week-05-hw-protein-design-part-ii
Protein Design Part 2
SOD1_A4V Mutated Code Used.
—> MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 1: PepMLM Generated Peptides
Summary
The four PepMLM-generated peptides were conditioned on the SOD1 A4V mutant sequence with a target length of 12 amino acids, with the exception of Peptide 2, which came out at 15 residues. Perplexity scores reflect the model’s confidence in each binder, where a lower score indicates higher confidence. Peptide 1 (WLYGAAGVRWGX) has the lowest perplexity at 13.06, making it the model’s most confident prediction, though it contains an X residue at the final position, which represents an unresolved or masked amino acid and should be noted as a potential issue before advancing it further. Peptides 2, 3, and 4 all cluster between 17 and 20, reflecting moderate confidence. The known binder FLYRWLPSRRGG is included as a structural and therapeutic benchmark and does not carry a perplexity score since it was not generated by PepMLM.
Results
| Peptide | Sequence | Length | Pseudo Perplexity | Source |
|---|---|---|---|---|
| 1 | WLYGAAGVRWGX | 12 | 13.06 | PepMLM |
| 2 | SRYDEYVVVVKAAKK | 15 | 17.72 | PepMLM |
| 3 | HRVYAVVVAWKK | 12 | 19.82 | PepMLM |
| 4 | WLYYAVALAWKE | 12 | 17.93 | PepMLM |
| 5 | FLYRWLPSRRGG | 12 | N/A | Known binder |
Part 2: AlphaFold3 Structural Evaluation
SOD1_A4V_Peptide
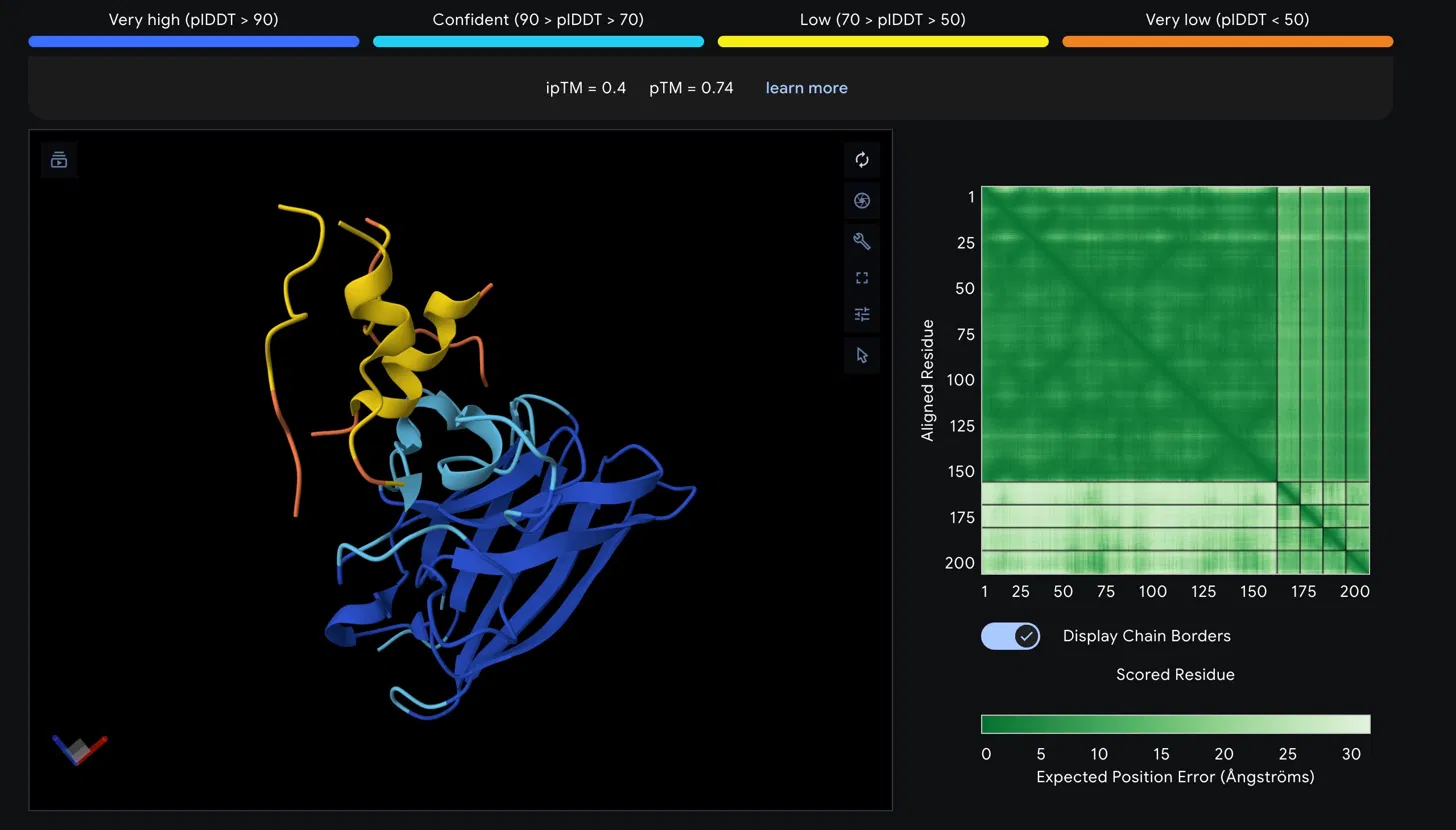
Scores
| Metric | Value | Interpretation |
|---|---|---|
| ipTM | 0.4 | Low confidence interface prediction |
| pTM | 0.74 | Reasonable overall fold quality |
Structural Observations
The SOD1 A4V beta-barrel is predicted with high confidence, appearing in blue and cyan in the structure viewer, consistent with the well-characterized immunoglobulin- like fold of SOD1. The peptide chain is rendered in yellow and orange, indicating low to very low pLDDT confidence in its predicted conformation. The peptide appears to associate loosely near the helical region at the top of the SOD1 structure rather than engaging the N-terminus directly where the A4V mutation at residue 4 is located. It does not appear to be buried at the dimer interface and is largely surface associated.
