Week 02 HW: DNA Read, Write, and Edit

Part 1: Benchling & In-silico Gel Art 🦠 Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks:
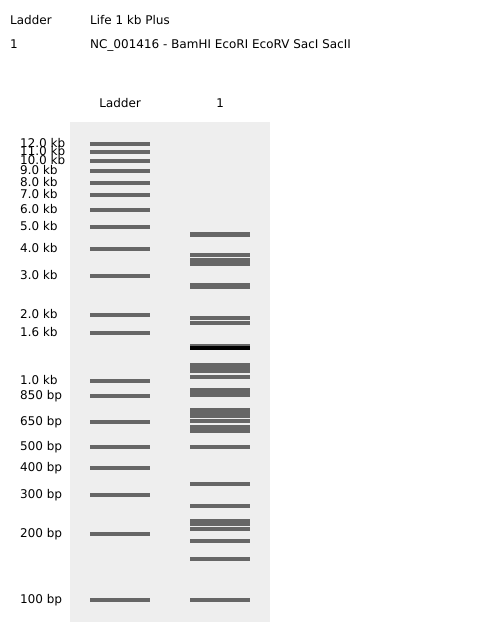
Part 3: DNA Design Challenge I chose poly(3-hydroxyalkanoate) polymerase / PHB synthase (PhaC) from Cupriavidus necator (UniProt accession P23608) because it is a key enzyme in microbial bioplastic production. PhaC catalyzes the polymerization of (R)-3-hydroxybutyryl-CoA monomers to form poly(3-hydroxybutyrate) (PHB), and engineered variants of PhaC are widely used to broaden substrate specificity and produce other polyhydroxyalkanoates (PHAs). I obtained the amino-acid sequence from UniProt (entry P23608) in FASTA format.
MA T G K G A A A S T Q E G K S Q P F K V T P G P F D P A T W L E W S R Q W Q G T E G N G H A A A S G I P G L D A L A G V K I A P A Q L G D I Q Q R Y M K D F S A L W Q A M A E G K A E A T G P L H D R R F A G D A W R T N L P Y R F A A A F Y L L N A R A L T E L A D A V E A D A K T R Q R I R F A I S Q W V D A M S P A N F L A T N P E A Q R L L I E S G G E S L R A G V R N M M E D L T R G K I S Q T D E S A F E V G R N V A V T E G A V V F E N E Y F Q L L Q Y K P L T D K V H A R P L L M V P P C I N K Y Y I L D L Q P E S S L V R H V V E Q G H T V F L V S W R N P D A S M A G S T W D D Y I E H A A I R A I E V A R D I S G Q D K I N V L G F C V G G T I V S T A L A V L A A R G E H P A A S V T L L T T L L D F A D T G I L D V F V D E G H V Q L R E A T L G G G A G A P C A A A A G L E L A N T F S F L R P N D L V W N Y V V D N Y L K G N T P V P F D L L F W N G D A T N L P G P W Y C W Y L R H T L P A E R A Q G T G Q A D R V R R A G G P G Q H R R P Y I Y G S R E D H I V P W T A A Y A S T A L L A N K L R F V L G A S G H I A G V I N P P A K N K R S H W T N D A L P E S P Q Q W L A G A I E H H G S W W P D W T A W L A G Q A G A K R A R P A N Y G N A R Y R A I E P A P G D T S K P R H
Although the genetic code is universal, most amino acids are encoded by multiple codons. However, different organisms do not use these synonymous codons at the same frequency. This phenomenon is known as codon bias. If a gene from one organism is expressed in a different host, the original codons may be rare in the new host, which can reduce translation efficiency, among other problems.
Codon optimization is necessary to modify the DNA sequence so that it uses codons preferred by the host organism. Optimizing codon usage can significantly increase protein expression levels, improve translation efficiency, enhance mRNA stability, and reduce the likelihood of misfolding or premature termination. For this project, I chose to optimize the phaC gene for Escherichia coli. I selected E. coli because it is the chassis organism we use at Bioplastix.
