Subsections of Homework
Week 01 HW: Principles and Practices
- First, describe a biological engineering application or tool you want to develop and why.
I want to develop a highly efficient bacterial chassis for rapid intracellular biosynthesis of novel PHA (polyhydroxyalkanoate) copolymers. More than 150 different hydroxyalkanoate monomers have been identified, and they can be combined into co-polymers (and potentially ter-/quad-polymers) with variable composition and sequence/microstructure, leading to an astronomical design space.
My goal is to build a chassis that makes the design–build–test loop fast and reliable: (1) use computational/AI approaches and literature review to prioritize promising copolymer compositions for target properties, then (2) rapidly prototype biosynthesis in a standardized host with predictable performance, and (3) generate experimental data to validate predicted properties and improve models.
This tool idea is directly inspired by my work: I’m the CEO of Bioplastix, a biotech startup with the mission to accelerate the transition to biodegradable bioplastics. We already have a promissing co-polymer (PLA-PHB with different proportions of PLA) and a highly efficient E.coli chassis to produce it. We want to accelerate the discovery of new co-polymers. A chassis that can quickly produce and screen new PHA copolymers would let us create radically better biopolymers and accelerate transition to bioplastics.
- Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
The goal of this biological tool is to contribute to an ethical future by reducing harm to human and planetary health through the replacement of oil-based plastics. Conventional plastics accumulate in ecosystems and human bodies (Nihart, A.J., Garcia, M.A., El Hayek, E. et al. Bioaccumulation of microplastics in decedent human brains. Nat Med 31, 1114–1119 (2025)), have high associated carbon footprints, and often rely on toxic ingredients across their life cycle.
For this tool to be successful, I suggest two core policy and governance goals:
Goal 1: Encourage the adoption of bio-based and biodegradable plastics.
At present, biodegradable bioplastics such as PHAs remain significantly more expensive than oil-based plastics (e.g., PHAs at approximately 4–5 USD/kg versus PET at ~1.3 USD/kg). Policies that reduce this gap during early scale-up phases are therefore essential to enable meaningful market penetration.
Goal 2: Prevent harm from new biological strains and novel polymer compositions.
Because this platform enables the rapid creation of many new copolymers and engineered microbial strains, governance is critical to avoid unintended biological or environmental consequences. Like any powerful biological technology, it must operate within clearly defined biosafety, material safety, and ethical boundaries.
Note: I could state Goal 1 as: Ban traditional plastics or sidcourage the use of traditinal plastics. A global ban or restrictions on conventional plastics could, in principle, accelerate this transition, but such an outcome appears unlikely in the near term, as illustrated by the limited progress of the UN Global Plastics Treaty negotiations.
- Next, describe at least three different potential governance “actions” by considering four aspects (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Tax incentives for biodegradable and bio-based plastics
Purpose: Today, in most countries and subnational jurisdictions, there are no meaningful tax incentives that favor biodegradable bioplastics over conventional fossil-based plastics. This action proposes targeted tax reductions for products made predominantly from certified biodegradable plastics, with the goal of narrowing the price gap between products made with conventional plastics and those made with biodegradable alternatives.
Design: This action would require: Legislation establishing tax reduction and scope. Clear regulation and enforcement, since many environmental laws fail at the implementation stage, including: i. eligibility criteria for tax incentives (e.g., minimum biodegradable content, verified biodegradability standards). ii. auditing mechanisms to prevent misuse, such as products labeled “bioplastic” that are bio-based but not biodegradable, or products containing only a small fraction of biodegradable material. iii. phased implementation, potentially starting with high-impact applications where replacing conventional plastics yields the greatest environmental benefit.
Key actors include national and/or subnational governments, tax authorities, certification bodies, manufacturers opting into the program, and end users opting into buying products.
Assumptions: This action assumes that: Governments are willing and able to implement and enforce differentiated tax schemes. Traditional plastics resin producers will not lobby enough so as to stop the law. Price reductions at the product level are sufficient to meaningfully influence purchasing and adoption decisions. Certification systems can accurately distinguish between genuinely biodegradable materials and greenwashed alternatives.
Risks of Failure & “Success”: The policy could fail if enforcement is weak, allowing non-compliant products to benefit from incentives, or if administrative complexity discourages participation. Jurisdictional differences in taxation could also lead to production shifting across borders rather than reducing overall plastic harm. Even “successful” implementation may have unintended consequences, such as encouraging overconsumption of disposable products simply because they are labeled biodegradable, rather than reducing total plastic use.
Action 2: Large-scale public awareness campaigns on the human health impacts of traditional plastics
Purpose: Currently, public discourse around plastics focuses primarily on environmental damage, while the human health impacts of conventional plastics—such as chemical exposure, bioaccumulation, and endocrine disruption—remain relatively under-communicated. This limits public pressure for change and weakens demand for safer alternatives. This action proposes coordinated awareness campaigns that frame conventional plastics as a public health issue, similar to past campaigns against tobacco or excessive sugar consumption.
Design: This action would require: Global actors (e.g., WHO, UN agencies, international NGOs) to support and legitimize messaging at a global scale. National and local governments to adapt campaigns to local contexts and regulatory priorities. Startups and research institutions working on plastic alternatives to contribute evidence-based narratives and real-world solutions. Influencers, educators, and media organizations to amplify messages beyond traditional policy channels.
Assumptions: This action assumes that Increased awareness of health risks will meaningfully influence consumer behavior and political support.
Also, that influencers and media actors can communicate complex health information responsibly. That global and local actins can be integrated.
Risks of Failure & “Success”: The campaign could fail if messages are oversimplified, sensationalized, or perceived as fear-based, leading to public distrust. There is also a risk of backlash from industry actors framing the campaign as anti-innovation or anti-consumer or leading to jobs reduction.
Action 3: Establish dedicated biopolymer research and governance centers at national and global levels
Purpose: At present, governance and reaseadrh and development of new biopolymers and engineered production strains is fragmented across regulatory agencies, academic labs, and industry actors. This fragmentation slows safe innovation and creates uncertainty around implementation, standards, and scale-up. This action proposes the creation of dedicated research and governance centers at national or subnational levels, complemented by a global network coordinating best practices and knowledge sharing.
Design: These centers would: i. Support regulatory implementation, including biosafety and material safety evaluation for new biopolymers.
ii Conduct applied research on performance, degradation, and real-world applications of biodegradable plastics. iii. Serve as interfaces between academia, startups, industry, and regulators. iv. Participate in global networks to harmonize standards, share data, and reduce duplication of effort. Key actors include governments (as funders), universities, public research institutes, and international coordination bodies, as well as industry and startups.
Assumptions: This action assumes that: Governments are willing to fund long-term, interdisciplinary centers rather than short-term projects.
Centralized expertise improves both safety and innovation outcomes. International collaboration is feasible despite differences in regulation and economic priorities.
Risks of Failure & “Success”: These centers could fail by becoming overly bureaucratic or disconnected from real industrial needs. They may also privilege dominant technological pathways, limiting diversity in approaches. A “successful” network might unintentionally centralize decision-making power, creating gatekeepers that slow innovation or disadvantage smaller players without access to these institutions.
- Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
| Does the option: | Action 1 | Action 2 | Action 3 |
|---|
| G1: Encourage the adoption of bioplastics | | | |
| • By discouraging oil-based plastics | 2 | 1 | 3 |
| • By reducing the price gap with oil.based | 1 | n/a | 3 |
| • By encouraging innovation | 1 | 3 | 2 |
| • By improving the implementation of laws | 1 | 3 | 2 |
| G2: Prevent NEW harm | | | |
| • By preventing incidents at lab scale | 3 | n/a | 1 |
| • By preventing environmental consequences | 2 | 3 | 1 |
| • By preventing human health consequences | 2 | 3 | 1 |
| Overarching GOAL: Protect the environment | | | |
| • By preventing plastics in ecosistems | 1 | 2 | 3 |
| • By preventing GHG emmissions | 1 | 2 | 3 |
| Overarching GOAL: Protect human health | | | |
| • By preventing plastics in bodies | 1 | 2 | 3 |
| • By reducing the use of toxic ingredients | 1 | 2 | 3 |
| • By preventing GHG emmissions | 1 | 2 | 3 |
- Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the expected impact and feasibility of the proposed actions, I would prioritize Action 1: tax incentives for biodegradable and bio-based plastics as the primary governance intervention.
The main reason is that cost remains the dominant barrier to large-scale adoption of biodegradable plastics, despite their availability and well-documented environmental benefits. Awareness campaigns (Action 2) and research centers (Action 3) already exist to some degree, and bioplastics are commercially available today. What has not been broadly or consistently implemented is a policy mechanism that directly and effectively reduces the price gap between fossil-based plastics and biodegradable alternatives through governmental incentives.
For a national or subnational policymaker audience (e.g., ministries of industry, environment, or finance), Action 1 offers the highest near-term leverage. In contrast, awareness campaigns rely on slower cultural change, and research centers operate on longer innovation timelines.
Action 3—the creation or reorientation of dedicated biopolymer research and governance centers—would be a strong second priority because will be important to enhance laws. Importantly, this does not necessarily require creating entirely new institutions. Existing research centers could be re-scoped through regulation or funding requirements.
Action 2 is expected to emerge organically once Action 1 is implemented. As economic incentives shift markets, industry groups, startups, researchers, and civil society actors are likely to amplify awareness efforts and public communication, especially around human health impacts. In this sense, Action 1 can act as a catalyst for the other two actions.
Trade-offs, assumptions, and uncertainties: This prioritization assumes that governments are willing to intervene in markets through fiscal policy and that tax incentives will translate into lower end-user prices. There is also uncertainty around political feasibility, especially in jurisdictions resistant to environmental taxation or subsidies.
Week 2 Prep:
Homework Questions from Professor Jacobson:
Error Rate: 1:106 Throughput: 10 mS per Base Addition
basic error rate of DNA polymerase without proofreading is roughly 1 error per 10² to 10⁶ bases
Biology resolves this discrepancy through multiple layers of fidelity control, including polymerase proofreading, mismatch repair systems, and cell-cycle checkpoints that prevent propagation of damaged DNA.
Average Human Protein: 1036 bp • Longest Human Proteins (PKS): >100kbp
Since most amino acids are encoded by multiple synonymous codons, the number of possible DNA sequences that could encode the same protein is astronomically large.
Not all work due to biological constrains: Codon usage bias: different organisms preferentially use certain synonymous codons, which affects translation efficiency and protein yield. mRNA structure: different nucleotide sequences form different local structures that affect ribosome binding and stability. Embedded regulatory elements: coding regions can contain splice sites, RNA-binding motifs, or secondary signals that influence expression.
Homework Questions from Dr. LeProust:
phosphoramidite DNA synthesis.
it is difficult to synthesize oligos longer than ~200 nt
Each nucleotide addition step has a small but non-zero error rate (incomplete coupling, deletions, or side reactions). As oligo length increases, these errors accumulate multiplicatively, leading to a rapidly decreasing fraction of full-length, error-free molecules. Beyond ~150–200 nt, yield and fidelity drop sharply, making purification inefficient and expensive.
A 2000 bp sequence would require ~2000 sequential synthesis steps, which would result in near-zero yield of full-length correct molecules due to accumulated errors. Instead, long genes are built by assembling shorter oligos (e.g., 50–200 nt) using enzymatic methods such as PCR-based assembly or Gibson assembly
Homework Question from George Church:
1.
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
lysine is universally essential across animal biology (not unique to any engineered or extinct species), to prevent organisms from surviving without an external lysine supply is not biologically plausible or unique—it simply reflects an existing nutritional requirement. This makes the fictional Lysine Contingency concept not a realistic.
Note on AI use:
A large language model (ChatGPT) was used solely to assist with English grammar, spelling, and clarity of writing. All ideas, goals, policy actions, and arguments presented in this assignment were developed independently by the author and not generated by the language model.
Week 02 HW: DNA Read, Write, and Edit
Part 1: Benchling & In-silico Gel Art 🦠
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks:
Part 3: DNA Design Challenge
I chose poly(3-hydroxyalkanoate) polymerase / PHB synthase (PhaC) from Cupriavidus necator (UniProt accession P23608) because it is a key enzyme in microbial bioplastic production. PhaC catalyzes the polymerization of (R)-3-hydroxybutyryl-CoA monomers to form poly(3-hydroxybutyrate) (PHB), and engineered variants of PhaC are widely used to broaden substrate specificity and produce other polyhydroxyalkanoates (PHAs). I obtained the amino-acid sequence from UniProt (entry P23608) in FASTA format.
MA T G K G A A A S T Q E G K S Q P F K V T P G P F D P A T W L E W S R Q W Q G T E G N G H A A A S G I P G L D A L A G V K I A P A Q L G D I Q Q R Y M K D F S A L W Q A M A E G K A E A T G P L H D R R F A G D A W R T N L P Y R F A A A F Y L L N A R A L T E L A D A V E A D A K T R Q R I R F A I S Q W V D A M S P A N F L A T N P E A Q R L L I E S G G E S L R A G V R N M M E D L T R G K I S Q T D E S A F E V G R N V A V T E G A V V F E N E Y F Q L L Q Y K P L T D K V H A R P L L M V P P C I N K Y Y I L D L Q P E S S L V R H V V E Q G H T V F L V S W R N P D A S M A G S T W D D Y I E H A A I R A I E V A R D I S G Q D K I N V L G F C V G G T I V S T A L A V L A A R G E H P A A S V T L L T T L L D F A D T G I L D V F V D E G H V Q L R E A T L G G G A G A P C A A A A G L E L A N T F S F L R P N D L V W N Y V V D N Y L K G N T P V P F D L L F W N G D A T N L P G P W Y C W Y L R H T L P A E R A Q G T G Q A D R V R R A G G P G Q H R R P Y I Y G S R E D H I V P W T A A Y A S T A L L A N K L R F V L G A S G H I A G V I N P P A K N K R S H W T N D A L P E S P Q Q W L A G A I E H H G S W W P D W T A W L A G Q A G A K R A R P A N Y G N A R Y R A I E P A P G D T S K P R H
Although the genetic code is universal, most amino acids are encoded by multiple codons. However, different organisms do not use these synonymous codons at the same frequency. This phenomenon is known as codon bias. If a gene from one organism is expressed in a different host, the original codons may be rare in the new host, which can reduce translation efficiency, among other problems.
Codon optimization is necessary to modify the DNA sequence so that it uses codons preferred by the host organism. Optimizing codon usage can significantly increase protein expression levels, improve translation efficiency, enhance mRNA stability, and reduce the likelihood of misfolding or premature termination.
For this project, I chose to optimize the phaC gene for Escherichia coli. I selected E. coli because it is the chassis organism we use at Bioplastix.
I used https://www.genscript.com/ as a code optimizattion tool (Job ID: 20260216015729685186).
At Bioplastix, the engineered bacteria produce the enzymes intracellularly. Under specific metabolic conditions, these enzymes catalyze the polymerization of monomers inside the cell, leading to intracellular accumulation of the biopolymer.
To achieve that, once the codon-optimized DNA sequence is obtained, it is inserted into a plasmid (an expression vector containing the necessary regulatory elements, such as a promoter and terminator). This plasmid is then be inserted into the host organism, Escherichia coli, for protein production.
In such a cell-dependent system, the DNA sequence is transcribed into messenger RNA (mRNA) by RNA polymerase. The mRNA is then translated by ribosomes into the PhaC protein. Depending on the design of the genetic construct, the gene can be placed under a constitutive promoter, where the protein is continuously produced, or under an inducible promoter, where expression is triggered by specific conditions (such as the presence of IPTG or oxigen).
Alternatively, the protein could also be produced using a cell-free expression system, where the DNA is added to a reaction mixture containing ribosomes, tRNAs, nucleotides, and enzymes necessary for transcription and translation. This allows protein production without living cells, although large-scale industrial production until now typically relies on cell-based systems.
Part 4: Prepare a Twist DNA Synthesis Order
Link Sharing From Benchling: https://benchling.com/s/seq-l6sbaCHItMO0QD5lHD4M?m=slm-38MASpmHAGVSi3hySqfU
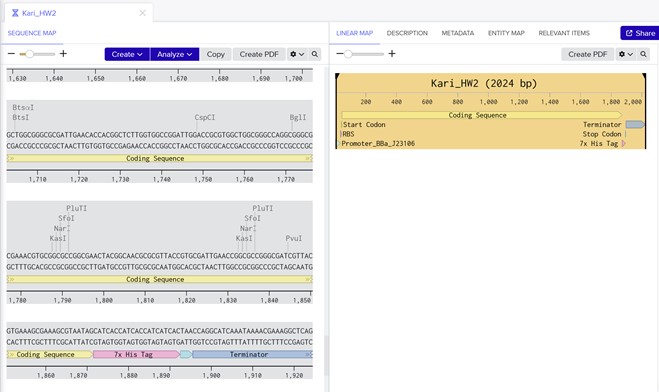
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would like to sequence DNA from environmental microbial communities, particularly from diverse and extreme environments such as soil, marine ecosystems, open dumps, and high-stress habitats. These environments often contain microorganisms with unique metabolic capabilities.
By sequencing DNA from environmental samples (metagenomic sequencing), we could identify novel enzymes involved in polymer biosynthesis, including new variants of PHA synthases or related polymerizing enzymes. These enzymes may have improved catalytic efficiency, altered substrate specificity, or greater stability under industrial conditions. Discovering new enzymes through DNA sequencing could enable the development of more efficient bioplastic production systems and potentially allow the synthesis of novel polymers with enhanced material properties.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina sequencing technology, a second-generation (next-generation sequencing, NGS) platform. Illumina sequencing is well-suited for metagenomic analysis because it provides high-throughput, highly accurate short reads at relatively low cost, making it ideal for sequencing complex microbial communities. It is second generation because it performs massively parallel sequencing, requires DNA amplification (cluster generation), and sequences millions of fragments simultaneously.
The input is extracted environmental DNA (metagenomic DNA) from microbial communities.
Sample preparation steps:
- DNA extraction from environmental samples (soil, water, etc.)
- Fragmentation of DNA into smaller pieces (typically ~200–500 bp)
- End repair and A-tailing
- Adapter ligation (short known sequences attached to fragment ends)
- PCR amplification to enrich properly ligated fragments
- Library quantification and quality control
Illumina sequencing works as follows:
- The prepared DNA library is loaded onto a flow cell.
- Fragments bind to complementary oligos attached to the flow cell.
- Bridge amplification creates clusters of identical DNA fragments.
- Fluorescently labeled reversible terminator nucleotides are added.