The Predicted Aligned Error matrix supports this interpretation. The large dark green block spanning SOD1 residues 1 to approximately 160 confirms strong internal positional confidence within the protein. The bottom right region corresponding to the peptide chain shows notably lighter green, indicating higher positional uncertainty. The off-diagonal inter-chain block between SOD1 and the peptide is also light green, reflecting weak confidence in the relative positioning of the two chains and consistent with the low ipTM score.
Assessment
An ipTM of 0.4 falls below the 0.5 threshold typically considered meaningful for protein-peptide interactions, suggesting this peptide does not form a confidently predicted stable complex with SOD1 A4V. The low pLDDT of the peptide further indicates its conformation is disordered in this predicted complex. This result would need to be weighed against the therapeutic property predictions from PeptiVerse before making any advancement decision.
Part 3: PeptiVerse Therapeutic Property Evaluation
Results Table
| Peptide | Sequence | Binding Affinity (pKd/pKi) | Solubility | Hemolysis | Net Charge (pH 7) | MW (Da) | GRAVY |
|---|---|---|---|---|---|---|---|
| 1 | WLYGAAGVRWGK | 6.641 (Weak) | 0.867 (Soluble) | 0.055 (Non-hemolytic) | +1.76 | 1363.6 | -0.09 |
| 2 | SRYDEYVVVVKAAKK | 6.453 (Weak) | 1.000 (Soluble) | 0.062 (Non-hemolytic) | +1.46 | 1755.0 | -0.41 |
| 3 | HRVYAVVVAWKK | 6.689 (Weak) | 0.970 (Soluble) | 0.093 (Non-hemolytic) | +2.84 | 1455.7 | +0.23 |
| 4 | WLYYAVALAWKE | 6.661 (Weak) | 0.760 (Soluble) | 0.110 (Non-hemolytic) | -0.23 | 1512.7 | +0.45 |
Summary
All four PepMLM-generated peptides passed the two most critical early-stage therapeutic thresholds: all are predicted to be soluble and non-hemolytic. This is an encouraging baseline, as poor solubility and hemolytic activity are among the most common reasons peptide candidates fail during preclinical screening. However, all four peptides were classified as weak binders against SOD1 A4V, with predicted binding affinities clustering narrowly between 6.45 and 6.69 pKd/pKi, indicating that none demonstrate strong predicted affinity for the target under current conditions.
