 4 hours to promote soluble expression of CotA-SUMO-His.The cells will be collected by centrifugation at 4,000 ×g for 10–15 min.Supernatants will be discarded, and cell pellets will be stored on ice or at −20 °C until lysis.4. Cell Lysis and His-Affinity Purification
4.1 Preparation of Buffers (from Tris Base and NaCl)
All buffers will be prepared from Tris base and NaCl and adjusted to the appropriate pH at room temperature.
Lysis/Binding Buffer
50 mM Tris-HCl, pH 8.0300 mM NaClWash Buffer
50 mM Tris-HCl, pH 8.0300 mM NaCl20–40 mM imidazoleElution Buffer
50 mM Tris-HCl, pH 8.0300 mM NaCl250–300 mM imidazole4.2 Cell Lysis
Following the expression, 5 mL of bacterial culture will be transferred into a centrifuge tube, and cells will be pelleted by centrifugation at 10,000 rpm for 2 minutes at room temperature.The supernatant will be discarded, and the cell pellet retained. A total of 1.67 mL S30 buffer will then be added to the pellet, and cells will be thoroughly resuspended by vortexing.The resuspended cells will be transferred into a bead-beating tube for mechanical disruption.Following bead beating, lysates will be clarified by centrifugation at 15,000 rpm for 30 minutes.The clear supernatant, representing the soluble cell extract containing CotA-SUMO-His and E. coli, will be retained while the pellet will be discarded.Cell extracts will be stored at −20 °C4.3 Purification of CotA-SUMO-His
A single Ni-NTA column will be equilibrated with 5–10 column volumes of lysis/binding buffer.The lysate will be loaded onto the column, allowing CotA-SUMO-His to bind via its His tag. The column will then be washed with 10–20 column volumes of wash buffer to remove non-specifically bound proteins.CotA-SUMO-His will be eluted using the elution buffer.4.4 SUMO and His Tag Removal Using the Same Column
His-tagged SUMO protease will be added at the recommended enzyme-to-substrate ratio, and the mixture will be incubated under the specified conditions (time and temperature) to cleave the SUMO-His tag from CotA based on the Sumo-protease kit instructions.Following cleavage, the reaction mixture will be reapplied to the same Ni-NTA column, which will be re-equilibrated with lysis/binding buffer.The His-tagged SUMO and His-tagged SUMO protease will bind to the resin, while de-SUMOylated, non-His CotA will be recovered.Native CotA protein is collected for the ABTS assay.5. Preparation of Assay Buffer and ABTS
Sodium acetate buffer solution will be prepared and adjusted to pH 4.5, which has been reported as near-optimal for CotA-type laccase activity using ABTS as substrate.A 0.1 M sodium acetate working buffer (pH 4.5) will be prepared for all activity assays.Fresh ABTS working solution will be prepared by diluting ABTS stock solution into sodium acetate buffer to the required final concentration ( 0.5–1 mM).ABTS solutions will be protected from light and used on the day of preparation.6. ABTS Activity Assay at 20 °C, 25 °C, and 35 °C
The assay will be performed in a 96-well microplate reader with absorbance.
6.1 Reaction Setup
For a representative well assay (total volume 200 µL):180 µL of 0.1 M sodium acetate buffer (pH 4.5)20 µL of purified CotA enzymeA blank will be included for each condition (20 µL of buffer)6.2 Absorbance Measurement
The absorbent plate reader will be set to 420 nm and equilibrated to the desired assay temperature (20 °C, 25 °C, or 35 °C).Buffer and ABTS solutions will be pre-equilibrated at the assay temperature before enzyme addition.Reactions will be initiated by the addition of the enzyme, rapidly mixed, and absorbance at 420 nm will be recorded immediately.Absorbance measurements will be collected every 30–60 seconds for 3–5 min, ensuring analysis is restricted to the initial linear phase of the reaction.7. Data Analysis and Validation
For each assay, absorbance at 420 nm will be plotted against time, and the initial linear rate will be determined as ΔA₄₂₀/min.Enzyme activity will be calculated using the extinction coefficient of the ABTS radical at 420 nm.Initial reaction rates obtained at 20 °C, 25 °C, and 35 °C will be compared to generate a temperature–activity profile at constant pH 4.5.In addition to spectrophotometric quantification, ABTS oxidation will also be assessed visually based on the intensity of the green-blue colour formed during the reaction. The degree of colour development ( how dark the oxidised ABTS solution becomes over time) will be used as a qualitative indicator of relative enzyme activity across temperatures, providing a complementary visual comparison alongside quantitative measurements.3.What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8.
