Week 04 HW: Protein Design - Part 1
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Assumptions: Meat ≈ 20% protein. 500 g meat → ~100 g protein. Average amino acid residue mass ≈ 100 Da ≈ 100 g/mol. So: 100 g ÷ 100 g/mol ≈ 1 mole of amino acid residues. 1 mole = 6.022 × 10²³ molecules. 👉 You ingest on the order of 6 × 10²³ amino acid residues in 500 g of meat.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish? Because digestion destroys biological structure. Proteins are hydrolyzed into amino acids. The original sequences (information) are lost. Your body reassembles amino acids according to human DNA instructions. Biological identity is encoded in sequence and genomic regulation — not in the raw amino acid building blocks. You absorb matter, not identity.
Note: While reading this response, I was prompted to ask ChatGPT the following question: “Do the 20 amino acids explain all the DNA of all species? Are there more evolved species that use more amino acids?” ChatGPT’s response was: Yes, the same 20 amino acids (with two rare exceptions) account for virtually all proteins in all known living organisms. There are no “more evolved” species that use a greater number of standard amino acids.
This follow-up helped clarify the universality of the genetic code and reinforced the idea that biological complexity arises from sequence variation and regulation, rather than from an expanded set of amino acid building blocks.
- Why are there only 20 natural amino acids? This is evolutionary, not chemical necessity. The 20 canonical amino acids: Provide sufficient chemical diversity: Hydrophobic, Polar, Charged, Aromatic, Reactive, Enable folding into complex structures.
Likely reflect optimization of the genetic code to minimize error impact. More than 500 amino acids exist in nature, but only 20 are universally encoded in the genetic code (with rare additions like selenocysteine). Evolution found a minimal but sufficient toolkit.
- Can you make other non-natural amino acids? Design some new amino acids. Yes. Synthetic biology routinely incorporates non-natural amino acids. Examples: A) Fluorinated phenylalanine. Replace para-H with F. Increased hydrophobicity. Enhanced stability. Altered electronic properties. B) Long-chain hydrophobic amino acid. Extend leucine side chain by two carbons. Stronger hydrophobic core packing. Enhanced β-sheet stabilization.
- Where did amino acids come from before enzymes that make them, and before life started? Prebiotic chemistry. Evidence includes: Miller–Urey experiments (electric discharge in reducing atmosphere). Meteorites (e.g., Murchison contains amino acids). Hydrothermal vent synthesis. Interstellar ice chemistry. Amino acids likely formed abiotically before enzymatic systems existed. Life inherited pre-existing chemistry.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Natural α-helices (L-amino acids) are: 👉 Right-handed. If composed of D-amino acids: 👉 The helix becomes left-handed. This inversion arises directly from chirality.
- Can you discover additional helices in proteins? Yes. Beyond α-helices: 3₁₀ helix, π-helix, Polyproline type II helix, Coiled-coils, Collagen triple helix. Protein design has also generated synthetic helices not found in nature. Helical geometry depends on backbone torsion angles and side-chain packing.
- Why are most molecular helices right-handed? Because life uses L-amino acids. Chirality at the monomer level restricts energetically favorable torsion angles, biasing right-handed helices. Handedness is an emergent property of stereochemistry.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? Because β-strands expose: Extended backbone hydrogen bond donors/acceptors. Flat, planar surfaces. They readily align and form intermolecular hydrogen bonds, leading to stacking and fibril formation. Their geometry promotes self-association. Primary forces: Backbone hydrogen bonding. Hydrophobic interactions. Aromatic stacking. Water exclusion (hydrophobic effect). Aggregation often lowers free energy significantly.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Because partially unfolded proteins expose aggregation-prone segments. These segments reorganize into cross-β structures: Highly ordered. Extremely stable. Resistant to degradation Examples: Alzheimer’s (Aβ), Parkinson’s (α-synuclein), Prion diseases. Amyloid represents a misfolded but energetically favorable state.
amyloid β-sheets can be used as materials. Amyloid fibrils have: High tensile strength, Nanofibrillar architecture, Structural robustness. They are being explored for: Hydrogels, Nanowires, Bio-scaffolds, Sustainable biomaterials. The main challenge is controlled assembly.
- Design a β-sheet motif that forms a well-ordered structure. Design principle: Alternate hydrophobic and polar residues to create amphipathic β-strands. Example motif: Val–Thr–Val–Thr–Val–Thr–Val–Thr This yields: One hydrophobic face. One polar face. Stable sheet stacking.