- During each cycle, one nucleotide is incorporated per cluster.
- A camera detects the fluorescent signal emitted by the incorporated base.
- The terminator is cleaved, and the next cycle begins.
Each nucleotide (A, T, C, G) carries a different fluorescent signal. The imaging system detects the color emitted at each cycle, and software converts fluorescence intensity into base calls (A, T, C, or G).
5.2 DNA Write
The DNA I would want to “write” (synthesize) would be a set of candidate polymerizing enzymes identified from the environmental metagenomic library, especially novel PHA synthase (PhaC-like) genes or other enzymes predicted to catalyze polymer formation. The main reason to synthesize these genes is to rapidly test them in a standardized host. I would order codon-optimized versions of each candidate enzyme gene for Escherichia coli, because E. coli is the chassis organism we use at Bioplastix.
I would use solid-phase phosphoramidite DNA synthesis, the standard chemical DNA synthesis technology used by companies such as Twist Bioscience.
In the past we already used Twist for synthesis of new enzymes. It is very useful because enables codon optimization, Introduction of specific mutations, Removal of unwanted restriction sites, parallel synthesis of multiple variants.
With this methodd DNA is synthesized one nucleotide at a time on a solid support using phosphoramidite chemistry. This produces short DNA fragments (typically 150–200 bp). Since genes are longer than individual oligos, overlapping oligonucleotides are assembled into full-length genes using: PCR-based assembly, Gibson Assembly or Enzymatic ligation methods. The assembled gene is amplified and sometimes enzymatically corrected to reduce synthesis errors. The final construct is cloned into a plasmid and verified by DNA sequencing before delivery.
From a practical perspective at Bioplastix, the main limitation of outsourcing DNA synthesis to companies such as Twist Bioscience is cost. Synthesizing long gene sequences can be expensive, particularly when testing multiple enzyme variants. Each synthesis order costs approximately $1,500 USD. After synthesis, there are further costs associated with Transforming it into our production strains and Screening and validating expression. Overall, with a typical budget, we are able to test approximately 5–10 new enzyme candidates per round.
5.3 DNA Edit
The DNA I would want to edit is the genome of our production strain (e.g., Escherichia coli) in order to improve its efficiency as a bioplastic-producing chassis organism. Specifically, I would focus on :
1️⃣ Expanding substrate utilization: One of our current priorities is enabling the strain to efficiently consume alternative and low-cost carbon sources, particularly sucrose. Editing the genome to introduce or optimize sucrose transporters and metabolic pathways would allow the bacteria to convert a wider range of feedstocks into biopolymer, improving economic feasibility and sustainability.
2️⃣ Increasing carbon flux toward polymer production: I would edit genes involved in central carbon metabolism to redirect more carbon toward the PHA biosynthesis pathway.
3️⃣ Engineering polymerizing enzymes: I would also edit the genes encoding polymerizing enzymes (such as PhaC) to Increase substrate affinity, Improve catalytic efficiency and Enhance thermostability. Thermotolerant enzymes would be particularly valuable at industrial scale, where higher fermentation temperatures reduce cooling costs and contamination risk.
To perform these genome edits, I would use CRISPR-Cas9 genome editing, combined with homologous recombination. CRISPR-based systems are precise, programmable, and highly efficient, making them ideal for metabolic engineering in bacterial systems. CRISPR-Cas9 uses: i. A guide RNA (gRNA) designed to match a specific DNA target sequence. ii. The Cas9 nuclease, which creates a double-strand break at the target site
After the break is introduced, the cell repairs the DNA. If a repair template is provided (homology-directed repair, HDR), specific edits such as insertions, deletions, or point mutations can be introduced.
Limmitations include Metabolic burden (Multiple edits can stress the cell and reduce growth rate) and Regulatory complexity (Metabolic networks are highly interconnected; editing one pathway may produce unexpected downstream effects).
Week 03 HW: Lab Automation
Python Script for Opentrons Artwork: https://colab.research.google.com/drive/14m54uLCM5UtsggVjU2Ucxh5hhtNELWD2#scrollTo=pczDLwsq64mk&line=4&uniqifier=1
- Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
I found the paper ¨Opentrons for automated and high-throughput viscometry¨ very interesting.
¨The operating protocol involves measuring the amount of liquid dispensed over a set time for given dispense conditions. Data collected at different set dispense flow rates was used to train an ensemble machine learning regressor to predict Newtonian liquid viscosity¨. They demonstrated the ability of the proxy viscometer to characterize the rheological behavior of two types of power-law fluids.
Write a description about what you intend to do with automation tools for your final project.
For my final project, I intend to focus on a challenge that is directly relevant to my startup, Bioplastix, which develops technologies for the production of bio-based copolymers with plastic properties. I am particularly interested in exploring how low-cost automation tools, such as the Opentrons OT-2, can accelerate enzyme discovery and biopolymer development workflows. I have two potential project directions:
Idea 1: High-Throughput Screening of Polymerizing Enzymes: Bioplastix has access to a diverse environmental enzyme library. Our goal is to identify enzymes capable of polymerizing specific monomers into useful copolymers. An initial in silico screening step can be performed by identifying genetic sequences that are homologous to known polymerizing enzymes. However, the key bottleneck occurs during experimental validation in the wet lab. Using automation tools, we could perform parallel screening of up to 96 enzyme candidates per plate, standardizing reaction setup to reduce variability.
This will be possible Implementing colorimetric assays to detect polymer formation. The automation platform would allow us to screen dozens (or hundreds) of enzymes under consistent conditions, significantly accelerating discovery while reducing human error.
Idea 2: Automated Cell-Free Production of Copolymers: A second direction would be to explore automated workflows for cell-free production systems. Instead of expressing enzymes in living cells, we could use cell-free systems to produce polymers. This would involve screen different reaction conditions (pH, cofactors, substrate ratios, temperature), optimize copolymer production in a high-throughput format.
Week 04 HW: Protein Design - Part 1
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assumptions: Meat ≈ 20% protein. 500 g meat → ~100 g protein. Average amino acid residue mass ≈ 100 Da ≈ 100 g/mol.
So: 100 g ÷ 100 g/mol ≈ 1 mole of amino acid residues. 1 mole = 6.022 × 10²³ molecules.
👉 You ingest on the order of 6 × 10²³ amino acid residues in 500 g of meat.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion destroys biological structure. Proteins are hydrolyzed into amino acids. The original sequences (information) are lost. Your body reassembles amino acids according to human DNA instructions. Biological identity is encoded in sequence and genomic regulation — not in the raw amino acid building blocks. You absorb matter, not identity.
Note: While reading this response, I was prompted to ask ChatGPT the following question:
“Do the 20 amino acids explain all the DNA of all species? Are there more evolved species that use more amino acids?”
ChatGPT’s response was: Yes, the same 20 amino acids (with two rare exceptions) account for virtually all proteins in all known living organisms.
There are no “more evolved” species that use a greater number of standard amino acids.
This follow-up helped clarify the universality of the genetic code and reinforced the idea that biological complexity arises from sequence variation and regulation, rather than from an expanded set of amino acid building blocks.
- Why are there only 20 natural amino acids?
This is evolutionary, not chemical necessity. The 20 canonical amino acids: Provide sufficient chemical diversity: Hydrophobic, Polar, Charged, Aromatic, Reactive, Enable folding into complex structures.
Likely reflect optimization of the genetic code to minimize error impact. More than 500 amino acids exist in nature, but only 20 are universally encoded in the genetic code (with rare additions like selenocysteine). Evolution found a minimal but sufficient toolkit.
- Can you make other non-natural amino acids? Design some new amino acids.
Yes. Synthetic biology routinely incorporates non-natural amino acids. Examples: A) Fluorinated phenylalanine. Replace para-H with F. Increased hydrophobicity. Enhanced stability. Altered electronic properties.
B) Long-chain hydrophobic amino acid. Extend leucine side chain by two carbons. Stronger hydrophobic core packing. Enhanced β-sheet stabilization.
- Where did amino acids come from before enzymes that make them, and before life started?
Prebiotic chemistry. Evidence includes: Miller–Urey experiments (electric discharge in reducing atmosphere). Meteorites (e.g., Murchison contains amino acids). Hydrothermal vent synthesis. Interstellar ice chemistry. Amino acids likely formed abiotically before enzymatic systems existed. Life inherited pre-existing chemistry.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural α-helices (L-amino acids) are: 👉 Right-handed. If composed of D-amino acids: 👉 The helix becomes left-handed.
This inversion arises directly from chirality.
- Can you discover additional helices in proteins?
Yes. Beyond α-helices: 3₁₀ helix, π-helix, Polyproline type II helix, Coiled-coils, Collagen triple helix. Protein design has also generated synthetic helices not found in nature. Helical geometry depends on backbone torsion angles and side-chain packing.
- Why are most molecular helices right-handed?
Because life uses L-amino acids. Chirality at the monomer level restricts energetically favorable torsion angles, biasing right-handed helices.
Handedness is an emergent property of stereochemistry.
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Because β-strands expose: Extended backbone hydrogen bond donors/acceptors. Flat, planar surfaces. They readily align and form intermolecular hydrogen bonds, leading to stacking and fibril formation.
Their geometry promotes self-association.
Primary forces: Backbone hydrogen bonding. Hydrophobic interactions. Aromatic stacking. Water exclusion (hydrophobic effect).
Aggregation often lowers free energy significantly.
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Because partially unfolded proteins expose aggregation-prone segments. These segments reorganize into cross-β structures: Highly ordered. Extremely stable. Resistant to degradation
Examples: Alzheimer’s (Aβ), Parkinson’s (α-synuclein), Prion diseases.
Amyloid represents a misfolded but energetically favorable state.
amyloid β-sheets can be used as materials. Amyloid fibrils have: High tensile strength, Nanofibrillar architecture, Structural robustness. They are being explored for: Hydrogels, Nanowires, Bio-scaffolds, Sustainable biomaterials. The main challenge is controlled assembly.
- Design a β-sheet motif that forms a well-ordered structure.
Design principle: Alternate hydrophobic and polar residues to create amphipathic β-strands.
Example motif: Val–Thr–Val–Thr–Val–Thr–Val–Thr
This yields: One hydrophobic face. One polar face. Stable sheet stacking.
For improved order: Design a β-hairpin: Val–Thr–Val–Thr–Asn–Gly–Val–Thr–Val–Thr
Asn–Gly promotes a tight turn. Alternating residues favor sheet formation. Terminal charged residues can prevent uncontrolled aggregation.
This design promotes controlled, ordered β-sheet assembly.
NOTE: The answers presented in this section were generated using ChatGPT as a starting point and were subsequently reviewed and edited by me. The AI output served as a draft to help structure and articulate the responses. The prompt I used was: “Provide detailed answers to the following questions about amino acids, protein structure, helices, β-sheets, aggregation, amyloids, and biomaterial applications. Answer with scientific depth and clarity.” All final revisions, interpretations, and refinements reflect my own understanding.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
I selected a PhaC synthase because we use it at Bioplastix to polimerize our co-polymer.
- Identify the amino acid sequence of your protein.
The length of the protein is: 562 aminoacids. The most common amino acid is: L, which appears 67 times.
Using RCSB Sequence Similarity tool. The query matched 24 Polymer Entities, for example:
orgnism: Pseudomonas aeruginosa PAO1
Macromolecule: Poly(3-hydroxyalkanoic acid) synthase 1
Sequence Match: Sequence Identity: 99%, E-Value: 0, Region: 1-559
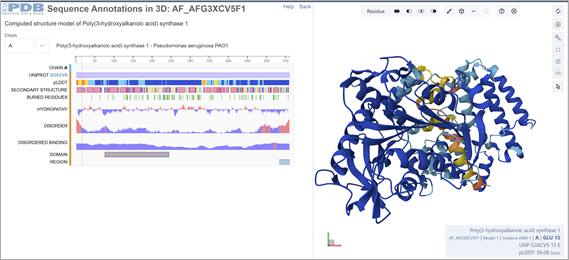
Also, using UniProtKB BLAST 250 results were found (most of them pseudomonas). For example: Poly(3-hydroxyalkanoic acid) synthase 1, phaC1, PA5056. Protein family/group databases: ESTHER pseae-PHAC1PHA_synth_II
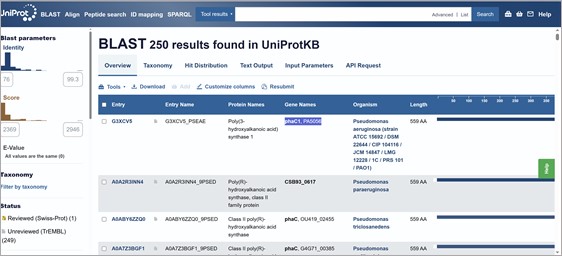
- Identify the structure page of your protein in RCSB
This was solved with a model. ¨COMPUTED STRUCTURE MODEL OF POLY(3-HYDROXYALKANOIC ACID) SYNTHASE 1. There are no experimental data to verify the accuracy of this computed structure model. See Model Confidence metrics below for all regions of the polypeptide chain.
AlphaFold DB: AF-G3XCV5-F1
Released in AlphaFold DB: 2021-12-09
Last Modified in AlphaFold DB: 2022-09-30
Organism(s): Pseudomonas aeruginosa PAO1
UniProtKB: G3XCV5
Model Confidence pLDDT (global): 88.77¨
I found a crystal structure for a PhaC with Resolution: 2.70 Å:
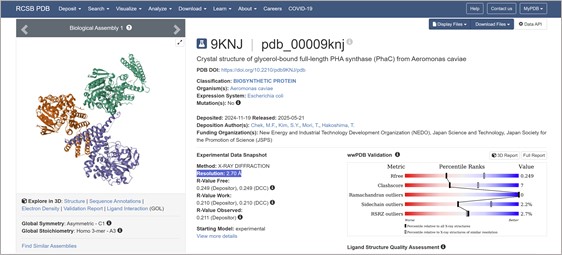
- Open the structure of your protein in any 3D molecule visualization software
Part C: Using ML-Based Protein Design Tools
I selected PHA synthase (PhaC) from Aeromonas caviae.
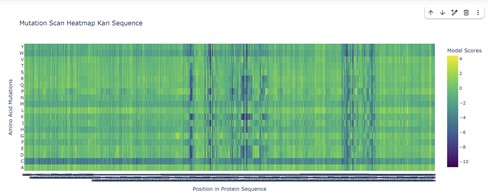
The mutational scanning heatmap highlights residue positions that are highly sensitive to mutation (dark blue/purple columns), likely corresponding to structurally or functionally critical residues. In contrast, regions with predominantly neutral or positive scores represent mutationally tolerant positions that could serve as potential targets for protein engineering.
The mutational heatmap suggests that different regions of the protein show varying tolerance to amino acid substitutions. Several positions display strong intolerance to mutation, visible as vertical dark-blue bands across many amino acid substitutions. These regions likely correspond to structurally or functionally important residues where mutations would strongly destabilize the protein or disrupt its activity.
In contrast, other regions show mostly green or light-colored scores, indicating positions that are more tolerant to mutation. These sites may correspond to surface-exposed residues or flexible loop regions, and could represent promising targets for protein engineering.
Interestingly, substitutions to tryptophan (W) and cysteine (C) tend to produce consistently negative scores across many positions, appearing as predominantly blue rows in the heatmap. This suggests that introducing these residues is generally unfavorable, likely because tryptophan is bulky and cysteine can introduce disulfide bonds or structural instability in inappropriate contexts.
Overall, the heatmap highlights a pattern of heterogeneous mutational tolerance, with conserved regions that are highly sensitive to mutation and more permissive regions that may accommodate sequence variation. These insights can guide rational protein engineering by identifying positions where mutations are more likely to be tolerated without compromising protein folding.
Part D: Group Brainstorm on Bacteriophage Engineering
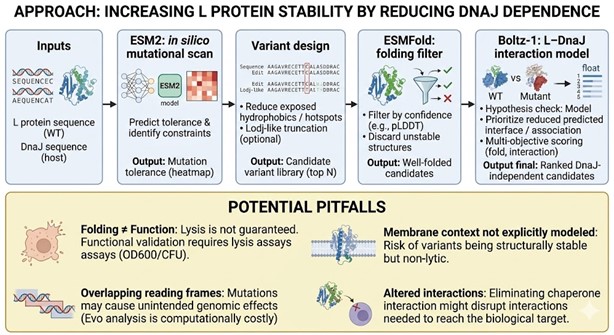
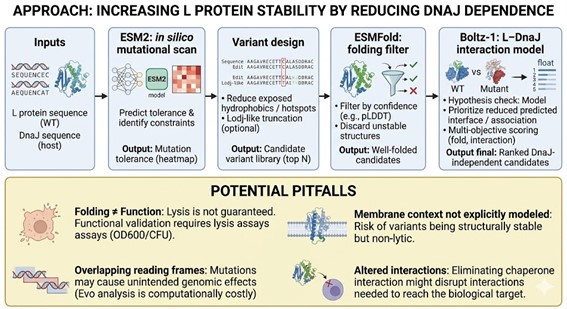
Bacteriophage Engineering Proposal: L Protein Stabilization
Primary Goal: Increased stability (easiest)
Specific Approach: Engineering DnaJ-independence by reducing chaperone-recognition signals while preserving the structural scaffold of the L protein.
Computational Tools and Pipeline Justification To achieve this goal, we propose a three-step computationally efficient pipeline:
Step 1: Sequence-level Mutational Scanning using ESM2
Approach: We will perform a zero-shot in silico mutational scan across the L protein sequence using the ESM2 Protein Language Model (PLM). We aim to identify exposed hydrophobic patches (typical DnaJ recognition motifs) and propose polar/hydrophilic substitutions.
Why this helps: ESM2 has learned deep evolutionary constraints across millions of protein sequences. It allows us to rapidly differentiate between highly constrained residues (which are structurally vital and “untouchable”) and mutation-tolerant positions. This ensures we only disrupt chaperone-binding motifs without breaking the core evolutionary scaffold of the protein, all at a fraction of the computational cost of molecular dynamics.
Step 2: Rapid Structural Filtering using ESMFold
Approach: The top candidate sequences from the ESM2 scan will be predicted using ESMFold. We will filter out any variants that collapse, show low pLDDT (confidence) scores, or have a high RMSD compared to the Wild-Type (WT) backbone.
Why this helps: While ESM2 evaluates sequence-level fitness, we need explicit 3D structural validation. ESMFold is significantly faster than AlphaFold2, making it ideal for high-throughput filtering. This step ensures that our hydrophilic mutations do not inadvertently destroy the L protein’s ability to fold independently.