To validate this aspect of my project, I used a variety of techniques. Starting with DNA construct design for a His‑tagged SUMO–CotA fusion in a pET backbone, using Benchling and databases such as NCBI/GenBank to source and align the Bacillus subtilis CotA sequence and ensure compatibility with E. coli BL21(DE3). I then designed and placed a Twist order for synthesis of the full expression cassette (T7 promoter, SUMO–CotA coding sequence, His tag, and kanamycin resistance), integrating what I had learned about plasmid architecture and expression hosts.
The planned validation workflow combines chassis selection (E. coli DH5α for plasmid propagation and BL21(DE3) for protein expression), bacterial culturing, and bacterial processing steps such as heat‑shock transformation, antibiotic selection, centrifugation, and plasmid minipreps to prepare and verify the construct.
4. You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data (e.g., using the Asimov Kernel or another simulation method).
Whilst I wait to conduct my lab experimentations, I decided to use alphafold to observe its predictions on the folding of the CotA Lacasse from Bacillus subtilis with and without the SUMOtag to evaluate whether it would affect the folding of the CotA.
CotA Lacasse amino acid sequence:
TLEKFVDALPIPDTLKPVQQSKEKTYYEVTMEECTHQLHRDLPPTRLWGYNGLFPGPTIE
VKRNENVYVKWMNNLPSTHFLPIDHTIHHSDSQHEEPEVKTVVHLHGGVTPDDSDGYPEA
WFSKDFEQTGPYFKREVYHYPNQQRGAILWYHDHAMALTRLNVYAGLVGAYIIHDPKEKR
LKLPSDEYDVPLLITDRTINEDGSLFYPSAPENPSPSLPNPSIVPAFCGETILVNGKVWP
YLEVEPRKYRFRVINASNTRTYNLSLDNGGEFIQIGSDGGLLPRSVKLNSFSLAPAERYD
IIIDFTAYEGESIILANSAGCGGDVNPETDANIMQFRVTKPLAQKDESRKPKYLASYPSV
QHERIQNIRTLKLAGTQDEYGRPVLLLNNKRWHDPVTEAPKVGTTEIWSIINPTRGTHPI
HLHLVSFRVLDRRPFDIARYQESGELSYTGPAVPPPPSEKGWKDTIQAHAGEVLRIAATF
GPYSGRYVWHCHILEHEDYDMMRPMDITDPHK
SUMO-CotA Lacasse amino acid fusion sequence:
GSLQDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGK
EMDSLTFLYDGIEIQADQTPEDLDMEDNDIIEAHREQIGGTLEKFVDALPIPDTLKPVQQ
SKEKTYYEVTMEECTHQLHRDLPPTRLWGYNGLFPGPTIEVKRNENVYVKWMNNLPSTHF
LPIDHTIHHSDSQHEEPEVKTVVHLHGGVTPDDSDGYPEAWFSKDFEQTGPYFKREVYHY
PNQQRGAILWYHDHAMALTRLNVYAGLVGAYIIHDPKEKRLKLPSDEYDVPLLITDRTIN
EDGSLFYPSAPENPSPSLPNPSIVPAFCGETILVNGKVWPYLEVEPRKYRFRVINASNTR
TYNLSLDNGGEFIQIGSDGGLLPRSVKLNSFSLAPAERYDIIIDFTAYEGESIILANSAG
CGGDVNPETDANIMQFRVTKPLAQKDESRKPKYLASYPSVQHERIQNIRTLKLAGTQDEY
GRPVLLLNNKRWHDPVTEAPKVGTTEIWSIINPTRGTHPIHLHLVSFRVLDRRPFDIARY
QESGELSYTGPAVPPPPSEKGWKDTIQAHAGEVLRIAATFGPYSGRYVWHCHILEHEDYD
I began by comparing the PDB structure and the predicted alpha fold of CotA Lacasse:
The AlphaFold prediction for CotA closely matches the experimentally determined CotA crystal structure (PDB ID: 7Y8C), supporting the reliability of the model. Additionally, the predicted structure has a high pTM score of 0.98, proving it as a high-confidence model.
