week-04-hw-protein-design-part-i
A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat?
(On average, an amino acid is ~100 Daltons)
Answer
1 Dalton ≈ 1 g/mol
Average amino acid ≈ 100 g/mol
If you eat 500 g of (pure) amino acids:
number of moles = Gm/ Tm = 500g/100g/mol
Using Avogadro’s number: 5×6.022×10^23 ≈ 3.0 × 10²⁴ molecules
So you consume roughly 3 septillion amino acid molecules.
Answer
Proteins are digested into individual amino acids in the stomach and small intestine.
Your body:
- Breaks proteins down.
- Absorbs amino acids.
- Reassembles them into human proteins according to your DNA.
Answer
Because they have been created by an intelligent design in such a way.
Answer
Yes. Scientists create non-natural amino acids using synthetic biology.
Examples of designs:
• A fluorescent amino acid (attach a fluorophore to side chain) • A metal-binding amino acid (add a bipyridine group) • A photo-switchable amino acid (add an azobenzene group) • A redox-active amino acid
These can:
- Expand protein function
- Create new biomaterials
- Enable bioelectronics
Answer
- Everything was created by the almighty God, who is an intelligent being.
Answer
Natural proteins use L-amino acids and form right-handed α-helices.
If you use D-amino acids, you would expect a left-handed α-helix.
The handedness flips due to stereochemistry.
Answer
Yes.
Beyond the α-helix, proteins contain:
3₁₀ helix
π-helix
Collagen triple helix
Structural biology and protein design can reveal or engineer new helix types.

Answer
Because biological systems predominantly use L-amino acids.
Their stereochemistry naturally favors right-handed packing for minimal steric clash and optimal hydrogen bonding.
What is the driving force for β-sheet aggregation? Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Design a β-sheet motif that forms a well-ordered structure.
Answer
Why β-sheets aggregate: β-strands expose backbone hydrogen bonding groups. They stack via intermolecular hydrogen bonds.
Driving force:
Hydrogen bonding
Hydrophobic interactions
π–π stacking (aromatic residues)
Amyloid diseases: Proteins misfold and form stable β-sheet fibrils.
Examples include:
Alzheimer’s disease
Parkinson’s disease
Amyloid β-peptides form cross-β sheet structures.
Materials applications: Yes — amyloid fibrils can be used as:
Nanowires
Hydrogels
Biocompatible scaffolds
- Design idea:
Create a repeating sequence like:
- Val–Ile–Val–Ile–Tyr–Val–Ile–Val