Step 3: Complex Modeling using Boltz-1
Approach: We will model the L protein + DnaJ complex for both the WT and our top folded mutant candidates. We will analyze the predicted interface contacts and Predicted Aligned Error (PAE) to assess binding affinity.
Why this helps: Folding correctly in isolation is not enough; we must explicitly prove reduced chaperone dependency. By comparing the mutant-DnaJ interface against the WT-DnaJ interface, we can prioritize variants that maintain a stable fold but show a significantly weakened or abolished interaction with the DnaJ chaperone.
Potential Pitfalls
- Pitfall 1: Overlapping Reading Frames and Genomic Constraints. Phage genomes are highly compact, meaning the DNA sequence encoding the L protein might also encode parts of other proteins or regulatory elements in alternative reading frames. Our targeted mutations could have unintended, fatal consequences for the phage’s overall viability. While genomic foundation models like Evo could assess these genome-wide constraints, their computational cost is prohibitive for our current scope.
- Pitfall 2: The Stability vs. Function Trade-off. ESMFold guarantees that the protein adopts a stable 3D conformation in solution, but it does not guarantee biological function (membrane lysis). Lytic activity heavily depends on complex factors like membrane insertion dynamics, oligomerization, and reaction kinetics. Furthermore, completely abolishing chaperone interaction might inadvertently prevent the L protein from being properly delivered to its target membrane.
Week 05 HW: Protein Design - Part 2
Part A. SOD1 Binder Peptide Design
The perplexity scores for the candidate peptides were:
‘SDGAVLLGSDGE’ (Candidate 1): 16.25
‘LLGSDGALQVGS’ (Candidate 2): 14.65
‘SGVAVLCSDGQG’ (Candidate 3): 25.34
‘AVGVCGVAVLGN’ (Candidate 4): 17.20
Lower perplexity scores suggest that the model finds a sequence more “familiar” or “expected,” potentially correlating with higher biological plausibility, conformational stability, or likelihood of interaction. Candidate 2, with a perplexity of 14.65, appears to be the most promising candidate from the mutated SOD1 sequence for further investigation, being closest to the known binding peptide’s score.
Candidate 1: ipTM = 0.52 pTM = 0.88
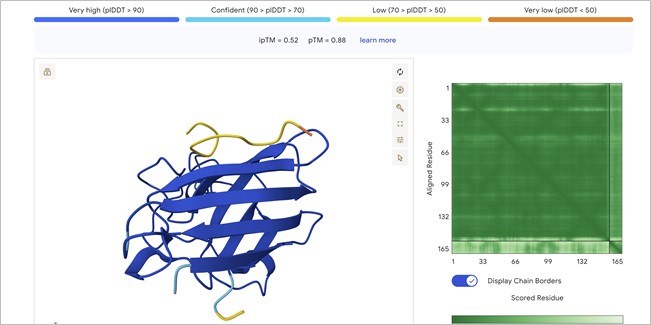
Localization: The peptide (the small chain with yellow and orange segments at the top) is localized near the N-terminus. It is observed to sit atop the beta-barrel, specifically in the region where the initial helix and the N-terminal end of the SOD1 (shown in dark blue) connect with the rest of the protein structure.Interaction with the beta-barrel: The peptide directly engages the $\beta$-barrel region. It appears to be “resting” on the upper beta-sheets, effectively acting as a molecular cap.Surface-bound vs. Buried: The peptide appears primarily surface-bound. While it is not deeply buried within the protein core, it maintains extensive contact with the exposed surface, a characteristic typical of stabilizing peptides.Relationship to A4V: Being situated at the top, near the start of the sequence, the peptide is in the immediate vicinity of residue 4. This suggests it may help “anchor” the N-terminus, preventing it from detaching or unfolding due to the destabilizing effect of the A4V mutation.
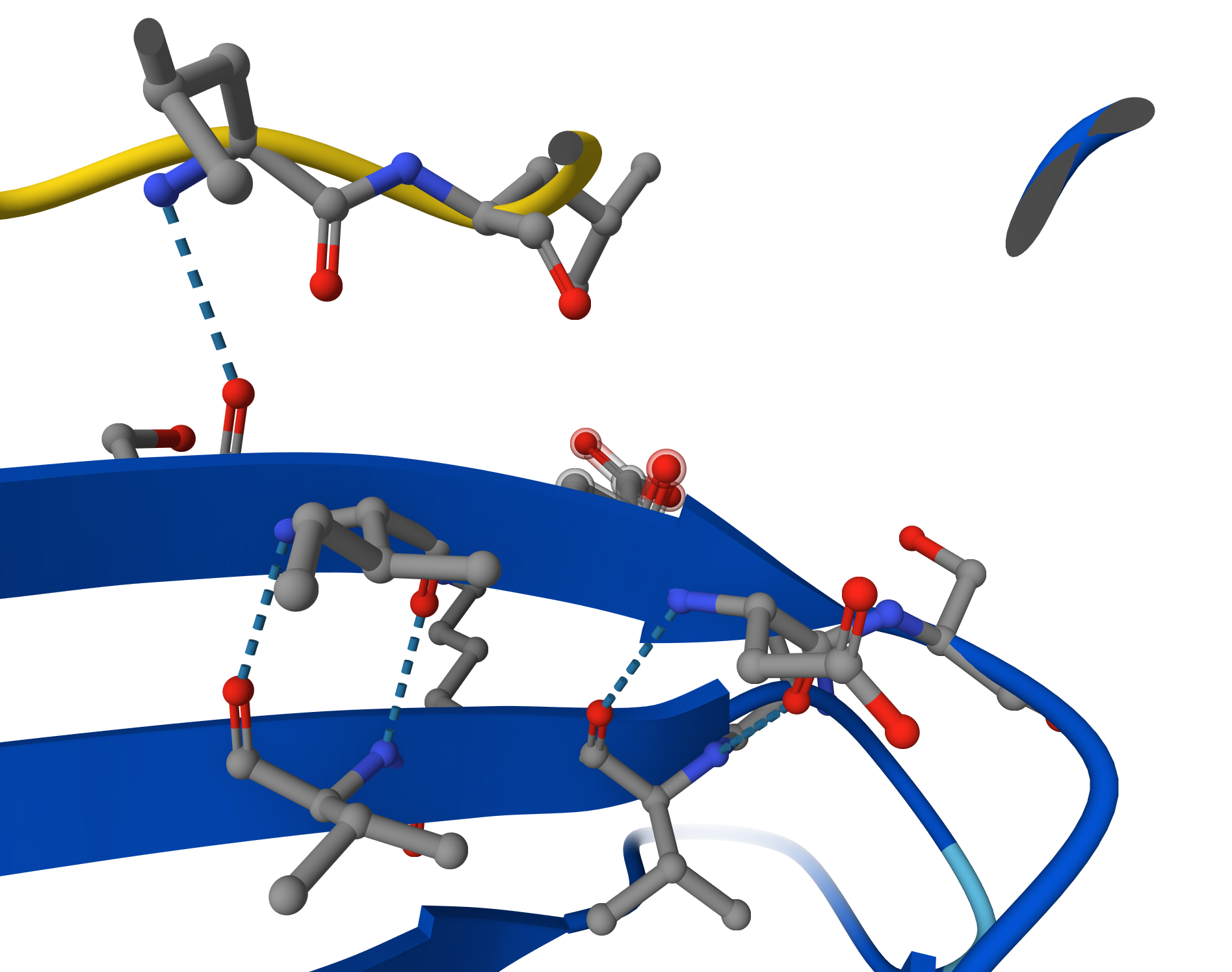
Upon closer inspection of the AlphaFold3 model, a clear hydrogen bond is visible between the peptide backbone and the SOD1 $\beta$-sheet. This specific inter-chain interaction confirms that the peptide is not just near the enzyme, but actively docking through electrostatic stabilization, which contributes to the observed ipTM of 0.52
Candidate 2: ipTM = 0.54 pTM = 0.89
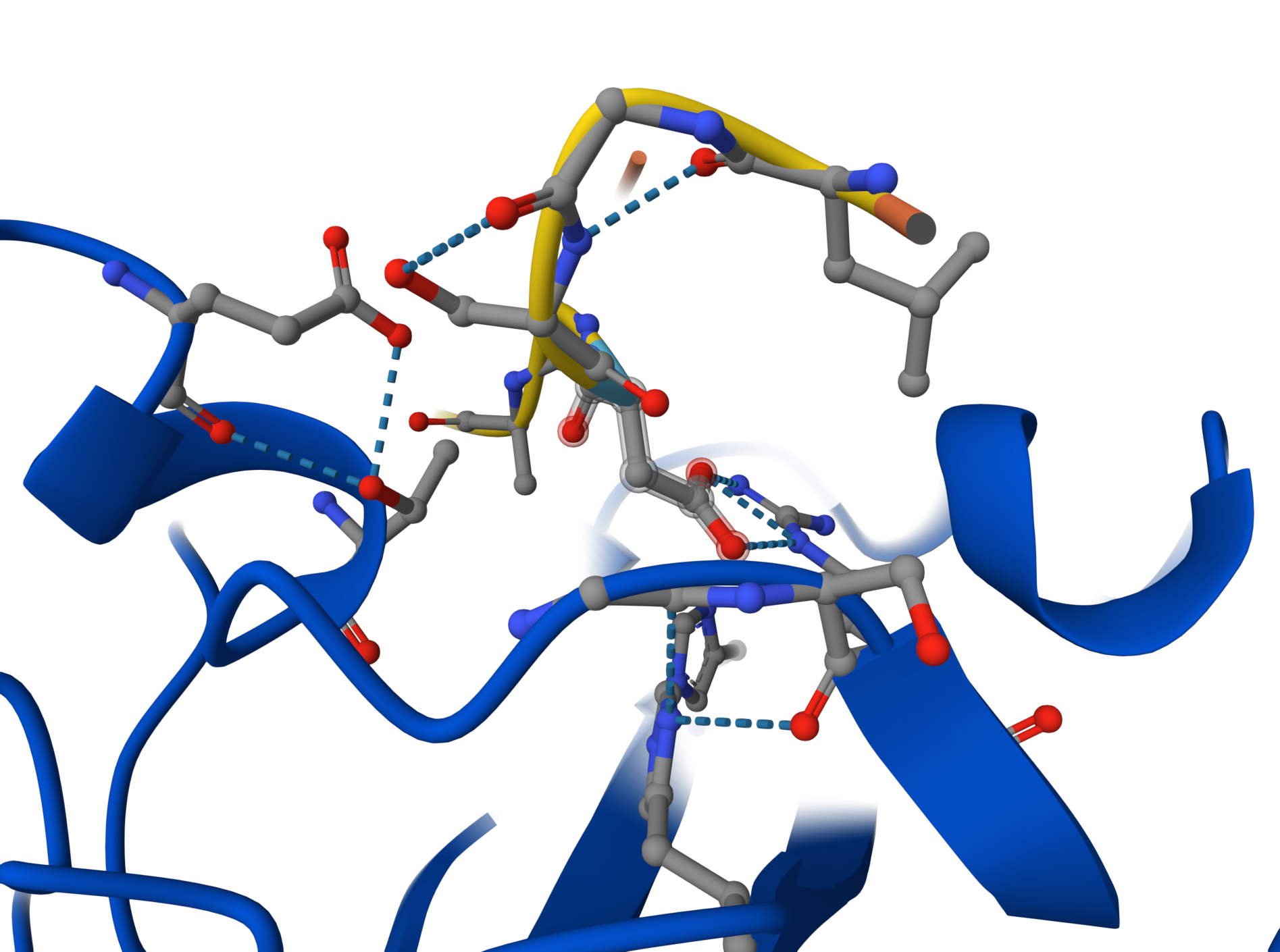
Although Candidate #2 shows a similar confidence score (ipTM = 0.54), the structural model reveals a significantly more robust interaction network. Detailed inspection shows multiple hydrogen bonds between the peptide backbone and the SOD1 surface loops, compared to the single-point attachment of previous candidate. This increased ‘molecular velcro’ effect likely provides better stabilization of the N-terminus, making this peptide a stronger therapeutic lead against the A4V mutation.
Candidate 3: ipTM = 0.35 pTM = 0.88
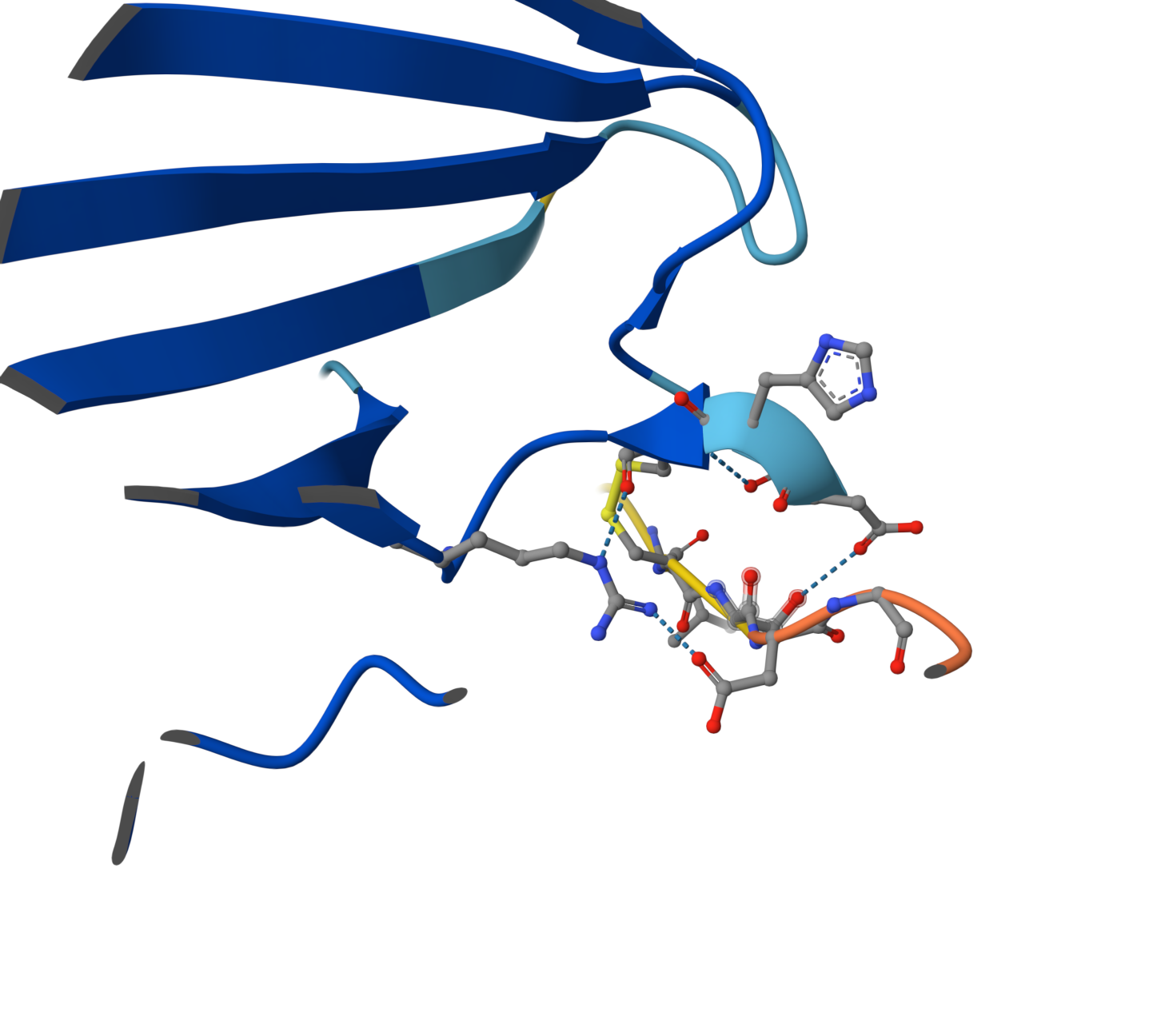
Although the global view suggests a weak interaction (ipTM ~0.35), detailed close-up inspection reveals a highly specific docking event. The peptide binds to a flexible disordered loop (cyan region) rather than the rigid $\beta$-barrel core. This interaction is mediated by a sophisticated hydrogen-bond network and includes crucial electrostatic contacts between a peptide Arginine ($Arg$) and acidic residues on the enzyme’s surface loop. This suggests the peptide acts as an allosteric stabilizer, reducing the flexibility of critical loops near the N-terminus. A key residue (appearing as a nitrogen-rich heterocycle) is perfectly positioned to stabilize the enzyme’s loop through electrostatic contacts.
Candidate 4: ipTM = 0.49 pTM = 0.89

ipTM of 0.49: By falling below the 0.5 threshold, AlphaFold is indicating a lack of confidence in the existence of a stable binding interface. Visual Evidence: The image shows the peptide (yellow chain) physically separated from the SOD1 enzyme (blue chain). Although the peptide attempts to adopt a self-folded conformation, there are no hydrogen bonds connecting it to the protein. pTM of 0.89: As in previous cases, this high value confirms that the SOD1 structure is correctly modeled and stable; the issue is strictly a lack of peptide affinity.
Why does it fail to bind? Despite having numerous Valines (V) and Glycines (G), this peptide appears to be excessively hydrophobic and prone to self-folding. Instead of targeting the SOD1 surface, the peptide prefers to interact with itself, remaining “afloat” in the solvent without engaging the target.
After a comprehensive analysis using PepMLM for generation and AlphaFold3 for structural validation, two distinct strategies for stabilizing the mutant SOD1 emerge:
Candidate 2 (LLGSDGALQVGS) - The Structural Lead: > With the lowest perplexity score (14.65) and a superior ipTM of 0.54, this peptide stands out as the most structurally stable binder. Its aliphatic composition allows it to dock firmly against the protein’s core, acting as a reliable “patch” for the hydrophobic vulnerability created by the A4V mutation. 💎
Candidate 3 (SGVAVLCSDGQG) - The “Dark Horse” Candidate: > While its global confidence metrics are lower, high-resolution inspection reveals a fascinating “allosteric” mechanism. Candidate 3 demonstrates a sophisticated hydrogen-bond network that specifically “clamps” onto disordered surface loops. By inmovilizing these flexible regions near the N-terminus, it could provide a unique form of protection against the unfolding process that leads to toxic aggregation. 💎
Final Recommendation: > While Candidate 2 is the primary choice for advancement toward therapy due to its overall stability, Candidate 3 warrants further investigation. Its ability to “freeze” specific protein loops offers a complementary approach to traditional binding, potentially providing a more nuanced way to rescue the native fold of the SOD1-A4V enzyme.
