Week 4 HW: Protein Design Part I

(American Chemical Society, 2023)
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Why do humans eat beef but do not become a cow, eat fish but do not become fish? Even though some may like to say you are what you eat, this really isn’t the case when it comes to eating an organism. Even though we consume its parts and convert protein into energy we are transforming energy into energy that we can use. One may argue we embody that organism to a degree, but we dont fully transform into that being. Your cells follow a genetic blueprint, and while other DNA enters your body, you do not absorb its genome, nor does it integrate into yours.
Why are there only 20 natural amino acids? Although the genetic code contains 64 possible codons, evolution stabilized around 20 canonical amino acids. This was not due to a strict numerical limitation, but because these 20 provide an optimal balance of chemical diversity, stability, and functional efficiency. Biochemical availability on early Earth also played a crucial role, early life utilized the amino acids that were most readily synthesized under prebiotic conditions. Mathematical analyses of the canonical set suggest that the 20 amino acids achieve a near-optimal coverage of chemical property space, maximizing structural and catalytic versatility while minimizing redundancy. Furthermore, the structure of the genetic code itself is organized in a way that reduces the impact of mutations: codons that differ by a single nucleotide often encode amino acids with similar physicochemical properties, thereby limiting potential damage to protein structure and function.
Can you make other non-natural amino acids? Design some new amino acids. Yes, and we already have. Scientists have even been able to genetically engineer organisms to expand the genetic code beyond the 20, by introducing new codons.() Some of the amino acids that have been synthesised include fluorescent amino acids, for example, to help scientists localise proteins in a cell and observe their interactions, improving observation.
Where did amino acids come from before enzymes that make them, and before life started? Amino acids can form spontaneously under prebiotic conditions through chemistry. Scientists have simulated Earth’s atmosphere using simple gases and found that amino acids can form through life’s basic chemistry.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? If you make an α-helix using D-amino acids, you would expect a left-handed twist, because the molecular building blocks themselves are mirrored. D-amino acids have the opposite chirality of the natural L-forms, so their backbone geometry favors a left-handed helix rather than a right-handed one.
Can you discover additional helices in proteins? Yes research continues to identify unsual or distrorted helical geometries. Scienttists have also designe helices from non antural amino acids, or even alternate hydrogen bonding patterns.
Why are most molecular helices right-handed? Most moleculart helices are right handed beacuse of chirality. The presence of molecular assymetry in L- amino acids determening helix twist direction, favouring a right handed direction. While left handed helices are possible, they are rare. An example of a left handed helices is a D helices, which its molecular building block is most stable and corresponds to a left hand.
Why do β-sheets tend to aggregate? β-sheets tend to aggregate because their extended structure exposes backbone hydrogen bond donors and acceptors, allowing strands from different proteins to easily form intermolecular hydrogen bonds. Their flat geometry also enables tight stacking into stable, repetitive “cross-β” structures, making aggregation energetically favorable.
What is the driving force for β-sheet aggregation? Backbone hydrogen bonds form between neighbouring β strands, satisfying exposed N-H and C=O groups. Hydrophobic chains pack together and repel water, thus loweing the systems free energy and agregates state is more stable than partially unfolded state.
Why do many amyloid diseases form β-sheets? Because the β-sheet structure is the most stable form in which misfolded proteins can agregate and be reorganized, creating a strong cross β structure.
Can you use amyloid β-sheets as materials? Amyloid β-sheets are one of the strongest and most stable protein structures in nature, making them greatly appealing in material architecture. Some of the properties that arise and be applied in material science include, tensile strength, chemical stability, self assembly in asolutions, heat resistance, and mechanical stiffness. Examples where nature uses the intelligent structure present in β-sheet scaffolidng is in silk fibroin and curli fibers in bacterial biofilms. Common applications of Amyloid β-sheets are hydrogels, drug delivery, nanofibres, tissue scafolidng, bioprinting, and even in conductive biofilms.
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization**
I will continue to explore reflectins and their biomechanical properties as they respond to stimuli and can serve as a beautiful template for material engineering and especially in applications such as smart textiles.