For improved order: Design a β-hairpin: Val–Thr–Val–Thr–Asn–Gly–Val–Thr–Val–Thr Asn–Gly promotes a tight turn. Alternating residues favor sheet formation. Terminal charged residues can prevent uncontrolled aggregation. This design promotes controlled, ordered β-sheet assembly.
NOTE: The answers presented in this section were generated using ChatGPT as a starting point and were subsequently reviewed and edited by me. The AI output served as a draft to help structure and articulate the responses. The prompt I used was: “Provide detailed answers to the following questions about amino acids, protein structure, helices, β-sheets, aggregation, amyloids, and biomaterial applications. Answer with scientific depth and clarity.” All final revisions, interpretations, and refinements reflect my own understanding.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it. I selected a PhaC synthase because we use it at Bioplastix to polimerize our co-polymer.
- Identify the amino acid sequence of your protein. The length of the protein is: 562 aminoacids. The most common amino acid is: L, which appears 67 times. Using RCSB Sequence Similarity tool. The query matched 24 Polymer Entities, for example: orgnism: Pseudomonas aeruginosa PAO1 Macromolecule: Poly(3-hydroxyalkanoic acid) synthase 1 Sequence Match: Sequence Identity: 99%, E-Value: 0, Region: 1-559
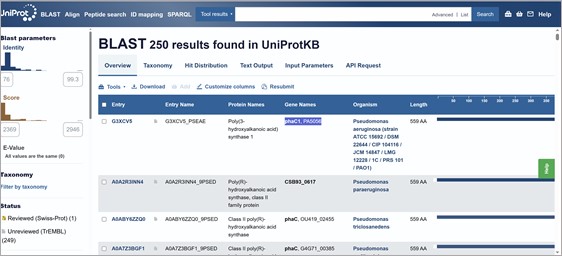
Also, using UniProtKB BLAST 250 results were found (most of them pseudomonas). For example: Poly(3-hydroxyalkanoic acid) synthase 1, phaC1, PA5056. Protein family/group databases: ESTHER pseae-PHAC1PHA_synth_II
- Identify the structure page of your protein in RCSB
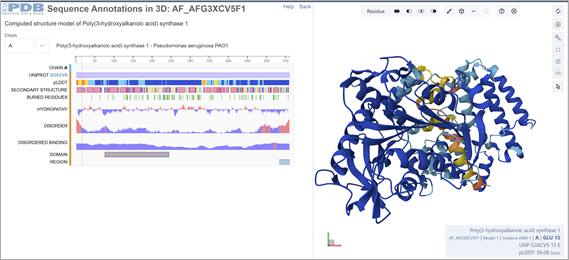
This was solved with a model. ¨COMPUTED STRUCTURE MODEL OF POLY(3-HYDROXYALKANOIC ACID) SYNTHASE 1. There are no experimental data to verify the accuracy of this computed structure model. See Model Confidence metrics below for all regions of the polypeptide chain. AlphaFold DB: AF-G3XCV5-F1 Released in AlphaFold DB: 2021-12-09 Last Modified in AlphaFold DB: 2022-09-30 Organism(s): Pseudomonas aeruginosa PAO1 UniProtKB: G3XCV5 Model Confidence pLDDT (global): 88.77¨
I found a crystal structure for a PhaC with Resolution: 2.70 Å:
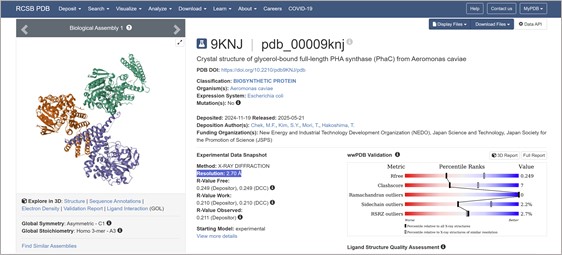
- Open the structure of your protein in any 3D molecule visualization software
Part C: Using ML-Based Protein Design Tools
I selected PHA synthase (PhaC) from Aeromonas caviae.
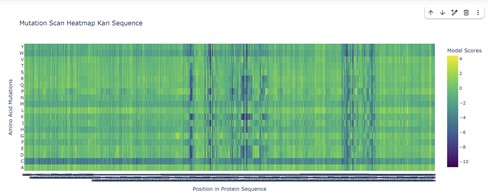
The mutational scanning heatmap highlights residue positions that are highly sensitive to mutation (dark blue/purple columns), likely corresponding to structurally or functionally critical residues. In contrast, regions with predominantly neutral or positive scores represent mutationally tolerant positions that could serve as potential targets for protein engineering.