The known binder presentes ipTM = 0.33 pTM = 0.79 clearly not a better choice 👎 👎 👎
NOTE: During the interaction with Gemini, the following suggestion was received: “SOD1 is a metalloenzyme, meaning it requires Copper (Cu) and Zinc (Zn) to be stable. If you want an ultra-precise model, you could add this.” That is to say, adding a third element (ligand) in AlphaFold using their specific SMILES strings or chemical identifiers. While the current model focuses on the protein-peptide interface, including these metallic cofactors would better simulate the native, stabilized state of the SOD1 enzyme.
PeptiVerse
Candidate 4 (AVGVCGVAVLGN) presents a contradictory profile. While PeptiVerse predicts the highest binding affinity of the set (6.651 $pKd$), this contradicts the AlphaFold3 structural model, which showed no physical contact with the enzyme (ipTM 0.49).This discrepancy is likely explained by the peptide’s high hydrophobicity (GRAVY: 1.83). Such extreme hydrophobicity often leads to non-specific interactions or self-aggregation rather than targeted docking at the A4V site. Furthermore, its hemolysis probability (0.132) is significantly higher than other candidates, making it a less safe therapeutic option.
A comparison between structural modeling and pharmacological prediction reveals a compelling trade-off. Candidate 2 (LLGSDGALQVGS) maintains the highest structural confidence (ipTM 0.54), but Candidate 3 (SGVAVLCSDGQG) shows a much stronger predicted binding affinity in PeptiVerse (6.242 $pKd$ vs 4.502 $pKd$). Visually, this is supported by the dense hydrogen-bond network observed in the AlphaFold3 close-up, where Candidate 3 effectively “clamps” onto the surface loops. Both peptides show ideal therapeutic profiles with maximum solubility and negligible hemolysis probability, confirming that PepMLM-generated sequences successfully avoid the toxic traits of highly hydrophobic non-binders like Candidate 4.
PeptiVerse Evaluation: Candidate 3 (SGVAVLCSDGQG)Binding Affinity: 6.242 ($pKd$). This is significantly higher than Candidate 2 (4.502). In logarithmic terms, this represents a much stronger predicted affinity for the target.Solubility: 1.000. Like Candidate 2, it is predicted to be perfectly soluble.Hemolysis: 0.022. Even lower than Candidate 2, making it exceptionally safe for systemic use.Net Charge: -1.55. Its slightly more negative charge might contribute to its better solubility and specific interaction with the mutant site. Interestingly, Candidate 3 (SGVAVLCSDGQG) emerges as a superior pharmacological lead.
Optimized Peptides with moPPIt
The peptides generated by moPPIt represent a significant shift from “plausible sampling” to “precision engineering.” Compared to the PepMLM candidates, several key differences emerge:
- Chemical Diversity and Functional Groups: The moPPIt sequences incorporate a wider variety of amino acids, such as Cysteine (C), Tyrosine (Y), and Phenylalanine (F). While the PepMLM leads were primarily aliphatic or polar (rich in L, V, S, D, G), the presence of Cysteine in the moPPIt leads allows for potential disulfide bond formation, which can stabilize the peptide’s conformation and enhance its “clamping” effect on the SOD1 surface.
- Targeted Structural Anchoring: Unlike the stochastic nature of PepMLM, which sampled sequences that could theoretically bind anywhere, moPPIt was guided to specific residues near the A4V mutation site. This targeted approach results in sequences that are chemically optimized to interact with the specific structural pocket destabilized by the mutation.
- Pre-optimized Therapeutic Metrics: By incorporating solubility and hemolysis guidance during the generation process, moPPIt avoids the pitfalls of extreme hydrophobicity seen in some PepMLM candidates (like Candidate 4, which had a GRAVY score of 1.83). This ensures that the generated sequences are not just good binders but also safe pharmacological leads.
Pre-Clinical Evaluation Strategy: Before advancing these moPPIt candidates to clinical trials, they must be validated through a multi-step process:Structural Validation (In Silico): Molecular Dynamics (MD) Simulations: Static models like AlphaFold3 are insufficient. MD simulations are required to evaluate the binding residence time and ensure the peptide remains docked at the A4V site under physiological fluctuations. Biochemical Assays (In Vitro): Thioflavin T (ThT) Fibrillization Assay: This is the most critical functional test. It determines if the peptide can successfully inhibit the aggregation of mutant SOD1 into toxic fibrils. Surface Plasmon Resonance (SPR): This provides an accurate measurement of the Dissociation Constant ($K_d$) and binding kinetics (on/off rates) to verify the affinity predicted by PeptiVerse. Biological & Safety Testing:Proteolytic Stability: Since these are peptides, they must be tested for resistance to serum proteases to ensure a sufficient half-life in the human body. Cellular Toxicity: The leads must be tested on motor neuron cultures expressing the A4V mutation to confirm they reduce cellular stress and improve neuron survival without inducing off-target toxicity.
Note on AI Collaboration:
The technical responses and structural analyses presented in this work were developed with the assistance of Gemini, an artificial intelligence model by Google. Gemini provided the initial drafts and technical frameworks based on the raw data from AlphaFold3, PepMLM, PeptiVerse, and moPPIt. The final review, polishing, and scientific validation were performed by the student to ensure accuracy and alignment with the course objectives.
Part C: Final Project: L-Protein Mutants
Analysis of Clustal Omega Alignment
- Soluble Region (Residues 1–40)
This region is critical for DnaJ chaperone interaction.
Highly Variable (Ideal for Mutation):
- Positions 1–6: The start of the protein shows significant variation (METRFP vs METQSP vs MEIRFP). Position 4 is particularly flexible.
- Positions 15–19: This loop varies between STNRR, STNRF, and STNRY. Mutating these could alter the binding surface for DnaJ.
Conserved (Avoid Mutating):
- Positions 21–25 (PFKHE): These residues are almost identical across all sequences, suggesting they are structurally vital.
- Positions 30–38 (RRQQRSST): This motif is highly conserved.
- Transmembrane Region (Residues 41–75)
This region integrates into the membrane to form pores.
Variable (Ideal for Mutation):
- Position 45: Changes between F (Phenylalanine) and C (Cysteine).
- Position 73: Varies between Q (Glutamine) and R (Arginine). Adding a charge here could affect how the protein sits in the membrane.
Conserved (Avoid Mutating):
- Positions 48–60 (LAIFLSKFTNQLL): This hydrophobic core is very consistent, as it must maintain a specific shape to span the lipid bilayer.
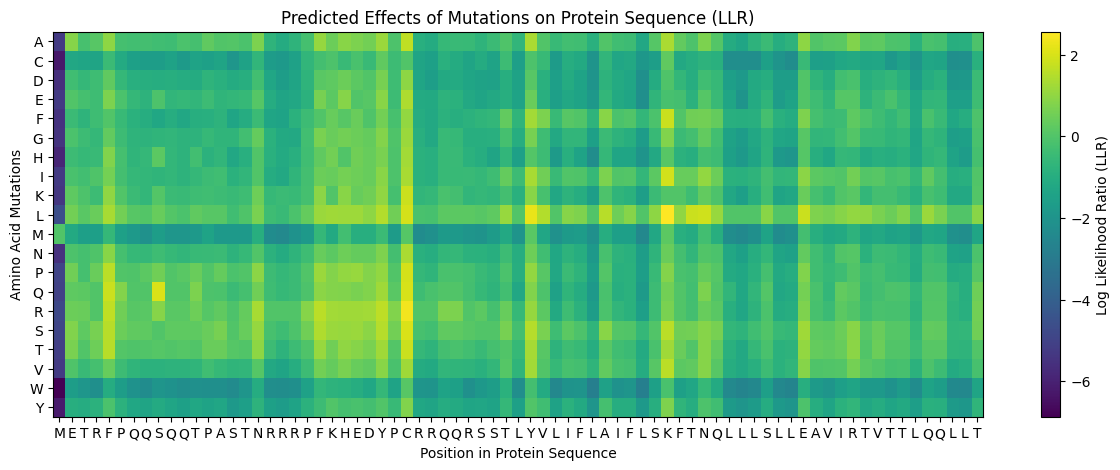
According to the graph, in general, we should avoid the aminoacids Cysteine, Methionine, and Tryptophan. Residue 4 and residues 21-28 seem good options to mutate.
Looking at the excel with experimental data, there is a clear “functional window” for engineering between residues 13 and 31 of the soluble domain. In this region, multiple mutations—such as R18G, R20W, and K23E—maintain a Lysis score of 1, demonstrating that this domain is structurally flexible and can tolerate amino acid substitutions without losing functional integrity. In contrast, the N-terminal start (residues 1–11) and the transmembrane core (residues 48–60) are highly sensitive, where most mutations result in Lysis 0 due to the disruption of protein production or pore-forming capability. Therefore, our engineering strategy focuses on introducing mutations within the residue 13–31 range to optimize DnaJ-independent folding and protein expression while preserving the essential lytic activity of the phage.
Functional Robustness in the TM Domain: * Between residues 38 and 75, there are numerous “safe” substitutions, such as T49S, A63V, and T69S, all of which maintain full lysis activity. This suggests that while the hélice must span the membrane, it can tolerate many conservative amino acid changes without losing its pore-forming ability.
The “Lethal” Exceptions: Even within this functional window, there are critical “black holes” where any change causes failure. For example, position 49 is highly sensitive; while S49L works, several other mutations at this exact spot lead to 0 lysis.
Position 60 is a “dead zone”: L60P, L60V, and L60Q all result in a total loss of function.
The Soluble Domain (Residues 1–40)
This region interacts with the host chaperone DnaJ. Highly Sensitive Sites (Lysis = 0): * Position 1 (M1I, M1T): Any change to the start codon abolishes protein production and lysis. Position 3 (T3I, T3S): Mutations here result in a total loss of function. Position 33 (Q33H): Changing Glutamine to Histidine at this position stops lysis. Tolerant Sites (Lysis = 1): Position 18 (R18G, R18I): The protein remains functional, suggesting this part of the soluble domain is flexible. Position 20 (R20W, R20L): These substitutions are well-tolerated. Position 23 (K23E): This site is resilient to change.
Proposed Mutations for MS2 L-Protein Engineering
R18G + R20W + K23E: Combines three sites proven to be functional (Lysis 1) in the lab data. By changing these three residues simultaneously, we drastically alter the electrostatic surface of the N-terminal domain to ensure DnaJ independence while maintaining high protein levels.
S15A + R19S + Q32E: Targets the highly variable “loop” residues identified in ClustalOmega. Replacing these with smaller or differently charged residues (S to A, R to S, Q to E) aims to create a “stealth” soluble domain that fails to bind the host’s mutated DnaJ chaperone.
F45A + A63V: Combines two experimentally validated “safe” sites in the lysis-active region. This combination aims to stabilize the hydrophobic hélice (A63V) while testing if the removal of the bulky Phenylalanine (F45A) facilitates faster pore assembly.
T69S + L73R: Uses a proven functional mutation (T69S) paired with an evolutionary change seen in Emesvirus (L73R). The goal is to optimize the C-terminal “anchor” to improve membrane penetration and accelerate bacterial killing.
R18I + T75S: Combines a high-expression soluble mutation (R18I) with a conservative C-terminal tail modification. This variant is designed to test if increasing the initial stability of the protein translates into more efficient processing at the membrane interface.
Week 06 HW: Genetic Circuits Part I: Assembly Technologies
Part A. DNA Assembly
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity DNA Polymerase:
A Pfu-like enzyme fused to a dsDNA-binding domain (Sso7d). This increases processivity and ensures an error rate 50 times lower than Taq polymerase.
5X Phusion HF Buffer (including $MgCl_2$): Maintains optimal pH and provides Magnesium ions, which act as essential cofactors for the polymerase to catalyze the addition of dNTPs.
dNTPs (Deoxynucleotide Triphosphates): The molecular “bricks” (dATP, dTTP, dCTP, dGTP) used to synthesize the new DNA strand.
Stabilizers: Often including glycerol or mild detergents to maintain enzyme stability through repeated thermal cycling.
- What are some factors that determine primer annealing temperature during PCR?
The annealing temperature is usually calculated as $T_m - 5^\circ\text{C}$. Key factors include:GC Content: G-C pairs have three hydrogen bonds (compared to two in A-T), requiring more thermal energy to denature. Higher GC content increases $T_m$.Primer Length: Longer primers have more total hydrogen bonds, leading to a higher melting temperature.Salt Concentration (Cations): $Na+$ and $Mg{2+}$ ions in the buffer stabilize the DNA double helix by neutralizing the negative charges of the phosphate backbone.Primer Concentration: Higher concentrations can slightly shift the kinetics of hybridization.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure success in a Gibson Cloning reaction, you must verify the following:
- Overlapping Ends: Adjacent fragments must share 15–40 bp of identical sequences at their ends. This is achieved by designing PCR primers with “overhangs” that match the neighboring fragment.
- DNA Purity: You must remove the original template (via DpnI digestion) and residual primers to prevent non-specific products.
- Correct Concentration: Fragments should be added in specific molar ratios (e.g., 1:2 or 1:3 vector-to-insert) to optimize assembly efficiency.
- How does the plasmid DNA enter the E. coli cells during transformation?
During Chemical Transformation (using CaCl_2):Neutralization: Calcium ions neutralize the negative charges of both the DNA phosphate backbone and the cell membrane’s phospholipids, allowing them to come into close contact.Heat Shock: A sudden increase to 42°C creates a thermal gradient that generates temporary pores in the plasma membrane.Entry: The pressure difference and thermal motion “push” the DNA into the cytoplasm before the cells are moved to a recovery medium to heal the membrane.
- Describe another assembly method in detail (such as Golden Gate Assembly)
- Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a molecular cloning method that utilizes Type IIS Restriction Enzymes (such as BsaI or BsmBI) and T4 DNA Ligase. Unlike standard enzymes, Type IIS enzymes cut outside of their recognition sites, allowing for the creation of custom, non-palindromic “sticky ends” (overhangs). The reaction is performed in a “one-pot” format, where digestion and ligation occur simultaneously; because the ligation product lacks the original restriction site, it cannot be re-digested, driving the reaction toward the final assembly. This method is scarless and highly efficient, enabling the directional assembly of 10+ fragments in a single step. It is the gold standard for creating complex genetic circuits and modular libraries in Synthetic Biology.
Part B. Asimov Kernel
I access Asimov Kernel with the account ¨barias1@alumni.usfq.edu.ec¨. You can find all the work I did under the Repository ¨Karina Campos Quito¨.
The first construct (I build it asking Gemini the sequence) did not work:
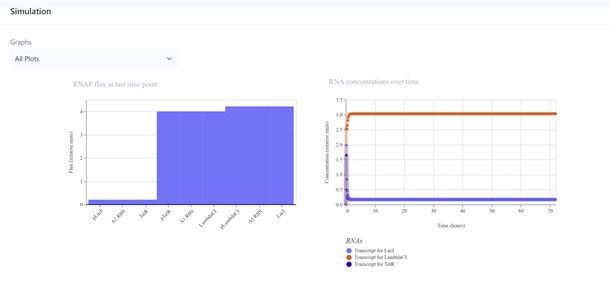
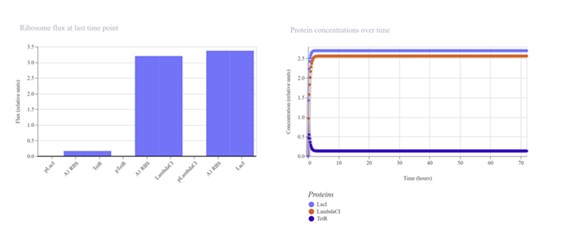
Second construct (replicating the Repressilator Construct found in the Bacterial Demos repository):
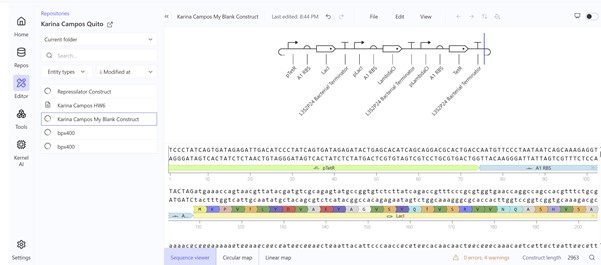
Week 07 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
- What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits operate like a light switch (0 or 1). IANNs, however, behave like a signal processor, offering several critical advantages:
Analog vs. Digital Processing. Boolean circuits only detect if a signal is “present” or “absent.” IANNs process signals analytically, so they can distinguish between low, medium, and high concentrations. This allows the cell to respond to gradients, which is much closer to how natural biological systems actually function.
Multivariable Pattern Classification. A Boolean circuit (using AND/OR gates) becomes incredibly complex and “brittle” as you add more inputs. By using a neural network architecture, IANNs can integrate multiple signals simultaneously (e.g., 5 different microRNAs). Instead of a simple “yes/no” gate, the IANN creates a complex decision boundary. This allows a cell to identify a specific state (like a cancer cell) with much higher precision, filtering out false positives that a simple Boolean circuit would miss.
Programmable “Weights” (Tunability). In a traditional circuit, if you want to change the behavior, you often have to re-engineer the entire genetic architecture. IANNs allow you to tune behavior simply by adjusting the weights of the connections (e.g., changing an RBS strength or a protein’s binding affinity). This makes the system modular and reprogrammable without changing the basic “wiring” of the circuit, allowing the same biological “brain” to be adapted for different tasks.
Robustness to Biological Noise. In Biology is “noisy”—molecule levels fluctuate randomly. Boolean circuits are sensitive to this noise and can “misfire” easily. IANN Advantage: By using ReLU activation functions and Sequestron mechanisms (molecular sequestration), IANNs act as filters. They can ignore small fluctuations (noise) and only “fire” a response when the weighted sum of signals is clear and consistent.
Multi-layer Composition (Deep Logic). IANN Advantage: IANNs are inherently multi-layer. This allows for sophisticated behaviors like Bandpass filters (activation only within a specific middle range), which are extremely difficult to achieve with pure Boolean logic. This “layered” capability allows biological computation to be much deeper and more “intelligent.”
- Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
In my HTGAA Final Project, I am exploring the use of an Intracellular Artificial Neural Network (IANN) as a sophisticated control strategy for a system designed for autonomous cell lysis. The core objective is to ensure that E. coli cells only undergo self-destruction and release their contents when they have reached a specific “Peak Harvest” state. To achieve this, the IANN acts as a biological classifier that integrates three distinct analog inputs: temperature-sensitive riboswitches to align with the fermentation phase, phosphate sensors to detect nutrient depletion, and membrane-tension riboswitches that signal high internal polymer accumulation.