Alternating hydrophobic residues promotes stacking and ordered β-sheet assembly.
B. Protein Analysis
I have chosen Herceptin (trastuzumab) for this section. Herceptin is a monoclonal antibody mainly involved in recognising cancer cells. It binds specifically to the HER2 receptor on cancer cells and blocks signaling pathways that promote tumor growth. I selected this protein because it is an important example of a therapeutic antibody widely used in breast cancer treatment.
Amino Acid Sequence (P04626-1)
CLICK HERE SEE THE SEQUENCE
MELAALCRWGLLLALLPPGAASTQVCTGTDMKLRLPASPETHLDMLRHLYQGCQVVQGNLELTYLPTNASLSFLQDIQEVQGYVLIAHNQVRQVPLQRLRIVRGTQLFEDNYALAVLDNGDPLNNTTPVTGASPGGLRELQLRSLTEILKGGVLIQRNPQLCYQDTILWKDIFHKNNQLALTLIDTNRSRACHPCSPMCKGSRCWGESSEDCQSLTRTVCAGGCARCKGPLPTDCCHEQCAAGCTGPKHSDCLACLHFNHSGICELHCPALVTYNTDTFESMPNPEGRYTFGASCVTACPYNYLSTDVGSCTLVCPLHNQEVTAEDGTQRCEKCSKPCARVCYGLGMEHLREVRAVTSANIQEFAGCKKIFGSLAFLPESFDGDPASNTAPLQPEQLQVFETLEEITGYLYISAWPDSLPDLSVFQNLQVIRGRILHNGAYSLTLQGLGISWLGLRSLRELGSGLALIHHNTHLCFVHTVPWDQLFRNPHQALLHTANRPEDECVGEGLACHQLCARGHCWGPGPTQCVNCSQFLRGQECVEECRVLQGLPREYVNARHCLPCHPECQPQNGSVTCFGPEADQCVACAHYKDPPFCVARCPSGVKPDLSYMPIWKFPDEEGACQPCPINCTHSCVDLDDKGCPAEQRASPLTSIISAVVGILLVVVLGVVFGILIKRRQQKIRKYTMRRLLQETELVEPLTPSGAMPNQAQMRILKETELRKVKVLGSGAFGTVYKGIWIPDGENVKIPVAIKVLRENTSPKANKEILDEAYVMAGVGSPYVSRLLGICLTSTVQLVTQLMPYGCLLDHVRENRGRLGSQDLLNWCMQIAKGMSYLEDVRLVHRDLAARNVLVKSPNHVKITDFGLARLLDIDETEYHADGGKVPIKWMALESILRRRFTHQSDVWSYGVTVWELMTFGAKPYDGIPAREIPDLLEKGERLPQPPICTIDVYMIMVKCWMIDSECRPRFRELVSEFSRMARDPQRFVVIQNEDLGPASPLDSTFYRSLLEDDDMGDLVDAEEYLVPQQGFFCPDPAPGAGGMVHHRHRSSSTRSGGGDLTLGLEPSEEEAPRSPLAPSEGAGSDVFDGDLGMGAAKGLQSLPTHDPSPLQRYSEDPTVPLPSETDGYVAPLTCSPQPEYVNQPDVRPQPPSPREGPLPAARPAGATLERPKTLSPGKNGVVKDVFAFGGAVENPEYLTPQGGAAPQPHPPPAFSPAFDNLYYWDQDPPERGAPPSTFKGTPTAENPEYLGLDVPV
Total Length: 1255 Most Common Amino Acid: Leucine(L)
It belongs to the immunoglobulin G (IgG1) subclass within the immunoglobulin superfamily. And it is part of the L-domian family. (Immunoglobulin Light-chain domain.)
Resolution: 4.36 Å, which shows low resolution of the model.
The crystal structure of trastuzumab bound to HER2 was solved in 2004.
Blast Analysis
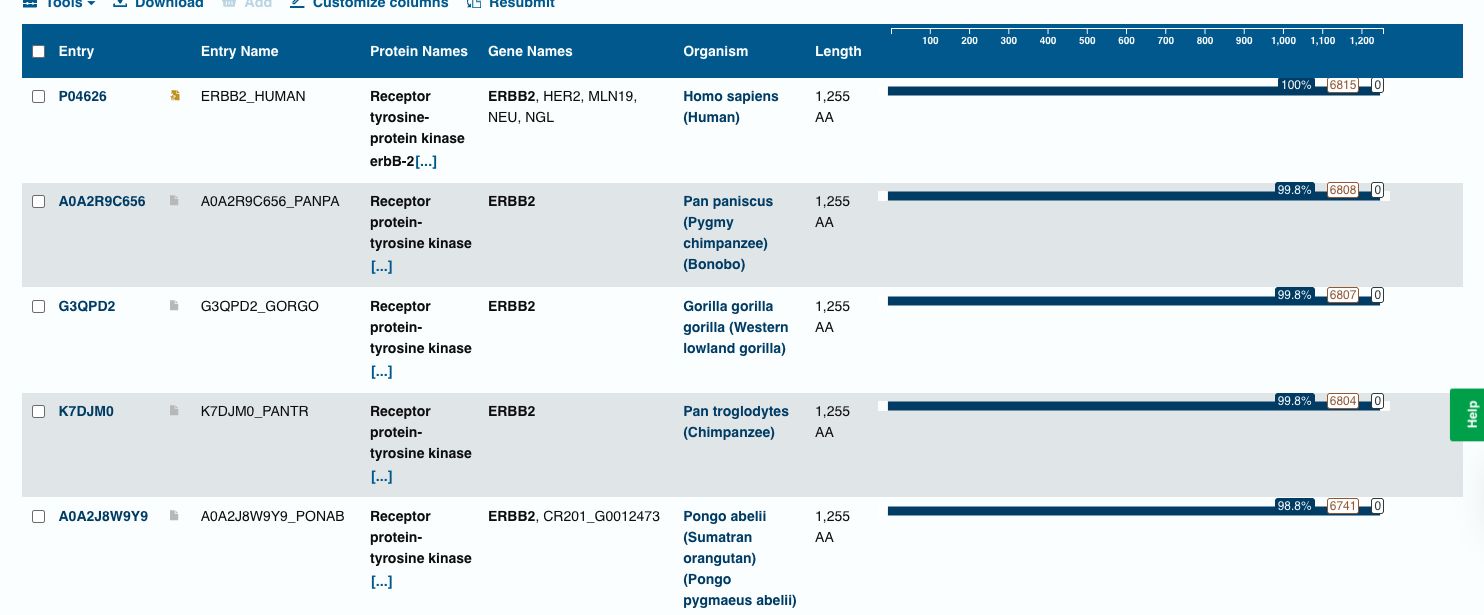
- The BLAST search identified homologous ERBB2 (HER2) protein sequences in several primates, including chimpanzee, bonobo, gorilla, and orangutan. These sequences show very high similarity (98–99% identity) with the query sequence, indicating that the HER2 receptor is highly conserved among mammals.