I then analysed both the CotA and SUMO-CotA structures from different angles:
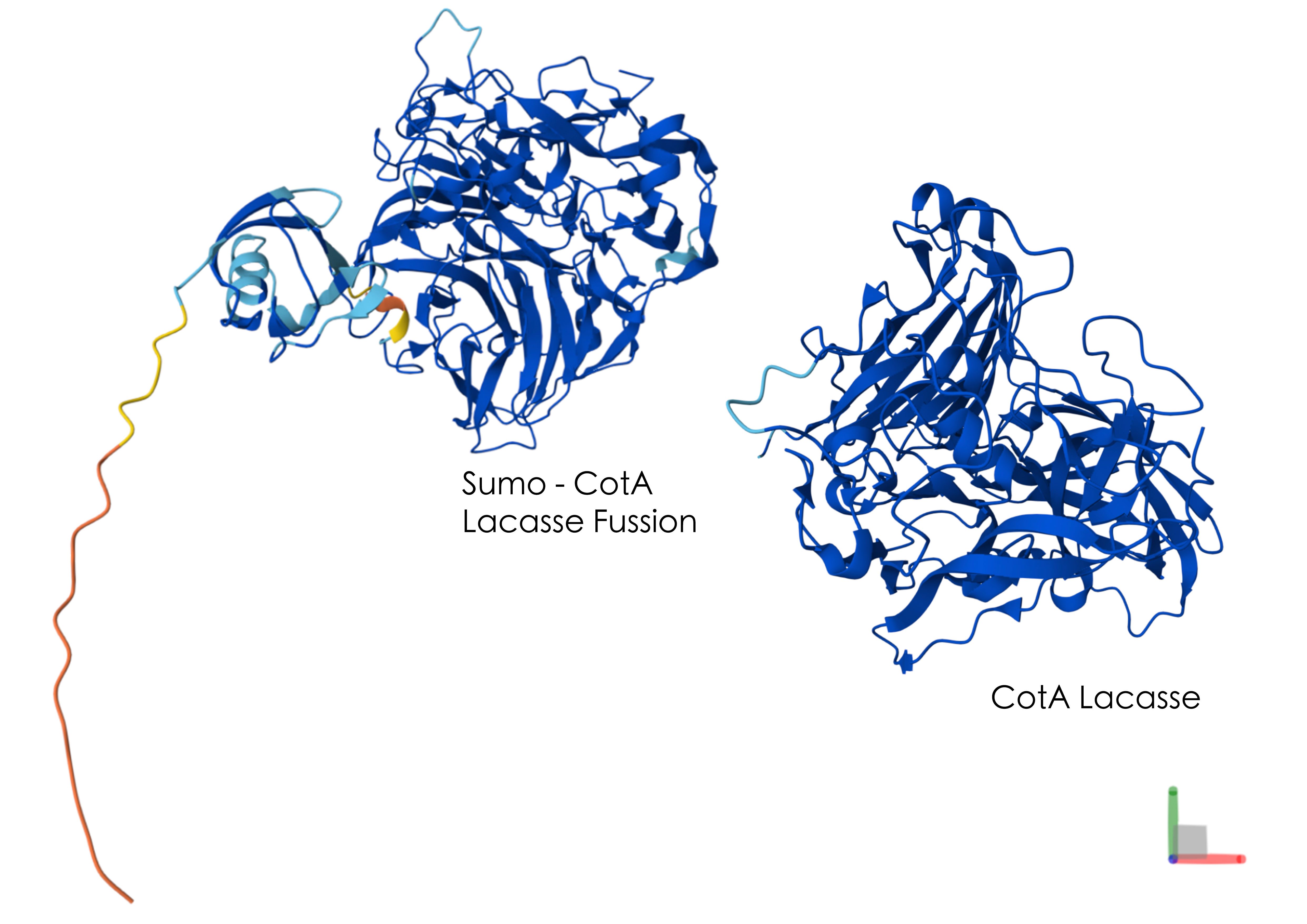
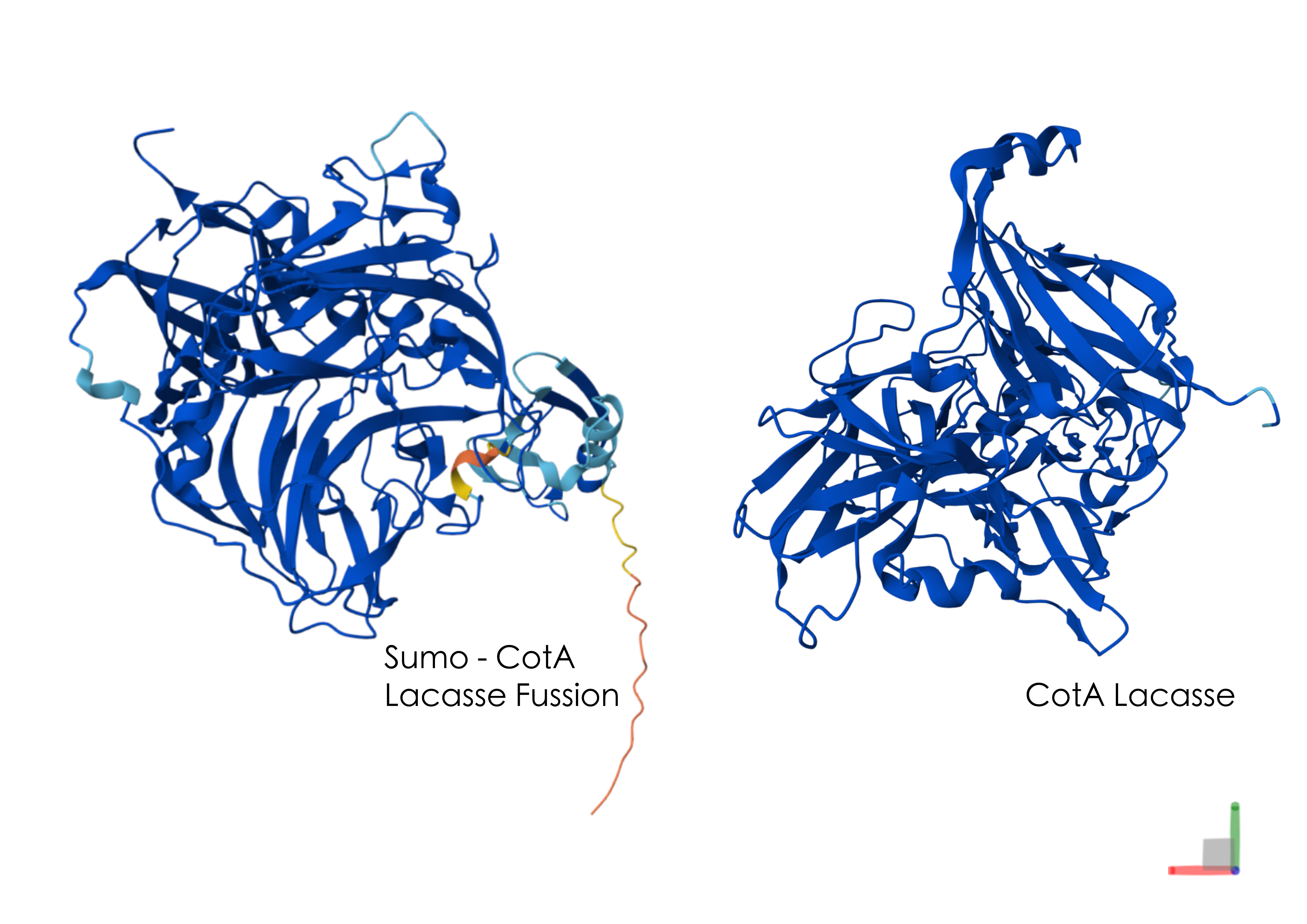
When I modelled the SUMO-CotA fusion as a single polypeptide, the prediction showed the CotA region still forming a globular laccase‑like core, but with an additional N‑terminal corresponding to the SUMO protein. This N‑terminal region appears as a flexible tail and a smaller folded domain attached to CotA. It shows a very low model confidence (orange), indicating that its exact position and conformation are uncertain.