I used https://www.genscript.com/ as a code optimizattion tool (Job ID: 20260216015729685186).
At Bioplastix, the engineered bacteria produce the enzymes intracellularly. Under specific metabolic conditions, these enzymes catalyze the polymerization of monomers inside the cell, leading to intracellular accumulation of the biopolymer.
To achieve that, once the codon-optimized DNA sequence is obtained, it is inserted into a plasmid (an expression vector containing the necessary regulatory elements, such as a promoter and terminator). This plasmid is then be inserted into the host organism, Escherichia coli, for protein production.
In such a cell-dependent system, the DNA sequence is transcribed into messenger RNA (mRNA) by RNA polymerase. The mRNA is then translated by ribosomes into the PhaC protein. Depending on the design of the genetic construct, the gene can be placed under a constitutive promoter, where the protein is continuously produced, or under an inducible promoter, where expression is triggered by specific conditions (such as the presence of IPTG or oxigen).
Alternatively, the protein could also be produced using a cell-free expression system, where the DNA is added to a reaction mixture containing ribosomes, tRNAs, nucleotides, and enzymes necessary for transcription and translation. This allows protein production without living cells, although large-scale industrial production until now typically relies on cell-based systems.
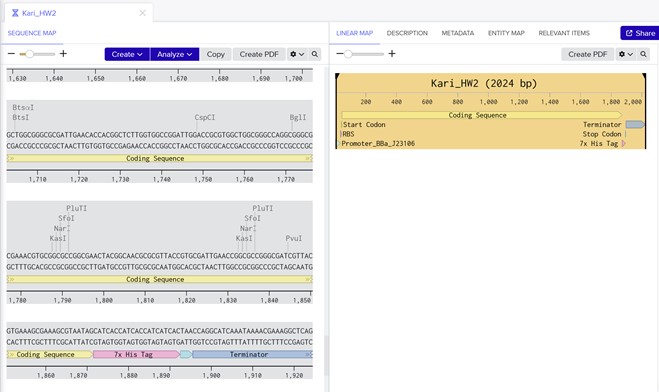
Part 4: Prepare a Twist DNA Synthesis Order
Link Sharing From Benchling: https://benchling.com/s/seq-l6sbaCHItMO0QD5lHD4M?m=slm-38MASpmHAGVSi3hySqfU
Part 5: DNA Read/Write/Edit 5.1 DNA Read (i) What DNA would you want to sequence (e.g., read) and why? I would like to sequence DNA from environmental microbial communities, particularly from diverse and extreme environments such as soil, marine ecosystems, open dumps, and high-stress habitats. These environments often contain microorganisms with unique metabolic capabilities.
By sequencing DNA from environmental samples (metagenomic sequencing), we could identify novel enzymes involved in polymer biosynthesis, including new variants of PHA synthases or related polymerizing enzymes. These enzymes may have improved catalytic efficiency, altered substrate specificity, or greater stability under industrial conditions. Discovering new enzymes through DNA sequencing could enable the development of more efficient bioplastic production systems and potentially allow the synthesis of novel polymers with enhanced material properties.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? I would use Illumina sequencing technology, a second-generation (next-generation sequencing, NGS) platform. Illumina sequencing is well-suited for metagenomic analysis because it provides high-throughput, highly accurate short reads at relatively low cost, making it ideal for sequencing complex microbial communities. It is second generation because it performs massively parallel sequencing, requires DNA amplification (cluster generation), and sequences millions of fragments simultaneously.
The input is extracted environmental DNA (metagenomic DNA) from microbial communities.
Sample preparation steps:
- DNA extraction from environmental samples (soil, water, etc.)
- Fragmentation of DNA into smaller pieces (typically ~200–500 bp)
- End repair and A-tailing
- Adapter ligation (short known sequences attached to fragment ends)
- PCR amplification to enrich properly ligated fragments
- Library quantification and quality control
Illumina sequencing works as follows:
- The prepared DNA library is loaded onto a flow cell.