Comparing these results to the AlphaFold3 structural data, the low ipTM score of 0.4 observed in Part 2 is broadly consistent with the weak binding predictions from PeptiVerse, suggesting that neither structural nor property-based evaluation strongly endorses any single peptide as a high confidence binder at this stage. No peptide was predicted to be hemolytic, meaning the structural uncertainty does not appear to stem from toxic or disruptive interactions with the target.
Among the four, Peptide 3 (HRVYAVVVAWKK) presents the strongest overall profile. It carries the highest predicted binding affinity at 6.689 pKd/pKi, the second highest solubility at 0.970, a low hemolysis probability at 0.093, and the highest positive net charge at pH 7 at 2.84, which may support favorable electrostatic interactions with SOD1 and aid membrane permeability. Its modest hydrophobicity score of 0.23 GRAVY suggests a reasonable balance between aqueous solubility and the hydrophobic contacts often required for stable protein binding. Peptide 2 shows perfect solubility at 1.000 and the lowest hydrophobicity, making it the safest from an aggregation standpoint, but its binding affinity is the weakest of the four at 6.453 pKd/pKi and its length of 15 residues may introduce additional pharmacokinetic challenges.
Peptide Selected for Advancement
Peptide 3: HRVYAVVVAWKK
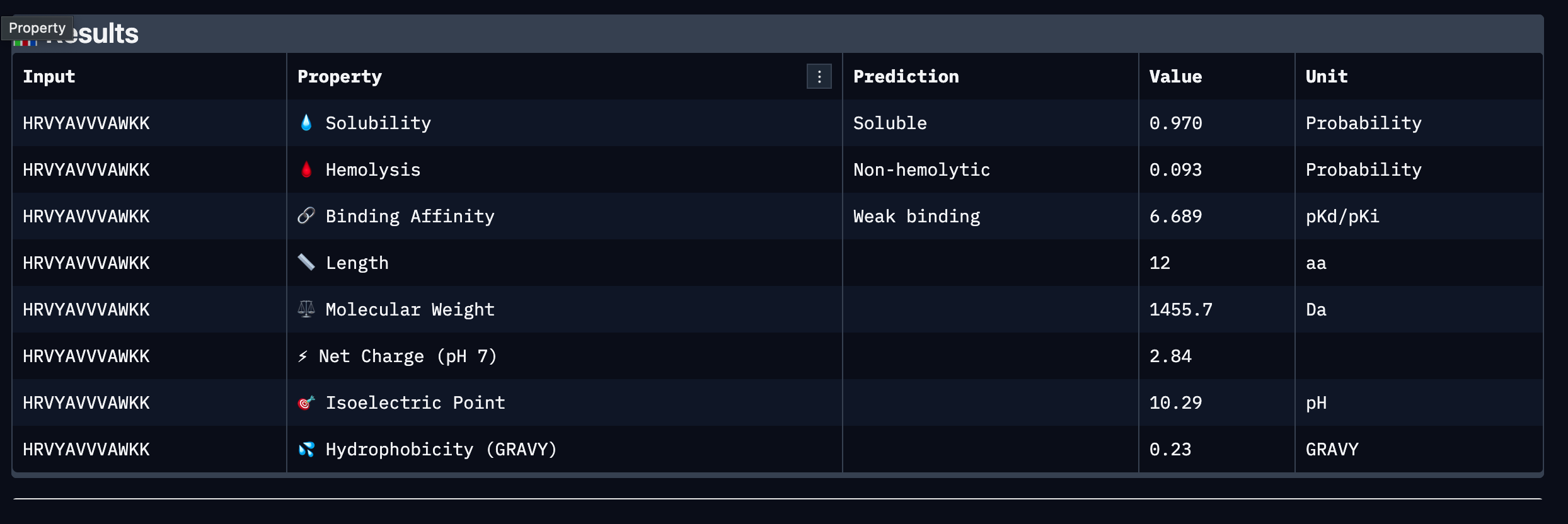
Peptide 3 is selected for advancement based on its combination of the highest predicted binding affinity among the four candidates, high solubility, low hemolytic risk, and a positively charged character at physiological pH that is consistent with favorable interactions at the SOD1 surface. While all peptides in this set are classified as weak binders and further optimization would be required, Peptide 3 represents the strongest starting point for motif-guided redesign using moPPIt in Part 4. Its valine-rich hydrophobic core may also provide a useful scaffold for introducing targeted contacts at the A4V mutation site or dimer interface.