The amino acid sequence of reflectin 3a
tr|Q6WDN6|Q6WDN6_EUPSC Reflectin 3a OS=Euprymna scolopes OX=6613 PE=4 SV=1 MNRYMNRFRNFYGNMCRNRNRGMMEPMSRMTMDFQGRYMDSQGRMVDPRYYDYYGRYNDY DRYYGRSMFNYGWMMDGDRYNRYNRWMDYPERYMDMSGYQMDMYGRWMDMQGRHCNPYSQ WMMYNYNRHGYYPNYSYGRHMFYPERWMDMSNYSMDMYGRYMDRWGRYCNPFYHYYNHWN RSGNNPGYYSYYYMYYPERYFDMSNWQMDMQGRWMDMQGRYCSPYWYNWYGRQMYYPYQN YYWYGRWDYPGMDYSNWQMDMQGRWMDMQGRYMDPWWMNDSYYNNYYN
The most frequent amino acid is Tyrosine with 58 in the sequence of 288 total residues.
(Use of AI, asking chat gpt to tell me how many of each amino acid in that sequence.)
Using UniProt BLAST with default parameters against UniProtKB, 5 significant sequence homologs were identified.
Reflectin belongs to the reflectin protein family including various types.
There is no refletcin 3a protein structure solved, and can be classifies as IDP- intrinsically dissordered protein.
REFLECTIN 3A STRUCTURE:
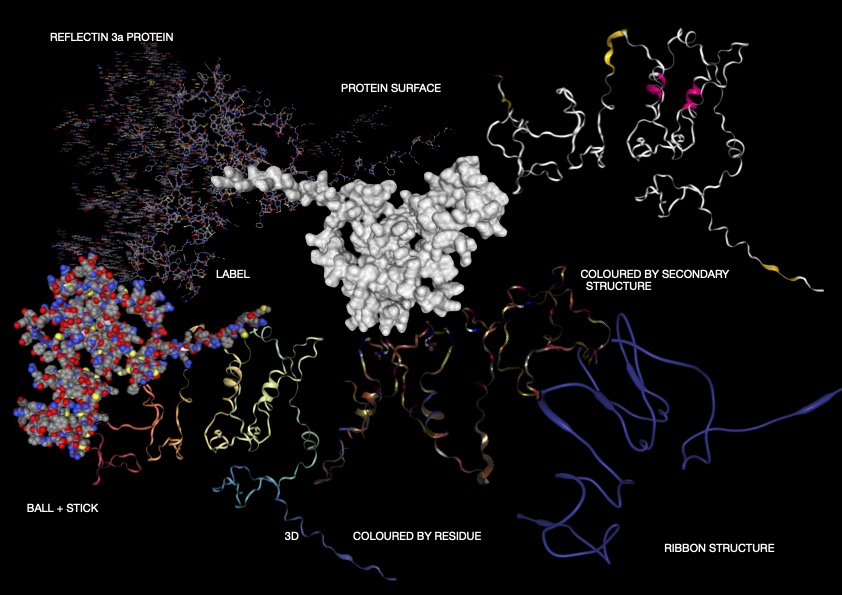
Figure 1
ANALYSIS:
As shown above (Figure 1), I visualised the Reflectin 3a protein in various forms. In the representation coloured by secondary structure, Reflectin appears to contain a mixture of structural elements, with some helical regions present alongside a larger proportion of flexible coil segments. The distribution of hydrophobic and hydrophilic residues appears relatively balanced; however, rather than forming a tightly packed globular structure with clear segregation, the residues are arranged in a more dispersed manner. While it may appear that hydrophobic residues are positioned toward the exterior and hydrophilic residues toward the interior, this organisation is not as clearly defined as in fully folded proteins, reflecting a less stable structural arrangement. Additionally, small cavities or regions resembling binding pockets can be observed, though these are likely transient and not rigidly maintained.
Overall, the 3D visualizations of Reflectin 3a reveal a predominantly extended and flexible structure with limited stable secondary elements, indicating that the protein is largely intrinsically disordered. The dominance of coil regions and absence of a compact globular core suggest that Reflectin does not adopt a fixed conformation but instead remains structurally dynamic. This flexibility, combined with its linear sequence organization and lack of rigid folding, supports its known function as a responsive biomaterial. Reflectin likely undergoes environmentally triggered conformational and assembly changes, enabling tunable optical properties such as those observed in cephalopod coloration.