PYMOL Analysis of Trastuzumab
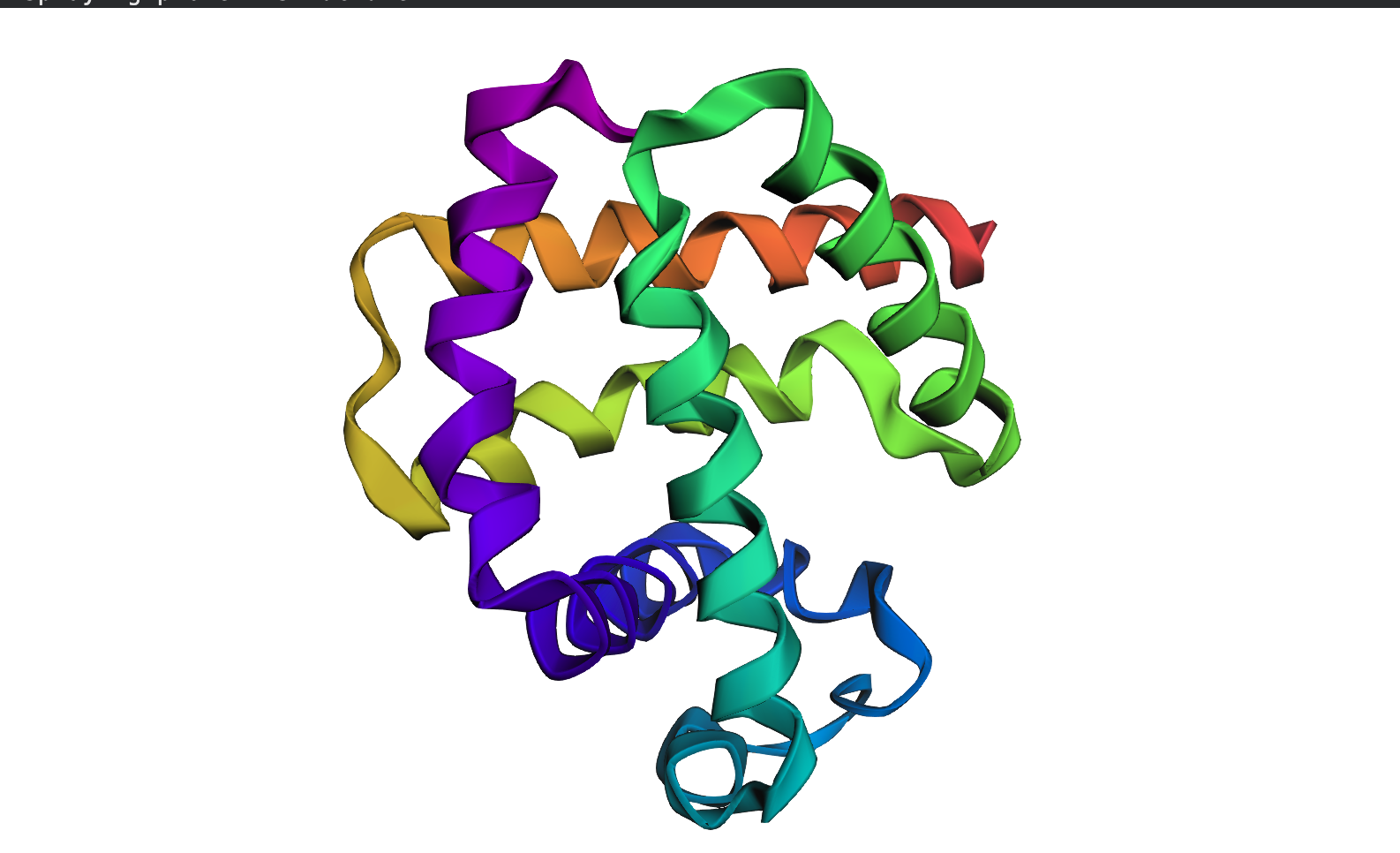
Ribbon Representation
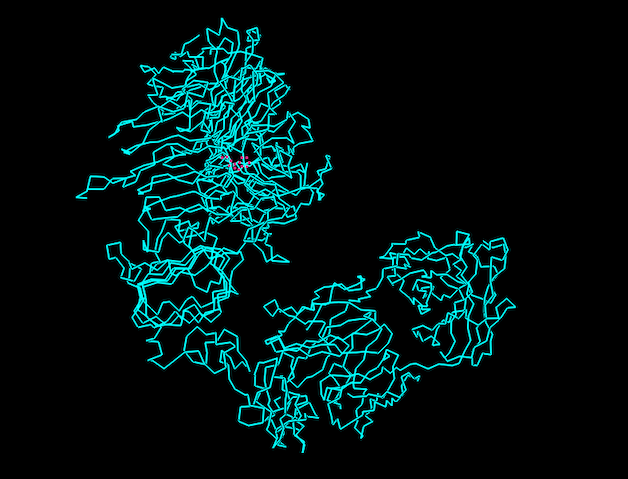
Ball and Stick
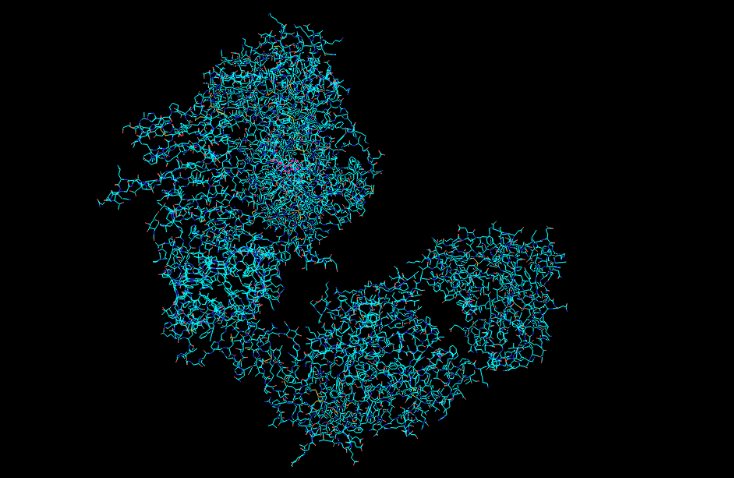