When the CotA portion of the fusion is overlaid with the CotA‑only model, the overall shape is different, suggesting that the tag may alter the relative arrangement of some β‑strands, α‑helices, or domains. Taken together, the models suggest that CotA can still adopt a laccase fold in the presence of the SUMO tag but that the fusion could alter local structure. Because structural prediction alone cannot tell me whether expression, solubility, or activity are improved or disrupted with a SUMO tag, these experimental questions need to be answered through laboratory work.
5. Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project.
One of the main challenges I anticipate with my experimental plan is achieving sufficient soluble expression of the CotA‑SUMO‑His fusion in E. coli BL21(DE3). With or without the SUMO tag, the CotA could misfold, which would reduce the amount of active enzyme available for ABTS assays and complicate the interpretation of weak colour changes.
Another likely difficulty is the incomplete binding or elution from the Ni‑NTA column. For example, if the His tag is not fully accessible, it could lead to low recovery and variable protein concentrations between preparations. I may also encounter problems with the ABTS readout, such as high background oxidation, which makes it hard to compare conditions accurately.
To address these possible issues, alternative strategies can be applied:
Varying induction temperature and time to improve foldingComparing the SUMO‑fused enzyme to a de‑SUMOed version after protease cleavage.Additionally, if ABTS signals are weak, I can:
Increase enzyme loadingExtend measurement timesOptimise the ABTS concentrationADDITIONAL INFORMATION
List all references in this assignment (bullet-point list)
Ardila-Leal, L.D., Monterey-Gutiérrez, P.A., Poutou-Piñales, R.A., Quevedo-Hidalgo, B.E., Galindo, J.F. and Pedroza-Rodríguez, A.M. (2021). Recombinant laccase rPOXA 1B real-time, accelerated and molecular dynamics stability study. BMC Biotechnology, 21(1). doi:https://doi.org/10.1186/s12896-021-00698-3.Ayilara, M.S. and Babalola, O.O. (2023). Bioremediation of environmental wastes: the role of microorganisms. Frontiers in Agronomy, 5. doi:https://doi.org/10.3389/fagro.2023.1183691.BenchChem (2025). Isopropyl β-D-1-thiogalactopyranoside (IPTG): A Comprehensive Technical Guide Chemical Properties and Structure.BenchChem Technical Support Team (2026). Application Notes: ABTS Assay for Laccase Activity. [online] Available at: https://pdf.benchchem.com/7949/Application_Notes_ABTS_Assay_for_Laccase_Activity.pdf [Accessed 12 Apr. 2026].Bhatt, K.K., Lily, M.K., Joshi, G. and Dangwal, K. (2018a). Benzo(a)pyrene degradation pathway in Bacillus subtilis BMT4i (MTCC 9447). Turkish Journal of Biochemistry, 43(6), pp.693–701. doi:https://doi.org/10.1515/tjb-2017-0334.BioTech (2026). Protein purification with PureCube Ni-NTA FPLC columns 1. Description. [online] Available at: https://cube-biotech.com/media/d9/6e/08/1775026826/PR162_Ni-NTA_FPLC_Column_Purification_%20Protocol_0001.pdf [Accessed 27 May 2026].Brissos, V., Pereira, L., Munteanu, F.-D., Cavaco-Paulo, A. and Martins, L.O. (2009). Expression system of CotA-laccase for directed evolution and high-throughput screenings for the oxidation of high-redox potential dyes. Biotechnology Journal, 4(4), pp.558–563. doi:https://doi.org/10.1002/biot.200800248.Butt, T.R., Edavettal, S.C., Hall, J.P. and Mattern, M.R. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification, 43(1), pp.1–9. doi:https://doi.org/10.1016/j.pep.2005.03.016.Dias, A.A., António J.S. Matos, Fraga, I., Sampaio, A. and Rui (2017). An Easy Method for Screening and Detection of Laccase Activity. The Open Biotechnology Journal, 11(1), pp.89–93. doi:https://doi.org/10.2174/1874070701711010089.Froger, A. and Hall, J.E. (2007). Transformation of Plasmid DNA into E. coli Using the Heat Shock Method. Journal of Visualized Experiments : JoVE, 6(6), p.253. doi:https://doi.org/10.3791/253.IQAir (2018). How does outdoor air pollution affect my indoor air quality? [online] @airvisual. Available at: https://www.iqair.com/gb/newsroom/how-does-outdoor-air-pollution-affect-my-indoor-air-quality [Accessed 23 Feb. 2026].JenaBios (n.d.). 4 benzenediol + O2 4 benzosemiquinone + 2 H2O Activity. [online] Available at: https://www.jenabios.de/wp-content/uploads/2016/03/Datenblatt_Laccase_Cu.pdf [Accessed 13 Apr. 2026].Lau, Y.-T.K., Baytshtok, V., Howard, T.A., Fiala, B.M., Johnson, J.M., Carter, L.P., Baker, D., Lima, C.D. and Bahl, C.D. (2018). Discovery and engineering of enhanced SUMO protease enzymes. The Journal of Biological Chemistry, 293(34), pp.13224–13233. doi:https://doi.org/10.1074/jbc.RA118.004146.Lei, L., Zhao, L., Hou, Y., Yue, C., Liu, P., Zheng, Y., Peng, W. and Yang, J. (2023). An Inferred Ancestral CotA Laccase with Improved Expression and Kinetic Efficiency. International Journal of Molecular Sciences, 24(13), pp.10901–10901. doi:https://doi.org/10.3390/ijms241310901.Lily, M.K., Ashutosh Bahuguna, Koushalya Dangwal and Garg, V. (2010). Optimization of an inducible, chromosomally encoded benzo [a] pyrene (BaP) degradation pathway in Bacillus subtilis BMT4i (MTCC 9447). Annals of Microbiology, 60(1), pp.51–58. doi:https://doi.org/10.1007/s13213-009-0010-y.Lily, M.K., Bahuguna, A., Dangwal, K. and Garg, V. (2009). Degradation of Benzo [a] Pyrene by a novel strain Bacillus subtilis BMT4i (MTCC 9447). Brazilian Journal of Microbiology, 40(4), pp.884–892. doi:https://doi.org/10.1590/s1517-83822009000400020.Liu, M., Chen, W.-J., Si, G., Yan, C., Song, H., Mishra, S., Ghorab, M.A. and Chen, S. (2025). Bacillus subtilis as a powerful weapon in the removal of environmental pollutants. Journal of Environmental Management, 396, p.127894. doi:https://doi.org/10.1016/j.jenvman.2025.127894.Mallah, M.A., Changxing, L., Mallah, M.A., Noreen, S., Liu, Y., Saeed, M., Xi, H., Ahmed, B., Feng, F., Mirjat, A.A., Wang, W., Jabar, A., Naveed, M., Li, J.-H. and Zhang, Q. (2022). Polycyclic aromatic hydrocarbon and its effects on human health: An overview. Chemosphere, 296, p.133948. doi:https://doi.org/10.1016/j.chemosphere.2022.133948.Margot, J., Bennati-Granier, C., Maillard, J., Blánquez, P., Barry and Holliger, C. (2013). Supporting Information: Bacterial versus fungal laccase: Potential for micropollutant degradation. AMB Express.Martins, L.O., Soares, C.M., Pereira, M.M., Teixeira, M., Costa, T., Jones, G.H. and Henriques, A.O. (2002). Molecular and Biochemical Characterization of a Highly Stable Bacterial Laccase That Occurs as a Structural Component of the Bacillus subtilis Endospore Coat. Journal of Biological Chemistry, 277(21), pp.18849–18859. doi:https://doi.org/10.1074/jbc.m200827200.Mo, Y., Hou Ip Lao, Sau Wa Au, Ieng Chon Li, Hu, J., Hoi Man Yuen, Wai Man Cheong, Lok, O. and Chi, L. (2022). Expression, secretion and functional characterization of three laccases in E. coli. Synthetic and Systems Biotechnology, 