PART C: Using ML-Based Protein Design Tools
Protein : Kalium Channelrhodopsin 1
8GI8_1|Chain A|Kalium Channelrhodopsin 1|Hyphochytrium catenoides (42384) MPFYDSRPPEGWPKGSINDMDYPLLGSICAVCCVFVAGSGIWMLYRLDLGMGYSCKPYKSGRAPEVNSLSGIICLLCGTMYAAKSFDFFDGGGTPFSLNWYWYLDYVFTCPLLILDFAFTLDLPHKIRYFFAVFLTLWCGVAAFVTPSAYRFAYYALGCCWFTPFALSLMRHVKERYLVYPPKCQRWLFWACVIFFGFWPMFPILFIFSWLGTGHISQQAFYIIHAFLDLTCKSIFGILMTVFRLELEEHTEVQGLPLNEPETLS
Mutation Scan
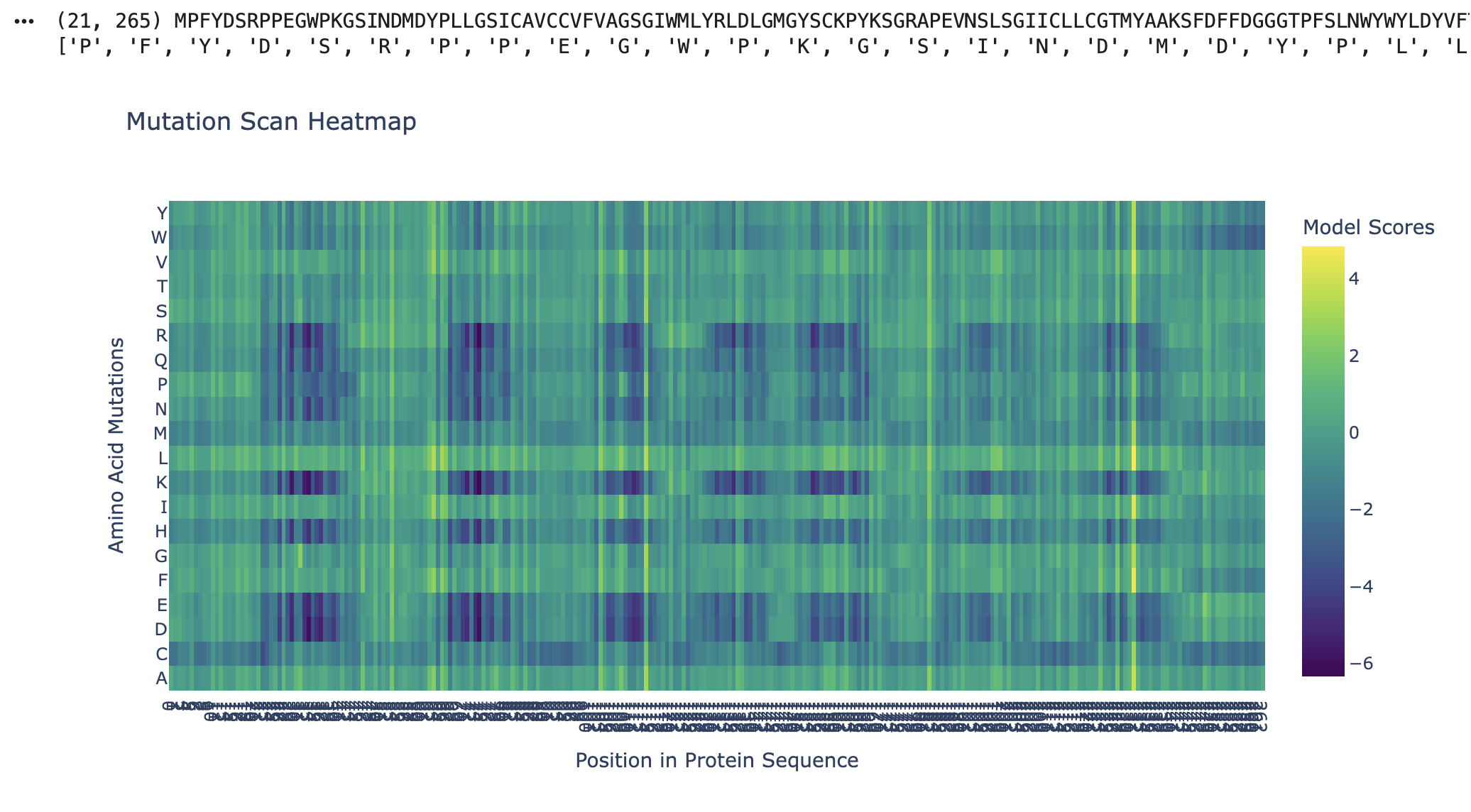
Figure 2
As seen in the mutation scan for Kalium Channelrhodopsin 1, many amino acids show dark purple bands. The protein likely resists mutation at these positions due to their functional roles, where mutations would destabilise the protein or its folding. Some of the functions of the residues in Kalium Channelrhodopsin 1 include retinal binding pockets, ion conduction pathways, and transmembrane helix packing residues. Rows (D, E, K, and R) correspond to the disruptive residues in hydrophobic regions, and since Kalium Channelrhodopsin 1 is a membrane protein, its helices are embedded in the membrane. Kalium Channelrhodopsin 1 is an evolutionary constraint, and this graph shows its intolerance to mutations due to its key roles relying on structure, such as retinal binding and ion conduction (RCSB PDB).