The mutational heatmap suggests that different regions of the protein show varying tolerance to amino acid substitutions. Several positions display strong intolerance to mutation, visible as vertical dark-blue bands across many amino acid substitutions. These regions likely correspond to structurally or functionally important residues where mutations would strongly destabilize the protein or disrupt its activity.
In contrast, other regions show mostly green or light-colored scores, indicating positions that are more tolerant to mutation. These sites may correspond to surface-exposed residues or flexible loop regions, and could represent promising targets for protein engineering.
Interestingly, substitutions to tryptophan (W) and cysteine (C) tend to produce consistently negative scores across many positions, appearing as predominantly blue rows in the heatmap. This suggests that introducing these residues is generally unfavorable, likely because tryptophan is bulky and cysteine can introduce disulfide bonds or structural instability in inappropriate contexts.
Overall, the heatmap highlights a pattern of heterogeneous mutational tolerance, with conserved regions that are highly sensitive to mutation and more permissive regions that may accommodate sequence variation. These insights can guide rational protein engineering by identifying positions where mutations are more likely to be tolerated without compromising protein folding.
Part D: Group Brainstorm on Bacteriophage Engineering
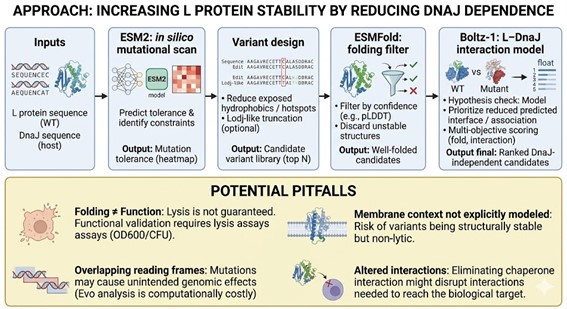
Bacteriophage Engineering Proposal: L Protein Stabilization Primary Goal: Increased stability (easiest)
Specific Approach: Engineering DnaJ-independence by reducing chaperone-recognition signals while preserving the structural scaffold of the L protein.
Computational Tools and Pipeline Justification To achieve this goal, we propose a three-step computationally efficient pipeline: Step 1: Sequence-level Mutational Scanning using ESM2 Approach: We will perform a zero-shot in silico mutational scan across the L protein sequence using the ESM2 Protein Language Model (PLM). We aim to identify exposed hydrophobic patches (typical DnaJ recognition motifs) and propose polar/hydrophilic substitutions. Why this helps: ESM2 has learned deep evolutionary constraints across millions of protein sequences. It allows us to rapidly differentiate between highly constrained residues (which are structurally vital and “untouchable”) and mutation-tolerant positions. This ensures we only disrupt chaperone-binding motifs without breaking the core evolutionary scaffold of the protein, all at a fraction of the computational cost of molecular dynamics. Step 2: Rapid Structural Filtering using ESMFold Approach: The top candidate sequences from the ESM2 scan will be predicted using ESMFold. We will filter out any variants that collapse, show low pLDDT (confidence) scores, or have a high RMSD compared to the Wild-Type (WT) backbone. Why this helps: While ESM2 evaluates sequence-level fitness, we need explicit 3D structural validation. ESMFold is significantly faster than AlphaFold2, making it ideal for high-throughput filtering. This step ensures that our hydrophilic mutations do not inadvertently destroy the L protein’s ability to fold independently. Step 3: Complex Modeling using Boltz-1 Approach: We will model the L protein + DnaJ complex for both the WT and our top folded mutant candidates. We will analyze the predicted interface contacts and Predicted Aligned Error (PAE) to assess binding affinity. Why this helps: Folding correctly in isolation is not enough; we must explicitly prove reduced chaperone dependency. By comparing the mutant-DnaJ interface against the WT-DnaJ interface, we can prioritize variants that maintain a stable fold but show a significantly weakened or abolished interaction with the DnaJ chaperone.
Potential Pitfalls
- Pitfall 1: Overlapping Reading Frames and Genomic Constraints. Phage genomes are highly compact, meaning the DNA sequence encoding the L protein might also encode parts of other proteins or regulatory elements in alternative reading frames. Our targeted mutations could have unintended, fatal consequences for the phage’s overall viability. While genomic foundation models like Evo could assess these genome-wide constraints, their computational cost is prohibitive for our current scope.
- Pitfall 2: The Stability vs. Function Trade-off. ESMFold guarantees that the protein adopts a stable 3D conformation in solution, but it does not guarantee biological function (membrane lysis). Lytic activity heavily depends on complex factors like membrane insertion dynamics, oligomerization, and reaction kinetics. Furthermore, completely abolishing chaperone interaction might inadvertently prevent the L protein from being properly delivered to its target membrane.