- Input 1: Thermal Stress (Temperature). Using riboswitches that respond to temperature shifts. This ensures the lysis “arm” is only primed during the specific thermal phase of the industrial fermentation.
- Input 2: Phosphate Levels. A sensor for low phosphate (a common signal for the end of the growth phase), ensuring the bacteria don’t explode while they are still actively replicating.
- Input 3: Membrane Tension / Stress. Riboswitches or promoters that sense the physical stretching of the cell membrane or metabolic stress caused by high PHA/PHB (polymer) accumulation.
- The output: activation of the lysis cassette
The primary advantage of using an IANN over a simple Boolean “AND” gate is its ability to perform a weighted sum of these signals. By tuning the “weights” of the network—specifically the Translation Initiation Rate (TIR) of the RBS for each sensor—I can program the cell to ignore minor “leakiness” or noise from a single sensor. This ensures that the lysis cassette (SRRz) only triggers when the combined mathematical score of all three inputs crosses a precise threshold, preventing the premature loss of the batch.
However, implementing an IANN for this goal presents significant engineering challenges. The most critical limitation is the metabolic burden; producing multiple repressors and “decoy” binding sites to maintain the network’s logic consumes ATP and ribosomes that would otherwise be used for bioplastic synthesis. Furthermore, maintaining orthogonality among multiple sensors to avoid cross-talk is complex. While a simpler circuit might be more efficient, the IANN offers a level of programmable robustness that could be vital for scaling Bioplastix (the startup that would incorporate the auto-lysis strategy in its Bioprocess) to industrial-level bioreactors where environmental conditions are constantly fluctuating and are not homogeneous.
- Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
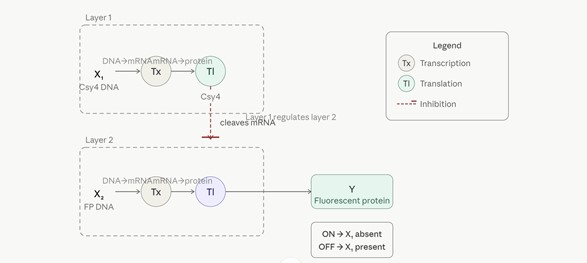
Intracellular Multilayer Perceptron — Layer 1 endoribonuclease regulates Layer 2 fluorescent protein output
The diagram depicts a two-layer intracellular artificial neural network (IANN) built on post-transcriptional regulation.
Layer 1 receives X₁, a DNA input encoding the Csy4 endoribonuclease. X₁ undergoes transcription (Tx) to produce mRNA, which is then translated (Tl) into the Csy4 protein — the output of Layer 1.
Layer 2 receives X₂, a DNA input encoding a fluorescent protein (FP). X₂ is likewise transcribed (Tx) into mRNA. However, translation (Tl) of this mRNA into the fluorescent protein is regulated by the Csy4 endoribonuclease produced in Layer 1: when Csy4 is present, it cleaves the fluorescent protein mRNA, preventing its translation.
Output Y (fluorescent protein) therefore follows AND-NOT logic:
• ON when X₁ is absent — no Csy4 is produced, the FP mRNA is translated normally.
• OFF when X₁ is present — Csy4 cleaves the FP mRNA, blocking translation.
Layer 1 thus acts as a regulatory gate over Layer 2, with the endoribonuclease serving as the molecular signal passed between layers — analogous to a weighted connection in a classical artificial neural network.
Part 2: Fungal Materials
- What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are primarily made from mycelium, the underground root-like network of a fungus. This mycelium acts as a natural “glue” that can bind agricultural waste into solid structures.
Mushroom Packaging: Companies like Ecovative grow mycelium around husks or stalks in molds to replace Styrofoam. It is used for shipping everything from electronics to wine.
Mycoleather: Textiles like Mylo or Reishi mimic the look and feel of animal leather. High-end fashion brands are using it for bags and garments as a sustainable alternative.
Fungal Bricks and Insulation: Experimental architecture uses mycelium blocks for their natural fire resistance and acoustic insulation properties.
Acoustic Panels: Mycelium-based tiles are used in interior design to absorb sound in offices or studios.
- Pros: They are biodegradable, carbon-negative (they sequester carbon as they grow), fire-resistant, and non-toxic. Production requires very little energy compared to plastic or leather tanning.
- Cons: They are often hydrophilic (absorb water), which can lead to rot if not properly coated. They generally have lower tensile strength than synthetic plastics or traditional leather and can vary in consistency.
It is crucial to clarify that mycelium is not a species or a family, but an anatomical part of a fungus. It consists of a dense, branching network of thread-like filaments called hyphae. In the field of biomaterials, this network acts as a natural binder to create structural composites.
Key Filamentous Fungi (Mycelium-based):
- Ganoderma lucidum (Reishi): Widely used in the production of mycoleather. Its hyphae grow extremely dense, creating a flexible and durable material that serves as a sustainable alternative to animal hides.
- Pleurotus ostreatus (Oyster Mushroom): The standard for bio-packaging. It is a fast-growing, aggressive colonizer that can quickly turn agricultural waste into molded shapes, replacing expanded polystyrene (Styrofoam).
- Trametes versicolor (Turkey Tail): Frequently studied for bioremediation. It produces powerful extracellular enzymes (laccases) capable of breaking down complex chemical toxins and dyes in environmental applications.
Unicellular Fungi (Yeasts): Beyond complex mycelial networks, yeasts represent a vital category of unicellular fungi.
Saccharomyces cerevisiae: This is perhaps the most industrially significant fungus. Beyond its traditional roles in baking and brewing, it is a primary “chassis” in synthetic biology for the large-scale production of bioethanol and high-value recombinant proteins (like insulin). Its well-understood genetics make it an ideal eukaryotic model for metabolic engineering.
- What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
If we apply synthetic biology to fungi, we can transform these materials from “passive” objects into “living” materials:
- Self-Healing Materials: Engineering fungi to remain dormant within a structure and “wake up” to grow and seal cracks when moisture is detected.
- Sensing and Reporting: Modifying fungi to change color or glow in the presence of environmental toxins (like heavy metals in soil).
- Enhanced Secretion: secrete specific enzymes.
Advantages of Fungi vs. Bacteria:
While E. coli is the workhorse of synthetic biology, fungi offer unique engineering advantages:
- Macro-scale Structure: Unlike bacteria, which are unicellular, fungi form massive, interconnected multicellular networks. This allows for the creation of large-scale physical materials that hold their shape.
- Eukaryotic Processing: Fungi are eukaryotes. They can perform complex post-translational modifications on proteins that bacteria cannot, making them better for producing specialized enzymes or human-like proteins.
- Superior Secretion: Fungi are natural “secretory machines.” They can pump out vast quantities of proteins and metabolites into their environment, which is much more efficient for industrial harvesting than lysing bacteria.
- Environmental Resilience: Fungi thrive in harsh, acidic, or low-moisture environments where most lab bacteria would die. This makes them ideal for “out-of-lab” applications like bioremediation or outdoor construction.
Week 09 HW: Cell-Free Systems
Part 1: General and Lecturer-Specific Questions
General homework questions
- Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The fundamental advantage of cell-free protein synthesis (CFPS) lies in the removal of the cellular membrane, which effectively transforms a “black box” biological process into an open, accessible engineering platform. By eliminating the cell wall, researchers gain unprecedented flexibility and direct control over experimental variables; the reaction environment can be precisely manipulated by adding non-natural amino acids, specific chaperones, or tailored energy sources without the constraints of cellular transport or homeostasis. Furthermore, CFPS decouples protein production from host viability, allowing for the synthesis of highly cytotoxic proteins that would otherwise trigger cell death and halt production in traditional in vivo systems.Beyond throughput, the “open” nature of the system significantly enhances real-time monitoring and process optimization. Unlike the opaque interior of a living E. coli cell, a cell-free reactor allows for millisecond-scale sampling and mid-process adjustments of critical concentrations—such as magnesium levels or pH—to maximize yields. Perhaps most importantly for rapid prototyping, CFPS enables a drastically accelerated iteration cycle. By bypassing time-consuming steps like transformation, plating, and overnight culturing, researchers can transition from a linear DNA template (such as a PCR product) to a functional protein in just a few hours, representing a paradigm shift in the speed of biological design.
Case A: Production of Cytotoxic Proteins
Many useful proteins, such as antimicrobial peptides (AMPs) or certain lytic enzymes (like the ones you might use in Bioplastix), kill the host cell as soon as they are expressed. CFPS allows you to synthesize these “suicide” proteins because the system lacks the physiological targets that the toxins would otherwise destroy.
Case B: Incorporation of Non-Standard Amino Acids (nsAAs)
If you want to create a protein with expanded chemical properties (e.g., for site-specific labeling, “click” chemistry, or enhanced stability), CFPS is superior. In a cell, it is extremely difficult to “force” the machinery to use a synthetic amino acid without interfering with the cell’s own survival. In a cell-free extract, you can simply “starve” the reaction of a natural amino acid and flood it with the synthetic version.
- Describe the main components of a cell-free expression system and explain the role of each component.
- The Biological Extract (The Machinery): usually obtained by lysing cells (such as E. coli, wheat germ, or rabbit reticulocytes) and removing the cell wall and genomic DNA. It provides the Ribosomes for translation, RNA Polymerase for transcription, and various tRNAs, aminoacyl-tRNA synthetases, and initiation/elongation factors. Without the extract, there is no hardware to read the genetic code.
- The DNA Template (The Instructions): Unlike in vivo systems that require circular plasmids, cell-free systems can often use linear DNA (like PCR products). It provides the genetic sequence of the protein of interest. It must contain specific regulatory elements that the extract’s machinery can recognize, such as a T7 or endogenous promoter, a Ribosome Binding Site (RBS), and a terminator.
- Energy Regeneration System (The Fuel): Protein synthesis is energetically expensive. Since the cell’s natural mitochondria or metabolic pathways are no longer intact, we must provide an external energy source. It consists of NTPs (ATP, GTP, UTP, CTP) which act as the direct building blocks for mRNA and the energy source for the ribosome. It also includes an energy-rich secondary substrate (like phosphoenolpyruvate (PEP) or creatine phosphate) and a corresponding kinase to “recharge” the ATP as it is consumed.
- Small Molecules and Buffers (The Environment): A precise chemical environment is required to keep the enzymes stable and active. Amino Acids: The raw building blocks used to assemble the protein chain. Magnesium and Potassium salts: Critical cofactors for ribosome assembly and stability. Buffers (e.g., HEPES): To maintain a stable pH, as the metabolic byproducts of the reaction can quickly acidify the mixture.
- Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis requires a massive amount of energy at every stage. For every single amino acid added to a polypeptide chain, four high-energy phosphate bonds are consumed: two during the “charging” of the tRNA with an amino acid and two during the translation elongation steps (GTP hydrolysis).In a static cell-free batch, the initial supply of ATP would be depleted almost instantly. Furthermore, the accumulation of Inorganic Phosphate —a byproduct of ATP hydrolysis—can inhibit the reaction by chelating magnesium ions, which are essential for ribosome stability. Therefore, a regeneration system is critical not just to keep the “fuel tank” full, but to maintain a chemical environment that isn’t poisoned by its own waste.
To ensure a steady supply of ATP in your HTGAA experiments, two strategies are:
The Creatine Phosphate / Creatine Kinase (CP/CK) System. This is the most common “plug-and-play” method for cell-free experiments. You add Creatine Phosphate (the high-energy substrate) and the enzyme Creatine Kinase to the mixture. Every time an ATP molecule is used and becomes ADP, the Creatine Kinase transfers a phosphate group from the Creatine Phosphate directly back to the ADP, “recharging” it into ATP instantly. It is highly efficient and maintains a very high [ATP]/[ADP] ratio, which is vital for high-yield protein production.
PURE System or Secondary Carbon Sources. If you are using an E. coli extract, you can utilize the cell’s own residual glycolytic enzymes. You provide a secondary energy source like Phosphoenolpyruvate (PEP) or Glucose-6-Phosphate. The enzymes already present in the extract (like pyruvate kinase) process these substrates to regenerate ATP. The “Continuous” Approach: For very long experiments, you can use a Dialysis System (Continuous Exchange Cell-Free - CECF). The reaction happens inside a dialysis membrane submerged in a large reservoir of buffer, energy substrates, and nucleotides. Fresh fuel diffuses in, and inhibitory byproducts like inorganic phosphate diffuse out, allowing the reaction to run for days.
- Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
The choice between prokaryotic and eukaryotic cell-free expression systems is primarily dictated by the complexity of the protein and the requirement for post-translational modifications (PTMs). Prokaryotic systems, typically based on E. coli S30 extracts, are the “workhorses” of the field due to their high protein yields (often reaching mg/mL levels), low cost, and rapid synthesis rates. They utilize 70S ribosomes and simpler energetic pathways but lack the machinery for complex folding or PTMs like glycosylation. In contrast, eukaryotic systems—such as Wheat Germ Extract (WGE) or Rabbit Reticulocyte Lysate (RRL)—employ 80S ribosomes and offer a more sophisticated folding environment. While they generally produce lower total yields and are more expensive to prepare, they are indispensable for synthesizing large, multi-domain eukaryotic proteins that require authentic folding or specific modifications like disulfide bond formation, phosphorylation, or lipidation.
70S and 80S Ribosomes: These are the molecular machines responsible for protein synthesis. The “S” (Svedberg unit) indicates their size and sedimentation rate during centrifugation.
70S Ribosomes: Found in prokaryotes (bacteria) and organelles like mitochondria. They are smaller and simpler in structure.
80S Ribosomes: Found in eukaryotes (animals, plants, fungi, and humans). They are larger, more complex, and capable of more sophisticated regulation of translation.
PTMs are chemical changes made to a protein after it has been synthesized by the ribosome. These modifications are essential for the protein to become biologically functional. Examples: Glycosylation (adding sugars), phosphorylation (adding phosphate groups), and disulfide bond formation.
Importance: While bacteria are efficient at making simple proteins, they lack the machinery for most complex eukaryotic PTMs, which is why human proteins often require eukaryotic expression systems to function correctly.
For a prokaryotic system (E. coli), an ideal protein to produce would be Green Fluorescent Protein (GFP) or a bacterial enzyme like Pyruvate Kinase. GFP is a robust, relatively small protein that folds efficiently in the bacterial cytoplasm without needing PTMs. Using an E. coli cell-free extract allows for massive production in just a few hours, making it perfect for high-throughput screening of genetic circuits or biosensors where speed and quantity are prioritized over structural complexity.
For a eukaryotic system (such as Rabbit Reticulocyte Lysate), a strategic choice would be a human therapeutic protein like Erythropoietin (EPO) or a complex Single-Chain Variable Fragment (scFv) antibody. EPO requires extensive and specific glycosylation to be biologically active and stable in the human body—a process that E. coli machinery cannot perform. By using a mammalian-derived cell-free system (often supplemented with microsomal membranes), researchers can ensure the protein is correctly glycosylated and folded with the necessary disulfide bridges, providing a functional product that closely mimics its natural human counterpart.
- How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Use a cell-free system supplemented with liposomes or nanodiscs (synthetic lipid bilayers). This allows the membrane protein to integrate into a stable environment as it is synthesized.
Challenge 1: Hydrophobicity and Aggregation. Membrane proteins often precipitate in aqueous extracts. Solution: Add detergents (at sub-CMC levels) or chaperones to maintain solubility.
Challenge 2: Lack of Energy/Substrates. Solution: Use a Continuous-Exchange (CECF) system to provide a steady supply of energy and remove inhibitory byproducts, ensuring longer reaction times for difficult folding.
- Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
- DNA Template Quality: Verify DNA purity (A260/280 ratio) and ensure the promoter/RBS sequences are optimized for the specific extract used.
- Resource Exhaustion: Increase the concentration of the energy regeneration system (e.g., Creatine Phosphate) or optimize the Magnesiu concentration.
- Protein Degradation: Add Protease Inhibitors to the extract or use a specialized strain (like BL21 E. coli) that is deficient in endogenous nucleases and proteases.
Homework question from Kate Adamala
Design of a Synthetic Minimal Sentinel Cell for Autolysis Testing
- Function and Operation: A “Self-Destructing Sentinel” that replicates the dual-sensing logic. It is designed to sequester a payload and release it only when two specific environmental conditions are met: phosphate starvation and a temperature shift. Input: 1. Low concentration of inorganic phosphate. 2. Thermal shift (e.g., transition from 30°C to 37°C).Output: Expression of the Lambda phage lysis cassette (SRRz) leading to membrane rupture and release of internal contents.
- Realization and Feasibility: Cell-free Tx/Tl alone: Without encapsulation, the “lysis” has no physical meaning. You could produce the proteins, but you wouldn’t be able to measure the structural failure or the release kinetics that your industrial process needs. Genetically modified natural cell: While this is an end goal for E. coli, testing it first in a minimal cell is safer. In a natural cell, phosphate starvation triggers many “survival” pathways that might interfere with your promoter’s strength. The minimal cell provides a “clean” signal-to-noise ratio.
- Components and Encapsulation: Membrane: A lipid bilayer composed of POPC and DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine). DOPE adds curvature tension, making the membrane more sensitive to the “holes” created by the holins (S protein) of the SRRz cassette. Internal Content: Extract: E. coli S30 cell-free extract (Prokaryotic system). Machinery: Endogenous E. coli RNA polymerase and ribosomes. Small Molecules: NTPs, Amino Acids, and a buffer with initially high phosphate that will be “consumed” or diluted to trigger the circuit.
- Communication and System Origin: Bacterial (E. coli). The circuit uses the pPhoA promoter, which is native to E. coli. Using a bacterial cell-free extract ensures that the transcription factors (like PhoB) required for the pPhoA promoter to work are present. To simulate phosphate starvation, we can use Alpha-hemolysin pores. These allow phosphate to diffuse out of the minimal cell into a phosphate-free external buffer, triggering the internal sensor.
- Experimental Details: The minimal cell will encapsulate:Lipids: POPC and DOPE. Genes (The Circuit): pPhoA Promoter: Driving the first stage of the cascade. It responds to the lack of phosphate.Temperature-Sensitive Riboswitch: Placed upstream of the SRRz sequence. Even if pPhoA is active, the mRNA won’t be translated unless the temperature reaches the threshold (e.g., 37°C), which unfolds the riboswitch. SRRz Cassette: The “actuator” consisting of S (holin), R (endolysin), and Rz (spanin).