7(1), pp.474–480. doi:https://doi.org/10.1016/j.synbio.2021.12.002.Nah, S.H., Kim, J.B., Chui, T., Suh, Y. and Yang, S. (2024). Enhanced Colorimetric Detection of Volatile Organic Compounds Using a Dye-Incorporated Photonic Crystal-Based Sensor Array. Advanced Materials. doi:https://doi.org/10.1002/adma.202409297.Nayra Niño (2024). Review Paper: Bacillus subtilis BMT4i a Bioremediation Agent for High-Molecular-Weight PAH…. [online] Medium. Available at: https://medium.com/insights-of-nature/review-paper-bacillus-subtilis-bmt4i-a-bioremediation-agent-for-high-molecular-weight-pah-f62438f1c574 [Accessed 2 Mar. 2026].NPU CHINA (2022). | NPU-CHINA - iGEM 2022. [online] Igem.wiki. Available at: https://2022.igem.wiki/npu-china/design#gene [Accessed 2 Mar. 2026].Patel, A.B., Shaikh, S., Jain, K.R., Desai, C. and Madamwar, D. (2020). Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Frontiers in Microbiology, 11(11). doi:https://doi.org/10.3389/fmicb.2020.562813.Promega Corporation (2013). Standard Transformation Protocol for Multiple-Use Cells E. coli Competent Cells: Multiple-Use Protocol. [online] Available at: https://www.promega.com/-/media/files/resources/protcards/e-coli-competent-cells-quick-protocol.pdf.Qiagen (2026). Ni-NTA Agarose Purification of xHis-tagged Proteins from E. coli under Native Conditions. [online] QIAGEN. Available at: https://www.qiagen.com/en-US/resources/download/Protocols/ni-nta-agarose-purification-of-6xhis-tagged-proteins-from-e-coli-under-native-conditions-en.Refaat, M., ElRakaiby, M.T., El Hariri El Nokab, M., Es Sayed, J., Elshewy, A., Sebakhy, K.O., Moneib, N., Wang, T., Smith, T.J. and Habib, M.H. (2024). Polymerization potential of a bacterial CotA-laccase for β-naphthol: enzyme structure and comprehensive polymer characterization. Frontiers in Microbiology, 15. doi:https://doi.org/10.3389/fmicb.2024.1501112.Shin-ichi Sakasegawa, Ishikawa, H., Imamura, S., Haruhiko Sakuraba, Goda, S. and Toshihisa Ohshima (2006). Bilirubin Oxidase Activity of Bacillus subtilis CotA. Applied and Environmental Microbiology, 72(1), pp.972–975.doi:https://doi.org/10.1128/aem.72.1.972-975.2006.Sumopro (n.d.). SUMOpro ® Gene Fusion Technology: NEW METHODS FOR ENHANCING FUNCTIONAL PROTEIN EXPRESSION AND PURIFICATION IN BACTERIA. [online] Available at: https://lifesensors.com/wp-content/uploads/2018/04/1000K_1000A_1001K_1001A_Ecoli_SUMOpro_Manual.pdf [Accessed 27 May 2026].Yuval Dorfan, Avichay Nahami, Morris, Y., Shohat, B. and Kolodkin-Gal, I. (2024). The Utilization of Bacillus subtilis to Design Environmentally Friendly Living Paints with Anti-Mold Properties. Microorganisms, 12(6), pp.1226.Zeng, J., Zhu, Q., Wu, Y. and Lin, X. (2016). Oxidation of polycyclic aromatic hydrocarbons using Bacillus subtilis CotA with high laccase activity and copper independence. Chemosphere, 148, pp.1–7. doi:https://doi.org/10.1016/j.chemosphere.2016.01.019.Zhou, Z., Yao, S., Ying, S., Yu, M., Song, Z., Sun, Y., Qian, L. and Zhang, Y. (2025). Engineering the Bacterial Laccase CotA for Functional Expression and Dye Decolorization Through Site-Directed Mutagenesis. Biology, 14(10), pp.1335–1335. doi:https://doi.org/10.3390/biology14101335.Create a supply list and budget for your project (bullet-point list)
The following table listing my supplies doesn’t take into consideration the delivery fees, division of supplies with other in-person lab students or what may already be available in the lab. Ultimately, the price should be reduced significantly from what is stated below.