Latent Space Visual
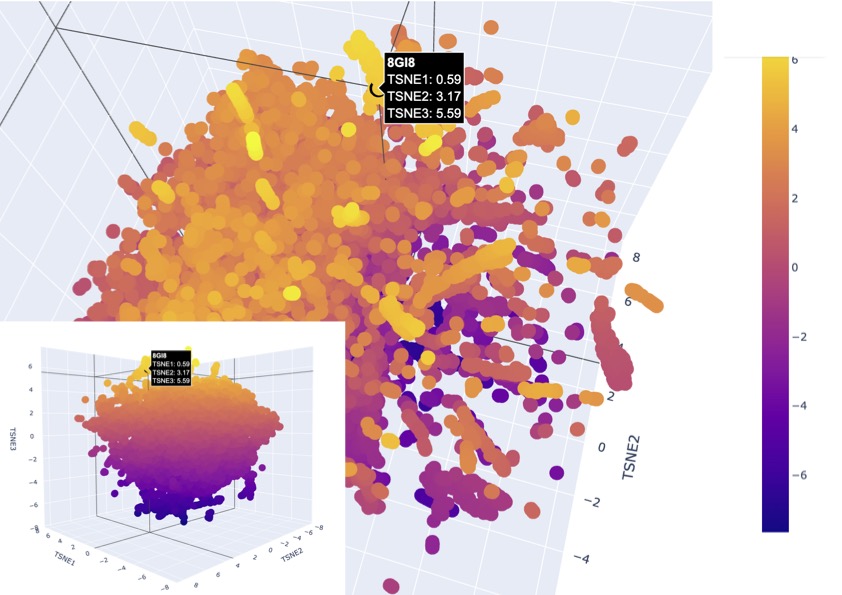
Figure 3
After many attempts, I finally found the protein. Some key errors in the code were missing the addition of:
#color it differently
fig_3d.add_scatter3d(
x=[my_point[“TSNE1”]],
y=[my_point[“TSNE2”]],
z=[my_point[“TSNE3”]],
marker=dict(
size=10, # Choose the dot size
color=“Black” # Choose a color
),
text=[“8GI8”],
hovertemplate="%{text}
TSNE1: %{x:.2f}
TSNE2: %{y:.2f}
TSNE3: %{z:.2f}
,before the protein synthesis, which specifically coloured the protein I was looking for.
The latent space visualisation shows that the protein is embedded within a dense cluster of neighbouring points rather than appearing as an outlier in a distinct class. This indicates that the protein shares strong similarity with a broader family of proteins in the dataset, suggesting conserved structural and functional features, as there are also no clear boundaries around the point. Additionally, the high density of surrounding points suggests that this region of the latent space corresponds to a well-represented and evolutionarily conserved protein family. Given that channelrhodopsins are membrane proteins with characteristic alpha-helical structures, the clustering supports that the model has correctly captured these features and placed the protein among structurally similar transmembrane proteins, existing as a larger family.