Protein Surface
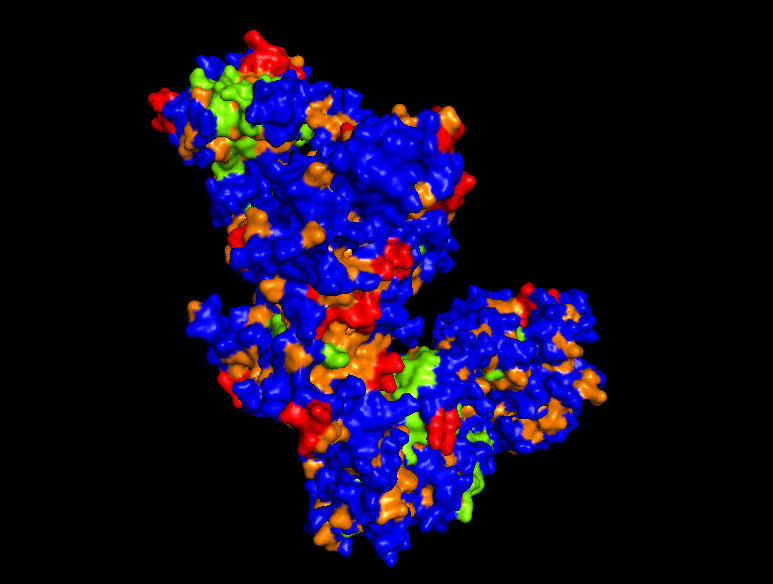
*Hydrophobic Region
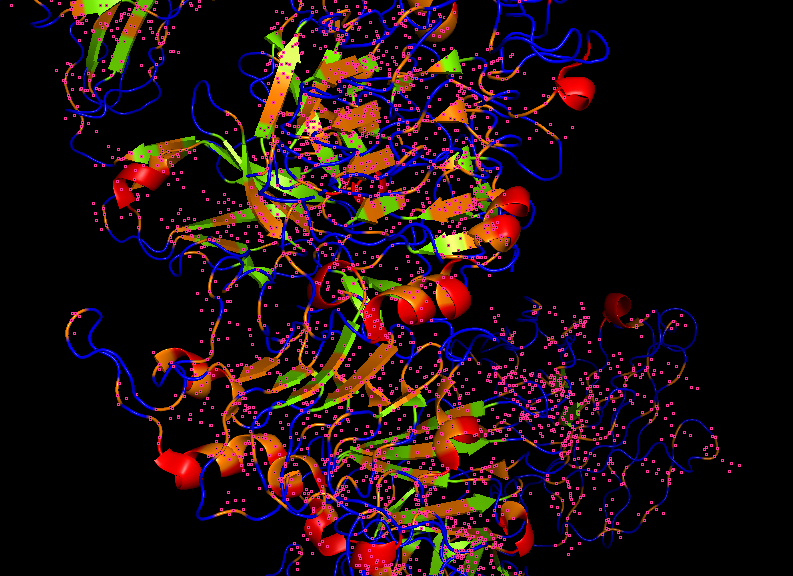
Secondary structures
C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