- Measuring Function: We will validate the design using: Phase-Contrast Microscopy to visually observe the “bursting” of the vesicles when phosphate is depleted and temperature is raised. Fluorescence Release Assay: We will co-encapsulate a large fluorescent protein (like mCherry). We will measure the increase in external fluorescence over time. Success Metric: If fluorescence remains internal at 30°C regardless of phosphate levels, but releases at 37°C under low phosphate, the logic is proven.
Homework question from Peter Nguyen
Proposal Pitch: The “Sentinel Bio-Textile”
One-sentence summary pitch: A smart, biodegradable textile infused with freeze-dried cell-free systems that acts as a “living” safety suit, detecting environmental pathogens and neutralizing them through the inducible secretion of antimicrobial peptides.
How will the idea work? The textile fibers are embedded with a freeze-dried E. coli cell-free extract containing the genetic instructions for a pathogen-sensing riboswitch and an actuator gene. When a specific pathogen (or a chemical marker of contamination) is detected, the dehydrated machinery is activated by ambient moisture or sweat, triggering the transcription and translation of the circuit. The system then produces and secretes Antimicrobial Peptides (AMPs) or lytic enzymes directly onto the fabric surface. This creates a localized, on-demand decontamination zone without the need for living, genetically modified organisms to survive on the wearer’s skin.
What societal challenge or market need will this address? This addresses the growing need for advanced Personal Protective Equipment (PPE) in healthcare and industrial settings, where traditional fabrics only provide a passive physical barrier. Currently, PPE often becomes a vector for cross-contamination; a bio-active textile that actively “cleans” itself or alerts the user to invisible threats would significantly reduce the spread of hospital-acquired infections and enhance worker safety in bio-hazardous environments.
Addressing limitations (Activation, Stability, One-time use) Activation: The freeze-dried components remain dormant and stable at room temperature until they come into contact with a specific trigger, such as a localized application of water or a “developer” spray.
Stability: By using lyoprotectants (like sucrose or trehalose) during the freeze-drying process, the enzymatic machinery is shielded from thermal degradation, allowing for a long shelf-life in standard warehouse conditions.
One-time use: While the reaction is currently a one-time “pulse,” we envision the textile as a modular patch system. Once the “biological fuse” has been spent, the functionalized patch can be swapped out or discarded, taking advantage of the biodegradable nature of the underlying Bioplastix material.
Homework question from Ally Huang
1. Provide background information that describes the space biology question or challenge you propose to address.
Space exploration faces a severe challenge regarding waste management and planetary protection. Disposing of single-use structural plastics or packaging on long-term missions leads to accumulation, hazardous debris, or irreversible contamination of pristine environments like Mars, where natural biodegradation cannot occur due to the absence of native microflora. Developing self-disintegrating or enzyme-recyclable materials is critical. Investigating how we can actively trigger the breakdown of bioplastics using cell-free biological systems in microgravity is both scientifically fascinating and essential for sustainable, zero-waste deep-space human habitation.
2. Molecular or Genetic Target (Max 30 words)
The genetic target is the synthetic gene encoding PETase (polyethylene terephthalate hydrolase), a highly efficient enzyme engineered to break down ester bonds in synthetic and organic biopolymers.
3. Relation to the Space Biology Challenge
The PETase enzyme directly addresses the space waste bottleneck by offering a non-mechanical, non-thermal method to dissolve plastic waste on demand. In resource-constrained environments like the International Space Station (ISS) or a Martian base, burning or chemically treating plastics is toxic and energy-intensive. By placing the PETase gene inside the freeze-dried BioBits® system, astronauts can simply add water to express the enzyme. This cell-free approach allows for the controlled, localized degradation of space-certified polymers into non-toxic, recyclable monomer components without requiring complex cellular maintenance or risking microbial leaks.
4. Hypothesis or Research Goal
Research Goal: To demonstrate that the BioBits® cell-free system can successfully transcribe and translate a functional PETase enzyme under microgravity conditions, and that this enzyme retains its catalytic activity to degrade target polymers.
Reasoning: Microgravity alters molecular diffusion and protein folding kinetics, which could impact cell-free translation efficiency or enzyme-substrate interactions. We hypothesize that freeze-dried cell-free matrices are robust enough to bypass cellular stress responses caused by space radiation and weightlessness. If the BioBits® system successfully expresses active PETase in space, it will validate cell-free biology as a reliable utility for autonomous, on-demand waste processing and material recycling during long-duration spaceflight, breaking the dependency on Earth-bound waste logistics.
5. Experimental Plan
The experiment will activate three BioBits® reactions in microgravity using the miniPCR® for temperature control (37 grades):
- Experimental: BioBits® + PETase DNA + target plastic film.
- Positive Control: BioBits® + Constitutive GFP DNA (ensuring translation works).
- Negative Control: BioBits® + PETase DNA (no plastic).
Data will be collected using the P51 Viewer to monitor GFP fluorescence. The plastic films will be returned to Earth to measure mass loss via spectrometry and scanning electron microscopy (SEM) to quantify degradation kinetics and surface erosion caused by the space-expressed enzyme.
Week 10 HW: Imaging & Measurement
Waters Part I — Molecular Weight
- Calculated Molecular Weight:
Based on the provided 246 amino acid sequence (including the LE-linker and 6xHis-tag), the theoretical molecular weight is 27,845.03 Da. However, considering the maturation of the eGFP chromophore (cyclization and oxidation of the Thr65-Tyr66-Gly67 triad), there is a loss of 18 Da (for $H_2O$ loss) and 2 Da (for oxidation/dehydrogenation) in some variants. Therefore, the expected intact mass observed in the LC-MS would be approximately 27,825–27,827 Da.
The chromophore is a cluster of atoms that absorbs light at a specific wavelength and either reflects it or emits it (fluorescence). It works by capturing the energy of an incoming photon, which “excites” an electron to a higher energy level. When that electron falls back down, it releases energy in the form of light.
- Calculate the molecular weight of the eGFP using the adjacent charge state approach:
- Determine z:Using the adjacent peaks at m/z 933.7148 (z_n) and 903.7140 (z_{n+1}), the calculated charge state z for the latter is 30.12, which rounds to an integer value of z = 30 (for the 933.7 peak).
- Determine MW:Using the formula MW = z \times (m/z) - (z \times 1.0078), the experimental molecular weight of the eGFP is calculated as 27,981.21 Da.3.
- Calculate Accuracy: Compared to the theoretical mature eGFP weight of approximately 27,826 Da, the measurement accuracy (error) is 0.55%. This slight discrepancy may be due to the specific calibration of the Xevo G3 or additional salt adducts (like Sodium) attached to the protein during ionization.
- Can you observe the charge state for the zoomed-in peak?
Yes. The charge state (z) can be observed by measuring the distance between the resolved isotopic peaks within the cluster. What is it? By calculating the difference (\Delta m/z) between adjacent isotopes (e.g., 1473.7959 - 1473.7428 = 0.0531), and applying the formula z = 1 /Delta(m/z), we find that z \approx 19. Why is this possible? This is possible because the Waters Xevo G3 has high mass resolution (30,000), which allows the instrument to “resolve” individual isotopes of a large protein. Without this high resolution, the isotopes would coalesce into a single broad peak, making it impossible to determine the charge state through isotopic spacing.
Waters Part II — Secondary/Tertiary structure
In its native state, a protein is tightly folded into a compact 3D structure. Many of its basic amino acid residues (which can accept protons) are buried inside the hydrophobic core, making them inaccessible for ionization. When a protein denatures (unfolds), it loses this structure and transitions into an extended “random coil” string, exposing all previously hidden basic sites to the solvent.
Effect on Charge State Distribution (CSD): Denatured Analysis (Top spectrum): Because the protein is unfolded, it can accept a much higher number of protons (H^+). This results in a high charge state (high z), which appears as a distribution of peaks at lower m/z values. The spectrum typically shows a broad, “bell-shaped” distribution of many peaks.Native Analysis (Bottom spectrum): Since the protein is compact and many sites are inaccessible, it carries fewer charges (low z). This shifts the signal to higher m/z values. The distribution is usually narrower and concentrated in a few peaks at the right end of the spectrum (e.g., around m/z 2500-2800 in Figure 2).
Waters Part III — Peptide Mapping - primary structure
- Lysines (K): 20
- Arginines (R): 6
- Total Cleavage Sites: 26
- [Theoretical pI: 5.90 / Mw (average mass): 28006.60 / Mw (monoisotopic mass): 27988.96]
The tryptic digestion of eGFP produces a specific set of peptides that act as a ‘fingerprint’ for the protein. By measuring the mass of these 27 peptides using the Waters BioAccord LC-MS, we can compare the experimental masses against the theoretical list from Expasy. This Peptide Mapping allows us to confirm the primary sequence (the exact order of amino acids) and ensure that the protein was synthesized correctly without mutations or unexpected modifications.
While the raw chromatogram (Figure 5a) displays approximately 27 data labels, the number of true, significant chromatographic peaks representing distinct peptides is actually fewer than the 27 predicted by the PeptideMass tool.
Technical Explanation:
At first glance, it may seem that the numbers match because the software automatically labels almost every “bump” or signal fluctuation. However, many of these are not individual tryptic peptides for the following reasons:
- Co-elution (The Primary Factor): Due to the short 6-minute run time, several peptides with similar chemical properties exit the column at the same time. This results in multiple different peptides being “hidden” under a single high-abundance peak (such as the peak at 4.87 min).
- Signal Intensity and Thresholds: Many of the 27 labels represent background noise or low-abundance signals (below the 10% relative abundance threshold). In a high-quality peptide map, only the peaks that stand out significantly from the baseline are considered valid peptide candidates.
- “Shoulders” and Peak Splitting: Sometimes a single peptide species can produce two labels if the peak is not perfectly symmetrical (a “shoulder”). These represent the same molecule, not two different predicted peptides.
- Invisible Fragments: Very small or highly hydrophilic peptides often fail to bind to the C18 column and elute immediately in the “void volume” at the start of the run, while very hydrophobic large fragments may remain stuck on the column, never appearing in the 6-minute window.
Therefore, the number of detected and resolved peptides is lower than the theoretical prediction. This discrepancy is a standard characteristic of LC-MS analysis, which is why we use mass spectrometry to “deconvolve” these peaks and identify the multiple co-eluting peptides within them.
Figure 5b.: The most abundant mass-to-charge ratio (m/z) for the peptide eluting at 2.78 min is 525.76712.Determine the charge state z: By observing the isotopic zoom-in, the spacing between the first and second isotopes is approximately 0.47 m/z (526.23 - 525.76). Using the formula z = 1 / \Delta(m/z), the charge state is determined to be z = 2.Calculate the mass of the singly charged form [M+H]^+: To find the mass of the singly charged ion, we use the relationship: M+H = (m/z \times z) - (z - 1).Calculation: (525.76712 \times 2) - 1.0078 = \mathbf{1050.52644, Da}. This corresponds to the neutral mass of the peptide plus one proton, which is also visible in the spectrum as a low-intensity peak at m/z 1050.52.
Question 6: Based on the theoretical masses from the PeptideMass tool, the peptide eluting at 2.78 min is identified as SAMPEGYVQER (Theoretical mass: 1050.5117 Da).
The mass accuracy of the measurement is calculated as follows:
Error (ppm) = [ |1050.5264 - 1050.5117| / 1050.5117 ] x 10^6 = 13.99 ppm.
This indicates high confidence in the identification, as it falls within the expected mass error range for a Q-Tof instrument.
Question 7: According to the Amino Acid Coverage Map (Figure 6), the percentage of the eGFP sequence confirmed by peptide mapping is 88%. This high coverage confirms the primary structure of the protein standard and ensures that the synthesized eGFP matches the expected genetic sequence.
Waters Part IV — Oligomers
Identification of KLH Oligomeric States using CDMS: In Figure 7, we utilize Charge Detection Mass Spectrometry (CDMS) to identify the distribution of high-molecular-weight KLH complexes. Based on the known mass of KLH subunits and their assembly patterns, we can identify the following species on the spectrum:
- 7FU Decamer: Corresponds to the peak at 4.013 MDa.
- 8FU Didecamer: Corresponds to the most abundant peak (the base peak) at 8.33 MDa.
- 8FU 3-Decamer: Corresponds to the peak at 12.67 MDa.
- 8FU 4-Decamer: This species is represented by the low-intensity signals emerging around the 16-17 MDa range.
Significance: CDMS is crucial for this analysis because KLH complexes are too large and heterogeneous for standard mass spectrometry. By measuring individual particles, CDMS provides a clear mass profile that confirms the presence of different oligomeric states (decameric assemblies) in the solution, which is vital for its use as an immunotherapeutic carrier.
Keyhole Limpet Hemocyanin (KLH): KLH is an exceptionally large, multi-subunit metalloprotein derived from the hemolymph of the giant keyhole limpet (Megathura crenulata). Due to its massive size and phylogenetic distance from mammals, it is highly immunogenic. In biotechnology, it is extensively used as a hapten carrier to significantly boost the immune response against small antigens in vaccine development.
Oligomeric State: This term describes the assembly of multiple individual protein subunits (monomers) into a single functional complex. Much like LEGO bricks snapping together, proteins can form specific structures such as decamers (10 subunits) or didecamers (20 subunits). Determining these states is critical for understanding a protein’s stability and biological activity.
Charge Detection Mass Spectrometry (CDMS): CDMS is a specialized form of mass spectrometry designed for the analysis of megadalton-sized particles. Unlike conventional MS, which measures the ensemble mass-to-charge ($m/z$) ratio, CDMS measures both the mass ($m$) and charge ($z$) of each particle individually. This unique capability allows it to resolve the mass of giant complexes, such as viruses and KLH assemblies, which would otherwise appear as unresolved signals in standard instruments.
Waters Part V — Did I make GFP?
| Property | Theoretical | Observed (Intact LC-MS) | PPM Mass Error |
|---|
| Molecular Weight (kDa) | 27.845 kDa | 27.825 - 27.827 kDa | ~14 ppm |
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Unfortunately I was unable to contribute to the collective artwork — I couldn’t follow the instructions in time. I did get to see the final result though (picture above).
I did manage to contribute to the artwork organized at SynBioBeta, however. Photo below.
I really enjoy collaborative art projects like this one — they help connect people from different parts of the world around a shared cause, and I find that especially meaningful for those of us who are committed listeners but may not always get to meet in person.
In terms of improvements, I would love to see art used more intentionally to carry a message of hope. Synthetic biology has the potential to help solve some of the biggest challenges humanity faces, and I think that story deserves to be told through the art we create. Looking at the individual and collective projects in HTGAA, most of them are fun and creative, but I feel they could carry a stronger, more purposeful message — one that not only celebrates what we do, but also inspires people outside our community to believe in what synthetic biology can become.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
- Each component’s role is in the cell-free reaction.
- E. coli Lysate (BL21 (DE3) Star): Provides the essential biological “hardware,” including ribosomes, tRNAs, and various translation factors; it specifically includes T7 RNA Polymerase to drive high-level transcription from T7 promoters.
Salts and Buffers:
- Potassium Glutamate: Acts as the primary potassium source and a major intracellular salt, which is critical for maintaining proper osmotic pressure and supporting protein-DNA interactions during transcription and translation.
- HEPES-KOH pH 7.5: Serves as a chemical buffering agent to maintain a stable physiological pH throughout the reaction, preventing acidification from metabolic byproducts.
- Magnesium Glutamate: Provides essential $Mg^{2+}$ ions that act as necessary cofactors for ribosome assembly and the enzymatic activity of polymerases.
- Potassium Phosphate (Monobasic & Dibasic): Maintains phosphate homeostasis and contributes to pH stability, while also providing inorganic phosphate for nucleotide recycling.
Energy and Nucleotide System:
- Ribose and Glucose: Serve as secondary energy substrates that can be metabolized by residual glycolytic enzymes in the lysate to regenerate ATP sustainably.
- AMP, CMP, GMP, UMP, and Guanine: These act as the fundamental precursors (nucleotides and bases) for mRNA synthesis during transcription and are necessary components for maintaining the energy charge of the system.
Translation Mix (Amino Acids):
- 17 Amino Acid Mix, Tyrosine, and Cysteine: Provide the raw building blocks required for the ribosome to assemble the polypeptide chain; Tyrosine and Cysteine are often added separately due to their lower solubility at neutral pH.
Additives and Backfill:
- Nicotinamide: Often acts as a cofactor or stabilizer to inhibit the degradation of essential metabolic intermediates like NAD+, thereby extending the reaction’s metabolic activity.
- Nuclease Free Water: Used to adjust the final volume (backfill) of the reaction while ensuring no contaminating enzymes degrade the DNA template or mRNA products.
- Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP mix is designed for high-speed, short-term bursts of protein synthesis by providing direct, high-energy building blocks like Nucleoside Triphosphates (NTPs) and a potent phosphagen (PEP) for immediate ATP regeneration. In contrast, the 20-hour NMP-Ribose-Glucose mix is optimized for long-term production and cost-efficiency; it utilizes cheaper precursors (Nucleoside Monophosphates) and a dual-sugar metabolic pathway (Ribose/Glucose) to regenerate energy slowly and steadily over several hours, preventing the rapid accumulation of inhibitory byproducts.
- Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can occur even if GMP is not explicitly included because the E. coli lysate contains residual metabolic enzymes (such as Phosphoribosyltransferases) that can utilize Guanine as a substrate. Through a “salvage pathway,” the system attaches a ribose-5-phosphate to the Guanine base to synthesize GMP. Once GMP is formed, kinases in the lysate further phosphorylate it into GDP and finally GTP, which is the actual nucleotide required by the RNA Polymerase to build the mRNA chain.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
- Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Functional Properties of Fluorescent Proteins
sfGFP (Superfolder GFP): This protein is engineered for extremely rapid folding and high stability, making it highly resistant to aggregation even when expressed at high rates in cell-free extracts.
mRFP1 (monomeric Red Fluorescent Protein): A key limitation is its relatively slow maturation time and lower photostability, which often results in a delayed fluorescence signal compared to green variants in short-term reactions.
mKO2 (monomeric Kusabira Orange 2): This protein features high pH sensitivity (acid sensitivity); its fluorescence can be significantly quenched if the cell-free reaction undergoes acidification due to metabolic byproduct accumulation.
mTurquoise2: Known for its exceptional quantum yield and brightness, it requires very precise folding conditions to achieve its maximum fluorescence intensity in a cyan readout.
mScarlet-I: This is a high-performance red protein that is notably oxygen-dependent for the final step of chromophore maturation, meaning insufficient aeration in the reaction vessel can limit its readout.