- Fragments bind to complementary oligos attached to the flow cell.
- Bridge amplification creates clusters of identical DNA fragments.
- Fluorescently labeled reversible terminator nucleotides are added.
- During each cycle, one nucleotide is incorporated per cluster.
- A camera detects the fluorescent signal emitted by the incorporated base.
- The terminator is cleaved, and the next cycle begins.
Each nucleotide (A, T, C, G) carries a different fluorescent signal. The imaging system detects the color emitted at each cycle, and software converts fluorescence intensity into base calls (A, T, C, or G).
5.2 DNA Write
The DNA I would want to “write” (synthesize) would be a set of candidate polymerizing enzymes identified from the environmental metagenomic library, especially novel PHA synthase (PhaC-like) genes or other enzymes predicted to catalyze polymer formation. The main reason to synthesize these genes is to rapidly test them in a standardized host. I would order codon-optimized versions of each candidate enzyme gene for Escherichia coli, because E. coli is the chassis organism we use at Bioplastix.
I would use solid-phase phosphoramidite DNA synthesis, the standard chemical DNA synthesis technology used by companies such as Twist Bioscience. In the past we already used Twist for synthesis of new enzymes. It is very useful because enables codon optimization, Introduction of specific mutations, Removal of unwanted restriction sites, parallel synthesis of multiple variants.
With this methodd DNA is synthesized one nucleotide at a time on a solid support using phosphoramidite chemistry. This produces short DNA fragments (typically 150–200 bp). Since genes are longer than individual oligos, overlapping oligonucleotides are assembled into full-length genes using: PCR-based assembly, Gibson Assembly or Enzymatic ligation methods. The assembled gene is amplified and sometimes enzymatically corrected to reduce synthesis errors. The final construct is cloned into a plasmid and verified by DNA sequencing before delivery.
From a practical perspective at Bioplastix, the main limitation of outsourcing DNA synthesis to companies such as Twist Bioscience is cost. Synthesizing long gene sequences can be expensive, particularly when testing multiple enzyme variants. Each synthesis order costs approximately $1,500 USD. After synthesis, there are further costs associated with Transforming it into our production strains and Screening and validating expression. Overall, with a typical budget, we are able to test approximately 5–10 new enzyme candidates per round.
5.3 DNA Edit The DNA I would want to edit is the genome of our production strain (e.g., Escherichia coli) in order to improve its efficiency as a bioplastic-producing chassis organism. Specifically, I would focus on :
1️⃣ Expanding substrate utilization: One of our current priorities is enabling the strain to efficiently consume alternative and low-cost carbon sources, particularly sucrose. Editing the genome to introduce or optimize sucrose transporters and metabolic pathways would allow the bacteria to convert a wider range of feedstocks into biopolymer, improving economic feasibility and sustainability.
2️⃣ Increasing carbon flux toward polymer production: I would edit genes involved in central carbon metabolism to redirect more carbon toward the PHA biosynthesis pathway.
3️⃣ Engineering polymerizing enzymes: I would also edit the genes encoding polymerizing enzymes (such as PhaC) to Increase substrate affinity, Improve catalytic efficiency and Enhance thermostability. Thermotolerant enzymes would be particularly valuable at industrial scale, where higher fermentation temperatures reduce cooling costs and contamination risk.
To perform these genome edits, I would use CRISPR-Cas9 genome editing, combined with homologous recombination. CRISPR-based systems are precise, programmable, and highly efficient, making them ideal for metabolic engineering in bacterial systems. CRISPR-Cas9 uses: i. A guide RNA (gRNA) designed to match a specific DNA target sequence. ii. The Cas9 nuclease, which creates a double-strand break at the target site
After the break is introduced, the cell repairs the DNA. If a repair template is provided (homology-directed repair, HDR), specific edits such as insertions, deletions, or point mutations can be introduced.
Limmitations include Metabolic burden (Multiple edits can stress the cell and reduce growth rate) and Regulatory complexity (Metabolic networks are highly interconnected; editing one pathway may produce unexpected downstream effects).