- Deep Mutational Scans
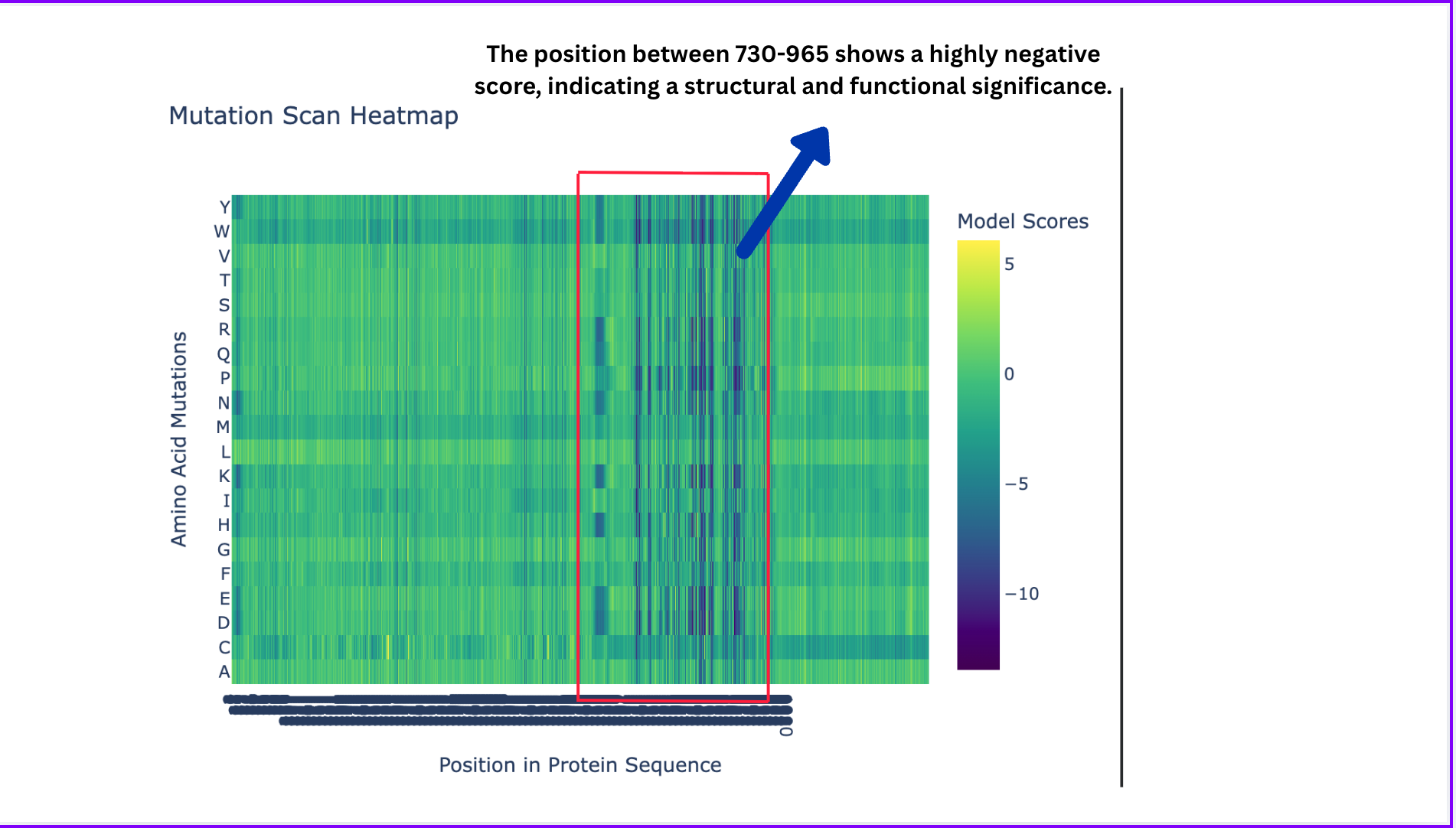
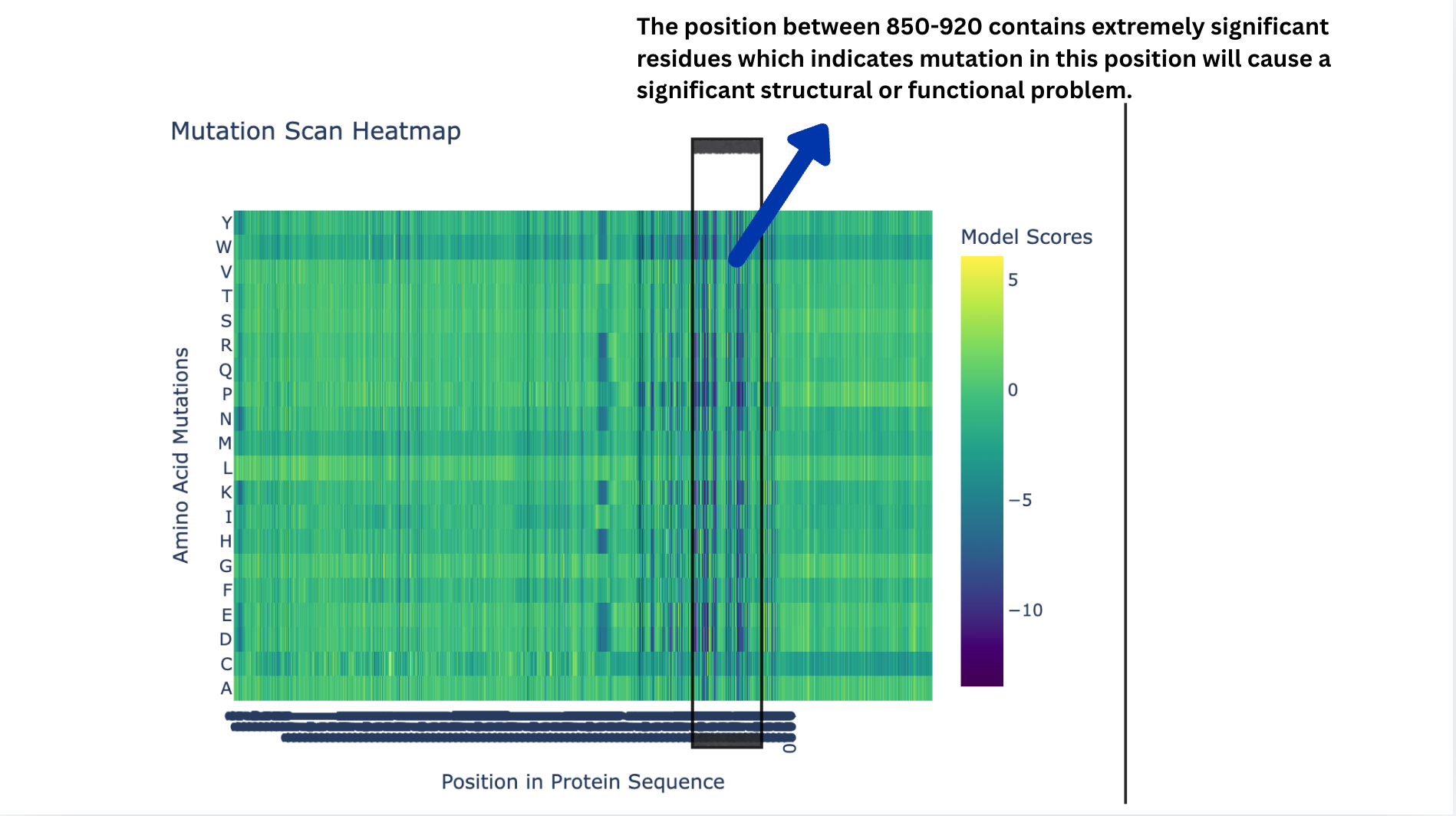
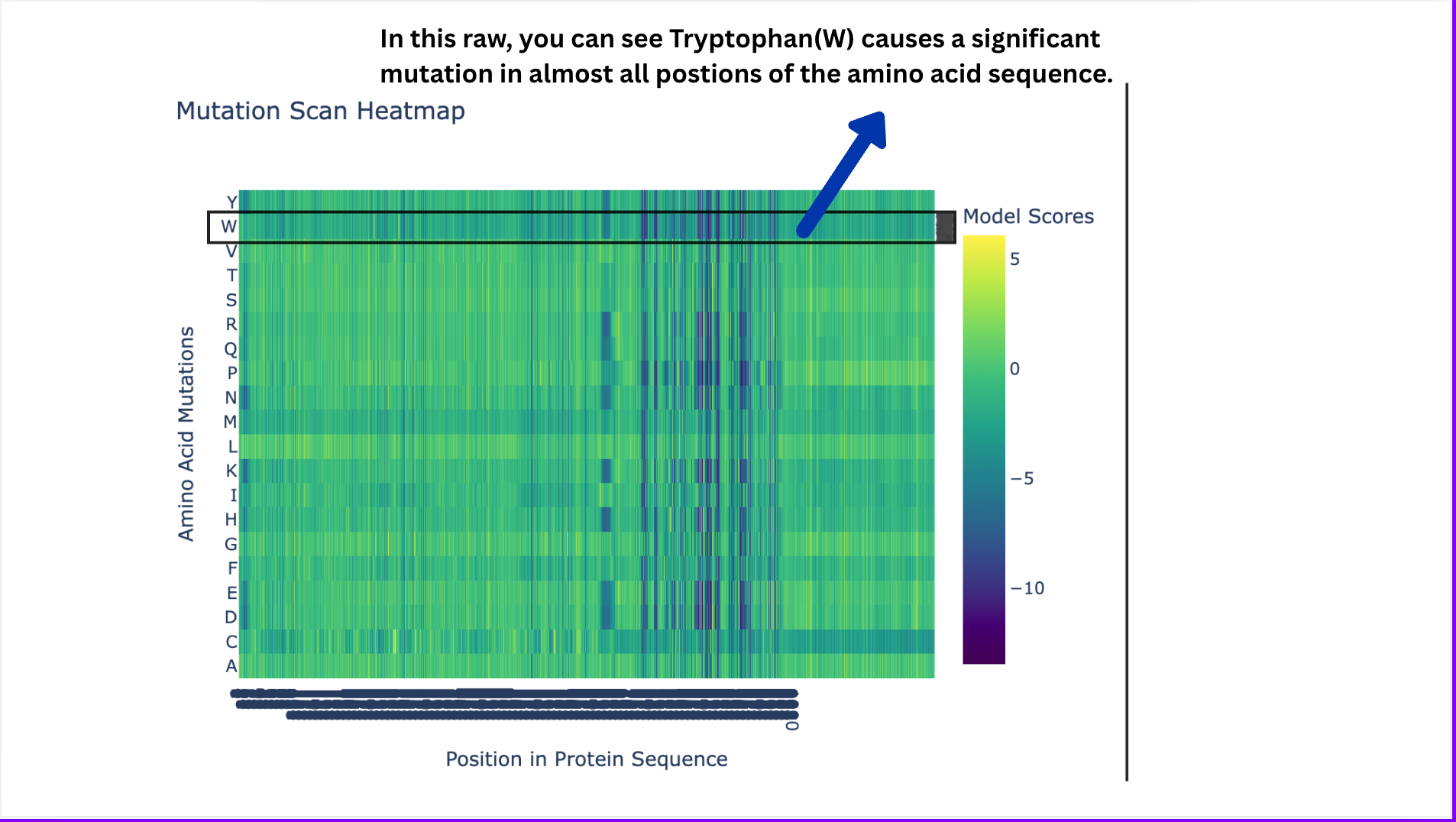
- Latent Space Analysis
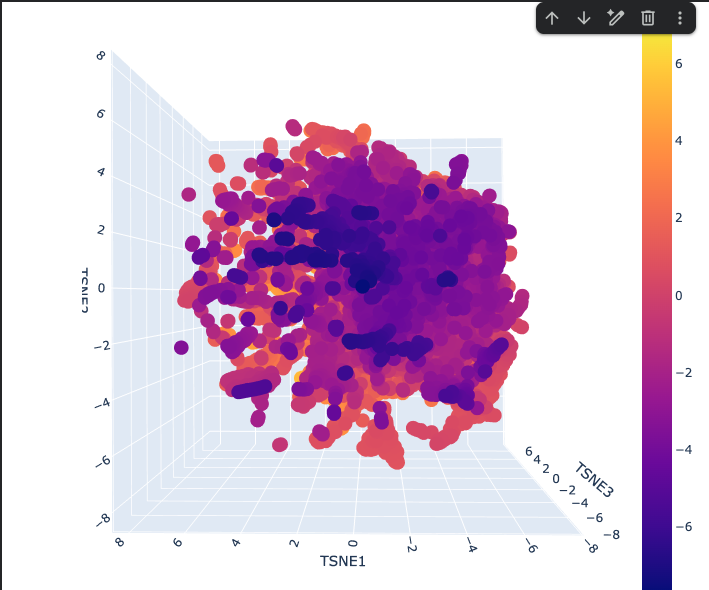
- The Latent space analysis shows the 3D representation of different proteins. This plot is a map of protein similarity — proteins close together are similar in sequence/function/structure, the dense center contains common proteins, and the scattered edges contain unusual ones. The color encodes an additional property (likely functional or structural) layered on top of the spatial layout.