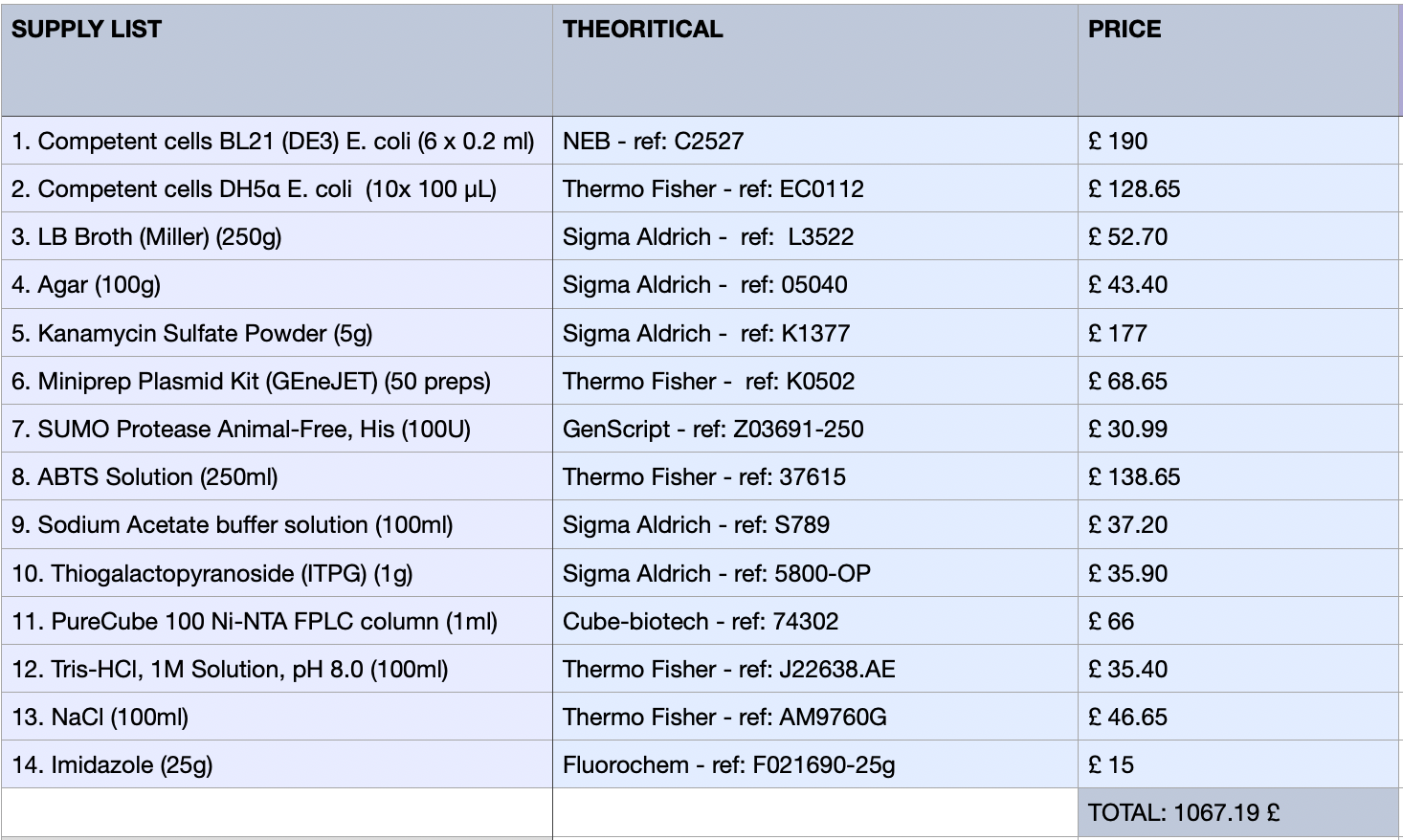
Group Final Project
![cover image]()
![cover image]()