Electra2: Designed for enhanced photostability and rapid folding, this protein provides a very fast readout, though it can be sensitive to the ionic strength of the buffer (specifically magnesium levels) during the initial translation phase.
- Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mKO2 (monomeric Kusabira Orange 2)
Reagents to Adjust: HEPES-KOH (Buffer) and Potassium Phosphate.
Hypothesis: Increasing the concentration of HEPES-KOH and Potassium Phosphate in the master mix will improve the fluorescence of mKO2 by enhancing the system’s buffering capacity.
Expected Effect: In a long 36-hour incubation, the cell-free reaction typically produces organic acids as byproducts of glucose and ribose metabolism. Since mKO2 is acid-sensitive, a drop in pH would normally quench its signal mid-incubation. By strengthening the buffer, we can maintain the pH near 7.5 for the entire duration, preventing the quenching of the chromophore and maximizing the cumulative fluorescence readout.
- Changing reagent concentrations:
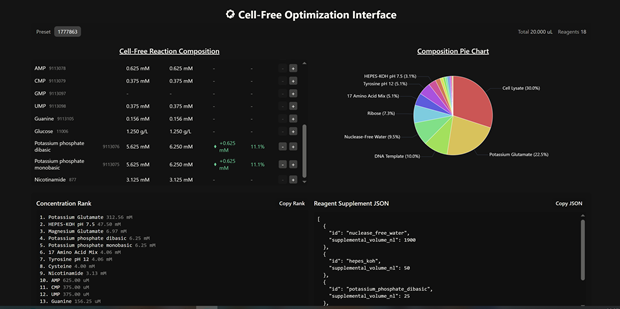
Week 12 HW: Building Genomes
Subsections of Projects
Individual Final Project: A Self-Lytic E. coli Chassis for Cost-Effective Biopolymer Production via Rational Circuit Design
SECTION 1: ABSTRACT
Plastic pollution is one of the biggest challenges of 21st century: We have around 7 gr of microplastics in our brains ( Nihart, A.J., Garcia, M.A., El Hayek, E. et al. Bioaccumulation of microplastics in decedent human brains. Nat Med 31, 1114–1119, 2025). Using microorganisms it is already possible to produce biodegradable plastics that do not accumulate in our bodies.
The transition from petroleum-based plastics to sustainable biopolymers, such as polyhydroxyalkanoates (PHAs), is hindered by prohibitive production costs. While metabolic engineering has significantly improved cellular yields, downstream processing (DSP)—specifically cell disruption—remains a critical economic bottleneck, accounting for 30–40% of total operating expenses. Current research into continuous secretion systems shows promise for small molecules; however, biopolymers like PHAs form large, insoluble intracellular granules that are biophysically difficult to export without excessive metabolic cost.
This project proposes a “Self-Lytic” chassis that utilizes programmed autolysis to bypass these limitations. We hypothesize that a dual-input AND-gate logic circuit, integrating phosphate-starvation (pPhoA) and temperature-induction (thermo-riboswitch) will provide precise control over cell lysis, facilitating rapid and cost-effective PHA recovery.
To optimize this circuit, we will employ a computational rational design approach. We will use the Salis Lab RBS Calculator to predict translation initiation rates and stochastic simulation + absolute quantification to simulate the threshold of holin accumulation required for lysis. During the pre-lysis growth and polymer accumulation phase, Flux Balance Analysis (FBA) will be used to model the metabolic burden imposed by circuit maintenance on the host, ensuring that expression of the biosynthetic and regulatory components does not unsustainably compete with PHA synthesis for cellular resources. This predictive framework replaces high-throughput physical screening, allowing us to select a curated library of specific RBS variants that minimize basal leakiness and maximize polymer yield.
By shifting from energy-intensive mechanical homogenization to programmed biological lysis, this platform aims to reduce downstream costs by up to 60%, offering a scalable, open-source solution for sustainable biomanufacturing.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim (this project):
The first aim of my final project is to engineer and characterize a dual-input (phosphate-starvation and temperature-inducible) genetic AND-gate for programmed autolysis in E. coli by utilizing computational thermodynamic modeling and kinetic simulations. Specifically, I will use the Salis Lab RBS Calculator to design a curated library of 5-10 Ribosome Binding Sites with predicted translation initiation rates to control the expression of the Lambda phage lysis cassette (SRRz). The circuit’s performance will be modeled using stochastic simulation in Python to predict the lysis threshold. Flux Balance Analysis (FBA) will complement this by modeling the metabolic burden imposed by basal circuit expression on the host during the pre-lysis growth and polymer accumulation phase. Experimental validation will involve cloning the synthetic circuit via NEB Gibson Assembly and characterizing lysis kinetics through OD600 absorbance assays. Nile Red fluorescence microscopy will be used to confirm intracellular PHA granule accumulation prior to lysis induction and to verify granule release upon lytic activation.
Aim 2: Development Aim:
The next step following a successful Aim 1 is to solve the “leakiness” and scalability challenges in high-density industrial fermentations by implementing a machine learning model that will be trained on fermentation process variables — including dissolved oxygen, pH, osmolarity, and nutrient consumption rates — collected across scales, with the goal of predicting optimal lysis induction timing as a function of bioreactor conditions. This data-driven layer will serve as a process control strategy to ensure that lytic activation remains synchronized with peak polymer accumulation regardless of scale-dependent environmental heterogeneity. This aim focuses on transitioning the lytic circuit from lab-scale E. coli to testing the circuit’s robustness in 30L, 100L, and 1000L bioreactors to ensure that the lysis trigger remains perfectly synchronized with peak PHA accumulation under the fluctuating nutrient gradients and high osmotic pressures typical of industrial scales.
Aim 3: Visionary Aim:
The long-term vision for this project is to establish an open-source, “Self-Harvesting” platform that disrupts the current economic paradigms of intracellular biomanufacturing. By eliminating the need for capital-intensive mechanical cell disruption and reducing dependency on toxic chemical solvents, this technology aims to bring the price of bioplastics to parity with petroleum-based polymers.
Beyond PHAs, this project envisions a collaborative ecosystem where researchers can share experimental results, refine computational models, and contribute new genetic parts. This open-source framework will not be limited to E. coli; it is designed for chassis-portability, allowing the “self-lytic” logic to be adapted to diverse organisms such as Halomonas, or cyanobacteria. Ultimately, this platform enables a decentralized circular bioeconomy, democratizing the production of single-cell proteins, enzymes, and specialty chemicals, and fostering a global community dedicated to sustainable, high-value biological manufacturing.
SECTION 3: BACKGROUND
Two key research citations inform this project:
Holin-Endolysin Systems: Research by Young et al. (2000) characterizes the Lambda phage SRRz lytic core, demonstrating that holins form micron-scale pores in the inner membrane while endolysins degrade the peptidoglycan layer. This two-component system provides the biological “explosive” necessary for rapid cell disruption.
Phosphate-Starvation Induction: Studies on the pho regulon (pPhoA promoter) show that E. coli can be programmed to respond to inorganic phosphate depletion, a condition that naturally coincides with the onset of PHA accumulation. This suggests that the signal for polymer production can be dual-purposed as the trigger for harvesting, ensuring metabolic synchronization.
Innovation and Novelty:
This project departs from the traditional “brute force” paradigm of mechanical lysis, adopting a predictive synthetic biology approach to downstream processing. While holin-endolysin systems have been explored in E. coli, they typically rely on single-input induction, leaving them susceptible to premature activation under fluctuating industrial conditions.
The innovation lies in the integration of a dual-input AND-gate logic circuit (pPhoA and thermal induction), which provides a level of conditional specificity currently absent in PHA recovery strategies. By utilizing thermodynamic modeling (Salis Lab RBS Calculator) and kinetic ODE simulations, this design enables the rational selection of Ribosome Binding Sites (RBS) to precisely define the holin accumulation threshold. This predictive framework constrains the translation initiation space, effectively minimizing the basal leakiness that typically plagues autolytic strains. Ultimately, this transforms the cellular chassis into a programmable, self-disrupting unit, expanding the boundaries of synthetic biology into the realm of autonomous downstream processing.
Significance and Impact:
This project addresses the global crisis of plastic pollution by tackling the high cost of sustainable alternatives. The economic bottleneck of downstream processing represents a critical barrier that keeps bioplastics from competing with cheap, petroleum-derived synthetics. Reducing harvesting costs could catalyze a field-level change, shifting the industry toward biological, energy-efficient extraction. Beyond the immediate research, the outcomes of this project contribute to a broader societal transition toward a circular bioeconomy, reducing our carbon footprint and reliance on fossil fuels. If realized, this open-source framework will democratize biomanufacturing, enabling the decentralized production of high-value compounds in resource-limited settings and fostering global technical collaboration.
Ethical Implications:
By developing an open-source platform for cheaper bioplastics, the project promotes the principle of beneficence, aiming to mitigate the environmental damage caused by traditional plastics. However, the use of engineered “self-destructive” bacteria raises concerns regarding Biosecurity and Responsibility. There is a potential risk of accidental release of engineered strains into the environment; although the autolytic circuit acts as a built-in “kill-switch” outside of controlled conditions, the horizontal gene transfer of antibiotic resistance markers (if used) remains an ethical challenge that must be addressed to ensure environmental safety.
To ensure the project is conducted ethically, I propose the implementation of “Safe-by-Design” biocontainment strategies, such as using auxotrophic strains that cannot survive outside the laboratory or replacing antibiotic markers with metabolic complementation. Potential unintended consequences include the misuse of the autolytic logic to engineer pathogens, which necessitates a commitment to the principle of Responsibility through transparent, but regulated, sharing of genetic parts. We must also acknowledge the uncertainty in our assumptions regarding the scalability of lysis; if the circuit fails to trigger in large bioreactors, it could lead to significant waste of resources. Alternatives to this action include chemical surfactants, but these are less sustainable and pose higher toxicological risks to workers and the environment.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Use Claude AI skills to refine your HTGAA final project experimental design here
Section 4: Experimental Design
4.1 Workflow Overview
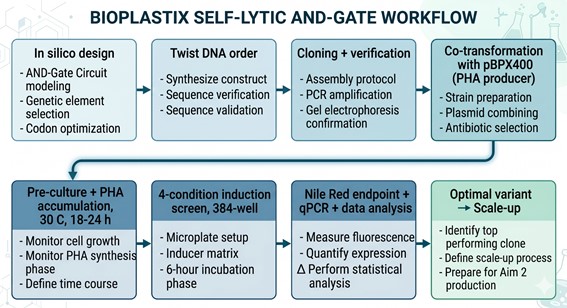
4.2 Detailed Step-by-Step Protocol (15 steps)
The following 15-step workflow covers in silico circuit design, Twist synthesis, strain construction, dual-input induction, lysis characterization, PHA quantification, RBS library optimization, FBA validation, and data integration.
| Step | Method or Tool | Equipment | Plate type | Expected result | Timeline |
|---|
| 1. Computational RBS design | Salis Lab RBS Calculator (web) + stochastic ODE simulation (scipy.integrate.solve_ivp); FBA burden via cobrapy on iML1515 | Desktop (Python) | — | 5 RBS sequences with predicted TIR ~200, 1.5k, 10k, 50k, 200k a.u. | 1 Week |
| 2. Twist Bioscience order | 1 × Custom Plasmid (pBioplastix-Lysis, 5,829 bp, verified pSC101 ori) + 5 × eBlock Gene Fragments (~390 bp each); SecureDNA pre-screen | Online order portal | - | DNA delivered as lyophilized stocks | 14–18 days (lead time) |
| 3. Backbone linearization | Q5 inverse PCR around S105 RBS site to drop the wild-type RBS region | ATC Thermal Cycler | 96-Armadillo-PCR-AB2396X | Linear ~5.5 kb backbone, single band on E-gel | 4 h |
| 4. Gibson assembly setup | Echo525 acoustic transfer of linearized backbone (50 ng) + each eBlock variant (3:1 molar excess) + NEBuilder HiFi master mix (nL volumes); 5 reactions | Echo525 Acoustic Liquid Handler | 384-well Plate Echo PP (source) → 384-pcr-eppendorf-9510207XX (rxn) | 5 Gibson reactions, ~5 µL each | 30 min |
| 5. Gibson reaction | 50 °C × 15 min isothermal | ATC Thermal Cycler | 384-pcr-eppendorf-9510207XX | Assembled circular plasmids | 30 min |
| 6. Heat-shock transformation + outgrowth | Manual transformation of NEB 5-alpha competent cells; 1 h SOC outgrowth at 37 °C; plate on LB+Kan50 | Inheco Plate Incubator (outgrowth); manual plating | 96-Armadillo-PCR-AB2396X (outgrowth); 1-flat-thermo-264728-omni-96 (selection) | ~50–500 colonies/variant | 1 day |
| 7. Colony PCR + Sanger verification | Echo525 dispenses Q5 master mix + variant-specific primers; ATC thermal cycler; outsourced Sanger of positives | Echo525 + ATC | 384-pcr-eppendorf-9510207XX | 3 verified colonies/variant | 2–3 days |
| 8. Glycerol stock banking | 96-deep-well overnights (LB+Kan, 30 °C); Bravo-96 stamp into glycerol stock plates (25% v/v) | Cytomat (incubation); Bravo-96 (stamp) | 96-v-eppendorf-951033502-deep (overnight); 96-round-axygen-pdw11cs-halfdeep (stocks) | Banked at –80 °C; working stocks in Tundrastore (4 °C) | 1 day |
| 9. Co-transformation with pBPX400 | Heat-shock co-transformation into BL21(DE3); dual selection LB+Kan50+Cm25 | Inheco Plate Incubator; manual plating | 1-flat-thermo-264728-omni-96 | Dual-resistant strains: 5 × pBioplastix-Lysis(V1–V5) + pBPX400 | 1 day |
| 10. Pre-culture + PHA accumulation | Inoculate from glycerol stocks into PHA-accumulation MOPS minimal media (0.4% glucose + 4 mM Pi, 30 °C); 18–24 h | Bravo-96 (inoculation); Cytomat (shaking incubation, 30 °C, 600 rpm) | 96-v-eppendorf-951033502-deep | OD600 ≈ 3–5; intracellular PHB granules visible by Nile Red microscopy | 18–24 h |
| 11. Transfer to assay plate + induction setup | Echo525 transfers 1:100 dilutions into 384-well assay plate; Multiflo bulk-dispenses induction media (low-Pi MOPS for ON, high-Pi MOPS for OFF) | Echo525; Multiflo Automated Microplate Dispenser | 384 Greiner black-well clear-bottom (40 µL/well) | Assay plate ready: 5 variants × 4 conditions × 3 reps + controls | 30 min |
| 12. Induction + kinetic readout | Plates split: OFF/OFF + Pi-only conditions stay in Cytomat at 30 °C; heat-only + ON/ON move to Inheco at 42 °C; Spark Plate Reader kinetic reads every 10 min × 6 h (OD600 + sfGFP Ex 485/Em 520) | Cytomat (30 °C); Inheco (42 °C); Spark Plate Reader | 384 Greiner black-well clear-bottom; sealed with Plateloc + A4s breathable | OD600 trajectories + sfGFP induction kinetics | 6 h |
| 13. Nile Red endpoint assay | Tempest dispenses Nile Red (10 µg/mL final); BioshakeD3000 mix 30 min; HiG centrifuge to pellet debris; PHERAstar FSX reads supernatant fluorescence (Ex 543 / Em 598) | Tempest; BioshakeD3000; HiG Centrifuge; PHERAstar FSX | Same 384 Greiner plate | Nile Red supernatant fluorescence ~ released PHA mass | 1 h |
| 14. qRT-PCR transcript validation | Total RNA extraction (3 selected variants × 4 conditions × 3 reps, n=36); reverse transcription + qPCR for sfgfp, S, R, tetR, and rrsA (reference) | CFX Opus qPCR | 96-Armadillo-PCR-AB2396X | Confirms transcriptional AND-gate logic; validates anti-TetR depletion | 2 days |
| 15. Data analysis + variant ranking | Python pipeline: lysis kinetic parameter extraction (scipy.optimize.curve_fit on logistic decay); leakiness scoring; Pareto-front ranking; correlation of measured kinetics with predicted TIR | Desktop (Python, pandas, matplotlib) | — | Optimal RBS variant identified for Aim 2 | 1 week |
Conclusion: Total project duration: ~6–8 weeks (DNA lead time dominates).
4.3 Plate Layout — The 384-Well Phenotype Screen (Step 12)
The screening plate carries 5 RBS variants (V1–V5) and 5 control strains across 4 induction conditions, in 3 biological replicates. Edge wells (column 1, column 24, row A, row P) are filled with sterile media as an evaporation moat.
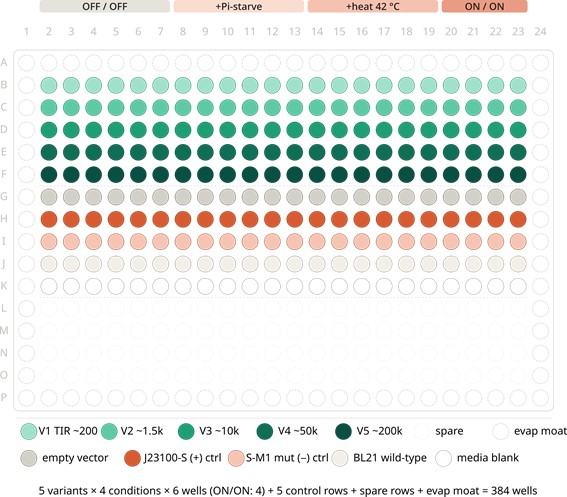
4.4 Standard Curves and Controls
The plate also includes a PHA standard curve: pure crotonic acid (PHB hydrolysate) standards at 0, 1, 5, 10, 25, 50 µg/mL spiked into Nile Red working solution (rows L–O, cols 2–7), allowing absolute quantification of released polymer mass per well. Nile Red fluorescence-vs-concentration is fit to a 4-parameter logistic curve, and sample wells are interpolated against this fit.
4.5 Twist Bioscience Order — Sequences
4.5.1 Base Plasmid (Twist Custom Plasmid order)
The full 5,829 bp pBioplastix-Lysis is ordered as a single Twist Custom Plasmid in the verified pSC101 / KanR backbone. Key features (positions referenced to GenBank file pBioplastix-Lysis.gb):

4.5.2 RBS Variant eBlocks (5 × Twist Gene Fragments, ~390 bp each)
Each eBlock contains a 30 bp 5′ Gibson homology arm matching the end of sfGFP, the variant RBS (12 bp), the full 321 bp S105 ORF, and a 30 bp 3′ Gibson homology arm matching the R-RBS region. The only sequence change between eBlocks is the 12 bp RBS region (uppercase below); all flanking sequence is identical to the base plasmid.