Explanation
Shape
One large continuous cloud — no hard separate clusters Reflects that protein sequence space is smooth and gradual, not divided into distinct categories
The Dense Purple Core
Where most proteins sit These are common, well-represented protein families that ESM2 has seen many times
The Scattered Orange/Yellow Periphery
Outlier proteins that are unusual or specialized Score higher on whatever the colorbar is measuring (likely a biological property or cluster score ranging from -7 to +7)
The Elongated Arms
Streaks radiating outward from the core Represent protein subfamilies that share a common origin but have diverged over evolution.
ESM fold Prediction
- N.B For this section, I selected Insulin because it is relatively smaller than HER2, which kept crashing while trying to predict how it folds.
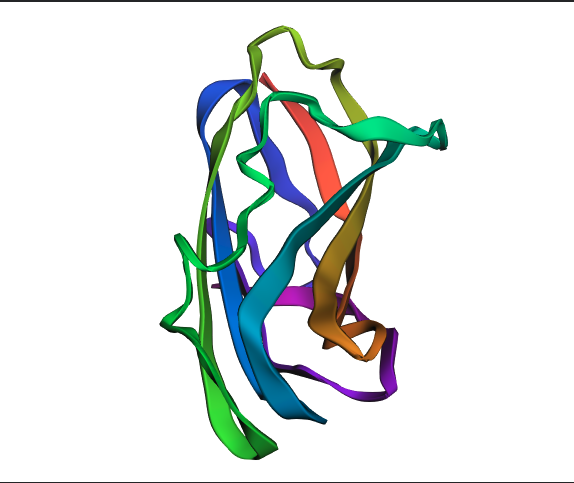
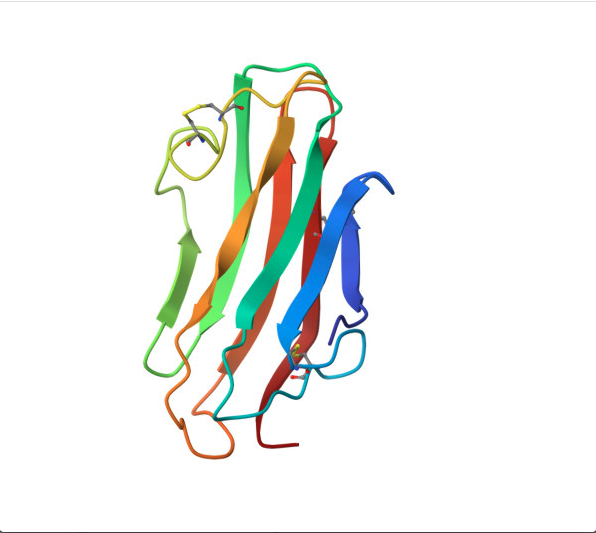
- ESMFold correctly predicted the beta sheet topology of insulin, identifying the major secondary structure elements consistent with the experimental RCSB structure. However, the predicted structure is notably more extended and loosely packed, with larger irregular loops compared to the compact real structure. This discrepancy is most likely due to insulin’s three disulfide bonds between Chain A and Chain B, which ESMFold does not explicitly model; these bonds are critical for anchoring the loops and achieving the tight globular shape seen in the experimental structure. The TM-score and RMSD would quantify this difference precisely, but visually, the fold class is correct while the fine-grained packing is not.
Reverse folding using ProteinMPNN.
For this part, I used the PDB file of the HER2 protein. After uploading the pdb file, a reverse folding was run, and 20 possible candidates for the actual sequence of the protein was predicted. Among the results, the one with the lowest log score was identified through manual screeing and was folded using the ESMfold model. The predicted sequence and the folded protein are attached below.
Predicted Structure
ALTPEQAALLAAAWAPVFADREANARAFVLDLFRAYPSLADLFPEFKGKTLEQIAASPALGPYAGAFADRLAQFVASSDNAAKMATFWENYANEHIRRGITASHFEQVRAVFPGFVASVAEPPPGAAAAWDQFWGGIIDALKKAGG
T=0.5, sample=0, score=0.9440, seq_recovery=0.4932
T = 0.5 (Temperature)
Controls how creative/diverse the designed sequence is 0.5 is moderate — balanced between staying close to original and exploring new sequences Lower (0.1) = conservative, Higher (1.0) = very adventurous
sample = 0
This is the first designed sequence (counting starts from 0) If you generated 10 sequences, you’d see sample=0 through sample=9 Each sample is an independent design attempt for the same backbone
score = 0.9440
Negative log likelihood — measures model confidence Lower = better — model is very confident this sequence fits your backbone Your score of 0.9440 is excellent — it’s below 1.0 which is better than your insulin results (1.06 and 1.08)
seq_recovery = 0.4932
49.32% of positions match the original protein sequence exactly Roughly 1 in 2 residues is identical to the original This is your best recovery so far — slightly higher than insulin’s ~46%