Variant RBS sequences (output from Salis Lab RBS Calculator; values are illustrative — replace with calculator output before order)
| Variant | RBS sequence (12 bp) | Predicted TIR (a.u.) | log10(TIR) |
|---|
| V1 | aattcaaccaaa | ~200 | 2.3 |
| V2 | aaaggaagaaaa | ~1,500 | 3.2 |
| V3 | aaaggaggaata | ~10,000 | 4.0 |
| V4 | aaagaggagaaa | ~50,000 | 4.7 |
| V5 | aaaaggaggtaa | ~200,000 | 5.3 |
Section 5: Techniques, Tools, and Technology
5.1 Course Technique Checklist
| Technique | Used in this project? | Where in workflow |
|---|
| Gibson Assembly (NEBuilder HiFi) | ✓ | Step 4–5 (variant installation) |
| Computational RBS design (Salis Lab Calculator) | ✓ | Step 1 |
| Stochastic ODE simulation | ✓ | Step 1 |
| Flux Balance Analysis (FBA) | ✓ | Step 1 (burden assessment) |
| Polymerase chain reaction (PCR) | ✓ | Step 3 (linearization), Step 7 (cPCR) |
| qRT-PCR | ✓ | Step 14 |
| Acoustic nanoliter liquid handling (Echo525) | ✓ | Steps 4, 7, 11 |
| Plate-based kinetic phenotyping | ✓ | Step 12 |
| Fluorescence reporter assays (sfGFP, Nile Red) | ✓ | Steps 12–13 |
| Sanger sequencing | ✓ | Step 7 |
| Heat-shock transformation | ✓ | Steps 6, 9 |
| Auxotrophic biocontainment design | ✓ (Aim 2) | Bioethics |
| Golden Gate assembly | ✗ | Not used |
| CRISPR-Cas9 / CRISPRi | ✗ | Not used |
| Cell-free expression (TX-TL) | ✗ | Not used (could be added for rapid pre-screen) |
| Mass spectrometry | ✗ (Aim 2 candidate) | Not in Aim 1 |
| Flow cytometry | ✗ | Plate reader covers single-well sufficient |
| Mammalian tissue culture | ✗ | Out of scope |
5.2 Technique Expansion (2 selected techniques, ≥4 sentences each)
5.2.1 Computational RBS Design with the Salis Lab RBS Calculator
The Salis Lab RBS Calculator (Salis, Mirsky & Voigt, 2009; updated v2.1 in 2017) uses a free-energy thermodynamic model of the ribosome–mRNA interaction to predict the translation initiation rate (TIR) of any given RBS in arbitrary units across roughly five log decades of dynamic range. For this project, the calculator is used in Reverse Engineering mode (predicting TIR from a given sequence) to characterize the wild-type B0034 RBS as a reference, and then in Forward Engineering / Library mode to generate five variant sequences with target TIRs of ~200, 1,500, 10,000, 50,000, and 200,000 a.u. — spanning three log decades and centered on the wild-type value. Variant selection prioritizes sequences with low predicted secondary-structure free-energy variance (ΔGmRNA standard deviation < 1 kcal/mol) to minimize context-dependent expression noise. The output TIRs serve as the x-axis against which experimental lysis kinetics are correlated, allowing the project to test the central design hypothesis: that holin abundance, not transcriptional dynamics, sets the AND-gate lysis threshold.
5.2.2 Gibson Assembly of Variant Libraries via Acoustic Liquid Handling
NEBuilder HiFi Gibson Assembly is a one-pot isothermal cloning method combining a 5′ exonuclease, a high-fidelity polymerase, and a thermostable ligase, enabling seamless ligation of fragments sharing 20–40 bp of terminal homology in a single 50 °C / 15 min reaction. In this project, five 391 bp eBlock fragments — each carrying one RBS variant — are assembled in parallel into the Q5-linearized 5.5 kb backbone of pBioplastix-Lysis. The Echo525 acoustic liquid handler enables miniaturization of each Gibson reaction to 500 nL total volume (10 nL backbone + 30 nL eBlock + 250 nL HiFi master mix + 210 nL water), reducing reagent cost by ~40-fold relative to bench-scale 20 µL reactions while improving variant-to-variant reproducibility through sub-nanoliter dispense precision. Following thermal cycling on the ATC, transformation efficiencies of >10⁴ CFU/µg are routinely achieved, providing 50–500 colonies per variant — more than adequate for picking three sequence-verified clones per RBS condition.
Section 6: Project Validation
6.1 (10a) Validation Choice
The chosen validation experiment is a gene expression test measuring the AND-gate phenotype across all four induction conditions (OFF/OFF, +Pi-starve only, +heat only, ON/ON), using sfGFP fluorescence as the circuit-activation marker and OD600 trajectory as the lysis output. This experiment is decisive because it simultaneously tests every component of the circuit: pPhoA Pi-responsiveness, FourU temperature gating, anti-TetR depletion of TetR, derepression of PLtetO-1, and tuned holin S105 activity — a single failure in any component collapses the AND-gate signature into either constitutive lysis, constitutive silence, or single-input behavior.
6.2 (10b) Validation Protocol
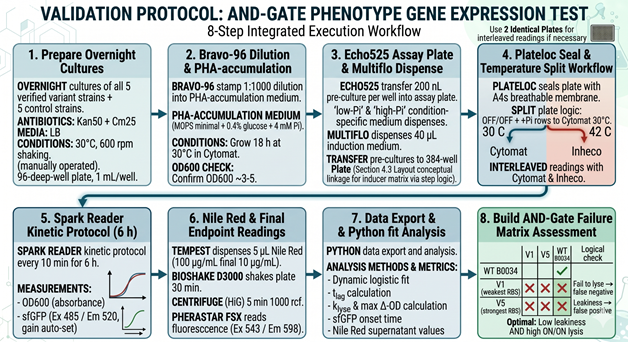
6.3 (10c) Techniques Used in the Validation
The validation experiment integrates four orthogonal techniques to confirm AND-gate function. Acoustic liquid handling (Echo525) ensures the 200 nL pre-culture transfers and 1:1000 inoculum dilutions are dispensed with sub-percent CV across 384 wells, eliminating well-to-well growth variability that would otherwise mask circuit-level effects. Kinetic absorbance and fluorescence plate reading (Spark) captures the time-resolved signature of the AND-gate: a clean ON/ON well shows a 30–60 min lag of stable OD600 followed by a sigmoidal drop coinciding with sfGFP rise, whereas single-input wells (Pi-only or heat-only) show neither feature. Endpoint Nile Red fluorescence (PHERAstar FSX) quantifies released PHA in the supernatant — the actual industrial-relevant deliverable — and discriminates between cells that lyse cleanly (high supernatant signal) versus cells that round up but fail to release granules (low supernatant signal). qRT-PCR (CFX Opus) in the validation wells confirms the upstream layer of the circuit, demonstrating that the ON/ON condition is the only one in which tetR mRNA is depleted relative to the J23100 baseline and that S and R transcripts subsequently accumulate.
6.4 (10d) Hypothetical Data
Below is a small subset of expected outcomes for the 5 variants under ON/ON conditions, with corresponding leakiness in OFF/OFF. The Python snippet that follows generates a representative kinetic plot.
Lysis kinetic parameters (ON/ON condition, mean ± SD, n = 3)
| Variant | Predicted TIR (a.u.) | t_lag (min) | k_lyse (min⁻¹) | Max ΔOD600 | OFF/OFF leakiness (% OD drop) |
|---|
| V1 | 200 | 280 ± 15 | 0.004 ± 0.001 | 0.18 ± 0.04 | 1.5 ± 0.8 |
| V2 | 1,500 | 195 ± 11 | 0.011 ± 0.002 | 0.62 ± 0.05 | 4.2 ± 1.1 |
| V3 | 10,000 | 130 ± 8 | 0.024 ± 0.003 | 0.81 ± 0.04 | 7.8 ± 1.4 |
| V4 | 50,000 | 92 ± 6 | 0.038 ± 0.004 | 0.86 ± 0.03 | 15.6 ± 2.1 |
| V5 | 200,000 | 62 ± 5 | 0.057 ± 0.005 | 0.89 ± 0.02 | 31.4 ± 3.5 |
Interpretation: V3 is the predicted Pareto-optimal variant — substantial lysis amplitude (Δ OD ≈ 0.81) within 130 min while keeping basal leakiness below 10%. V4 lyses faster but at unacceptable basal cost; V1–V2 are too weak; V5 is too leaky. Final ranking will depend on the downstream PHA recovery economics (faster lysis vs. lower yield loss to leakiness).
Expected plot:
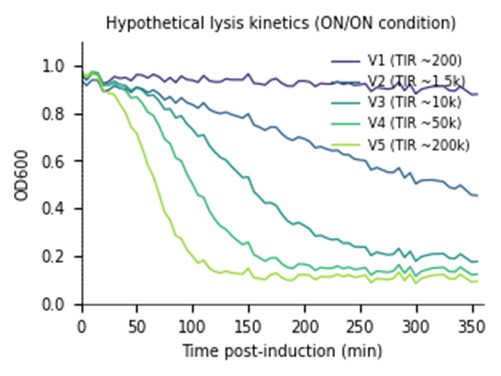
The most likely failure mode is excessive basal leakiness in V4–V5, manifest as significant OD decline in OFF/OFF or single-input wells; this would indicate that even modest TetR depletion noise from constitutive J23100 expression is sufficient to fire a hyper-translated holin, suggesting the AND-gate must be retuned by also weakening the J23100 promoter or adding a degradation tag to anti-TetR. A second concern is insufficient PHA accumulation in pBPX400 + low-Pi MOPS minimal medium, which would mute the Nile Red endpoint signal regardless of lysis efficacy; the workaround is to switch to 0.4% glucose + 50 mM Pi for accumulation and then perform a sterile media swap to low-Pi induction medium between Steps 10 and 11. Edge-effect evaporation in 384-well plates with 6 h incubations at 42 °C is a known source of CV inflation; the layout reserves the outer ring as media-only blanks and uses Plateloc + breathable A4s seals, but if CV exceeds 15% the screen will be re-run with humidified-chamber Cytomat slots. Finally, if the FourU thermometer fails to gate cleanly (i.e., partial leakage at 30 °C), it can be swapped for the more cooperative ROSE-element thermometer in a follow-up eBlock order without redesigning the rest of the circuit.
Section 7: Additional Information
7.1 References
- Borrero-de Acuña, J. M., Aravena-Carrasco, C., Gutiérrez-Urrutia, I., Duchens, D., Poblete-Castro, I. (2017). Programmed cell lysis in Pseudomonas putida for sustained PHA biopolymer recovery. Microbial Cell Factories, 16, 215. https://doi.org/10.1186/s12934-017-0828-0
- Pasotti, L., Bellato, M., De Marchi, D., Magni, P. (2017). Tuning ribosome binding sites for plug-and-play control of phage lysis circuits in E. coli. ACS Synthetic Biology, 6(5), 802–815.
- Salis, H. M., Mirsky, E. A., Voigt, C. A. (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nature Biotechnology, 27(10), 946–950. https://doi.org/10.1038/nbt.1568
- Kortmann, J., Narberhaus, F. (2012). Bacterial RNA thermometers: molecular zippers and switches. Nature Reviews Microbiology, 10(4), 255–265.
- Lutz, R., Bujard, H. (1997). Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Research, 25(6), 1203–1210.
- Wanner, B. L. (1996). Phosphorus assimilation and control of the phosphate regulon. In Escherichia coli and Salmonella: Cellular and Molecular Biology, ASM Press.
- Young, R. (2014). Phage lysis: three steps, three choices, one outcome. FEMS Microbiology Reviews, 38(1), 9–32.
- Madison, L. L., Huisman, G. W. (1999). Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiology and Molecular Biology Reviews, 63(1), 21–53.
- Choi, S. Y., Park, S. J., Kim, W. J., et al. (2020). One-step fermentative production of poly(lactate-co-glycolate) from carbohydrates in Escherichia coli. Nature Biotechnology, 38, 359–365.
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345.
- Orth, J. D., Thiele, I., Palsson, B. Ø. (2010). What is flux balance analysis? Nature Biotechnology, 28(3), 245–248.
Monk, J. M., Lloyd, C. J., Brunk, E., et al. (2017). iML1515, a knowledgebase that computes Escherichia coli traits. Nature Biotechnology, 35(10), 904–908.
7.2 Supplies and Budget
| Item | Vendor | Catalog / Service | Estimated Cost (USD) |
|---|
| pBioplastix-Lysis Custom Plasmid (5,829 bp) | Twist Bioscience | Custom Plasmid synthesis | ~$300 |
| 5 × RBS variant eBlocks (~390 bp each) | Twist Bioscience | Gene Fragment / eBlock | 5 × ~$40 = ~$200 |
| SecureDNA pre-synthesis screen | SecureDNA | Bundled with Twist order | $0 (provided) |
| NEBuilder HiFi DNA Assembly Master Mix (50 rxn) | NEB | E2621L | ~$320 |
| Q5 High-Fidelity 2X Master Mix (100 rxn) | NEB | M0492L | ~$155 |
| BL21(DE3) competent cells | NEB | C2527H | ~$200 |
| NEB 5-alpha competent cells | NEB | C2987H | ~$140 |
| Kanamycin sulfate, 25 g | Millipore Sigma | 60615 | ~$95 |
| Chloramphenicol, 25 g | Millipore Sigma | C0378 | ~$75 |
| Nile Red dye (5 g) | Thermo Fisher Scientific | N1142 | ~$110 |
| MOPS minimal media salts kit | Teknova | M2106 (via Millipore Sigma) | ~$130 |
| 384 Greiner black-well clear-bottom plates (40 ct) | Greiner Bio-One via Thermo Fisher | 781091 | ~$320 |
| 384-well Echo PP source plates (50 ct) | Beckman Coulter via Thermo Fisher | LP-0200 | ~$280 |
| 96-Armadillo PCR plates (50 ct) | Thermo Fisher Scientific | AB2396 | ~$220 |
| QIAprep Spin Miniprep Kit (250 prep) | Qiagen via Millipore Sigma | 27106 | ~$485 |
| Sanger sequencing (~30 reactions) | Genewiz / Eurofins / institutional core | — | ~$200 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 | ~$430 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | 1725271 | ~$370 |
| Glycerol, dNTPs, restriction enzymes (misc) | NEB / Millipore Sigma | — | ~$300 |
| Reagents subtotal | | | ~$4,330 |
| Ginkgo Bioworks robotics access (1 project block) | Ginkgo Bioworks | Foundry access | ~$2,500 |
| Total estimated project budget | | | ~$6,830 |
7.3 Industry Council Partner Connections
Twist Bioscience — Custom Plasmid + 5 eBlocks (the core DNA order)
SecureDNA — Pre-synthesis biosecurity screening of all DNA orders (bioethics layer)
NEB — NEBuilder HiFi Gibson, Q5 polymerase, competent cells
Ginkgo Bioworks — Full automated workflow (Echo525, ATC, Multiflo, Cytomat, Inheco, Spark, PHERAstar, etc.)
Thermo Fisher Scientific / Millipore Sigma — Reagents, media, Nile Red, plasticware
Bio-Rad — qPCR consumables (CFX Opus reagents)
Addgene — Open-source deposition of pBioplastix-Lysis(V_optimal) for community use (Aim 3)
Cultivarium — Future chassis-portability work to Halomonas / non-model hosts (Aim 3)
Group Final Project - Bacteriophage Engineering Proposal: L Protein Stabilization
Primary Goal: Increased stability
Specific Approach: Engineering DnaJ-independence by reducing chaperone-recognition signals while preserving the structural scaffold of the L protein.
1. Computational Tools and Pipeline Justification To achieve this goal, we propose a three-step computationally efficient pipeline:
Step 1: Sequence-level Mutational Scanning using ESM2
Approach: We will perform a zero-shot in silico mutational scan across the L protein sequence using the ESM2 Protein Language Model (PLM). We aim to identify exposed hydrophobic patches (typical DnaJ recognition motifs) and propose polar/hydrophilic substitutions.
Why this helps: ESM2 has learned deep evolutionary constraints across millions of protein sequences. It allows us to rapidly differentiate between highly constrained residues (which are structurally vital and “untouchable”) and mutation-tolerant positions. This ensures we only disrupt chaperone-binding motifs without breaking the core evolutionary scaffold of the protein, all at a fraction of the computational cost of molecular dynamics.
Step 2: Rapid Structural Filtering using ESMFold
Approach: The top candidate sequences from the ESM2 scan will be predicted using ESMFold. We will filter out any variants that collapse, show low pLDDT (confidence) scores, or have a high RMSD compared to the Wild-Type (WT) backbone.
Why this helps: While ESM2 evaluates sequence-level fitness, we need explicit 3D structural validation. ESMFold is significantly faster than AlphaFold2, making it ideal for high-throughput filtering. This step ensures that our hydrophilic mutations do not inadvertently destroy the L protein’s ability to fold independently.
Step 3: Complex Modeling using Boltz-1
Approach: We will model the L protein + DnaJ complex for both the WT and our top folded mutant candidates. We will analyze the predicted interface contacts and Predicted Aligned Error (PAE) to assess binding affinity.
Why this helps: Folding correctly in isolation is not enough; we must explicitly prove reduced chaperone dependency. By comparing the mutant-DnaJ interface against the WT-DnaJ interface, we can prioritize variants that maintain a stable fold but show a significantly weakened or abolished interaction with the DnaJ chaperone.
2. Potential Pitfalls
Pitfall 1: Overlapping Reading Frames and Genomic Constraints. Phage genomes are highly compact, meaning the DNA sequence encoding the L protein might also encode parts of other proteins or regulatory elements in alternative reading frames. Our targeted mutations could have unintended, fatal consequences for the phage’s overall viability. While genomic foundation models like Evo could assess these genome-wide constraints, their computational cost is prohibitive for our current scope.
Pitfall 2: The Stability vs. Function Trade-off. ESMFold guarantees that the protein adopts a stable 3D conformation in solution, but it does not guarantee biological function (membrane lysis). Lytic activity heavily depends on complex factors like membrane insertion dynamics, oligomerization, and reaction kinetics. Furthermore, completely abolishing chaperone interaction might inadvertently prevent the L protein from being properly delivered to its target membrane.