ESM Protein Fold
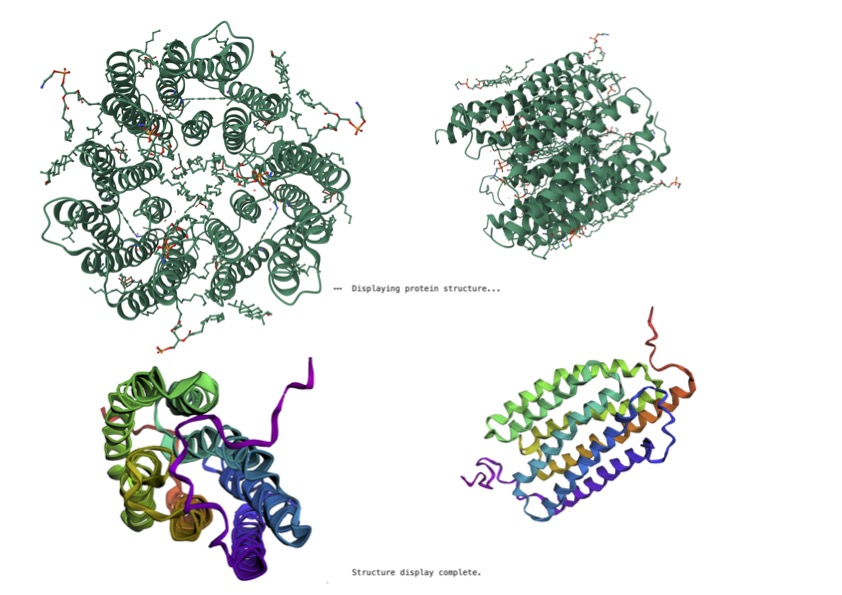
Figure 4
Kalium Channelrhodopsin VS ESM Predicted Fold Protein
The ESM-predicted structure represents the protein as a single subunit, whereas the reference structure appears oligomeric, consisting of multiple interacting chains. This indicates that the model captures the fold of an individual protein chain but does not fully reproduce the higher-order quaternary structure of the native assembly. In addition, the reference structure is more compact and ordered, with clearer symmetry and well-defined alpha-helical regions. The prediction successfully captures the overall secondary structure, particularly the alpha-helical architecture, but shows reduced accuracy in loop placement and local packing. These differences suggest that while the model is effective at predicting the general fold and topology of the protein, it is less precise in reconstructing flexible regions and inter-subunit interactions, which are important for full structural and functional accuracy.
Inverse Sequence Output:
Generating sequences…
tmp, score=1.9742, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 INDMDYPLLGSICAVCCVFVAGSGIWMLYRLDLGMGYSCKPYKSGRAPEVNSLSGIICLLCGTMYAAKSFDFFDGGGTPFSLNWYWYLDYVFTCPLLILDFAFTLDLPHKIRYFFAVFLTLWCGVAAFVTPSAYRFAYYALGCCWFTPFALSLMRHVKERYLVYPPKCQRWLFWACVIFFGFWPMFPILFIFSWLGTGHISQQAFYIIHAFLDLTCKSIFGILMTVFRLELEEHTEVQGL T=0.1, sample=0, score=1.0180, seq_recovery=0.3208 SADLPWPLLAAVFLVTALIVGGTGLLILYLTRRGLCWTRRPWPNGRPPWWVLLPGLICIVTGLLFLLAALDAAYGGVHPFSVLRVLFLPDLLTIPLNILALCLLLKLPFPLLFFLLALLTVLCFVLSWLTKSPLRWVWYALGMLFFIPMYILLKKLAKKEYKKLSPKTRVFFKLIMIITFGLYPLYPVLWVLSFYGLGIISLGAFYVIMAVLNLLIFAVNGLLMALYLKSLNHERLCKGK
After many attempts, I finally got an inverse sequence. The inverse protein design results show that the model can generate a significantly different amino acid sequence (sequence recovery ~32%) while maintaining compatibility with the original protein structure. This suggests that the overall fold of the protein is robust to sequence variation and that multiple sequences can encode the same structural architecture. Notably, the designed sequence retains a high proportion of hydrophobic residues, consistent with the protein’s transmembrane nature, indicating that the model preserves key biophysical constraints required for membrane insertion. The improved score of the designed sequence further suggests that it is a stable alternative to the original sequence, highlighting the ability of inverse design methods to explore diverse sequence solutions for a given structural fold.
Amino acid Probabilities
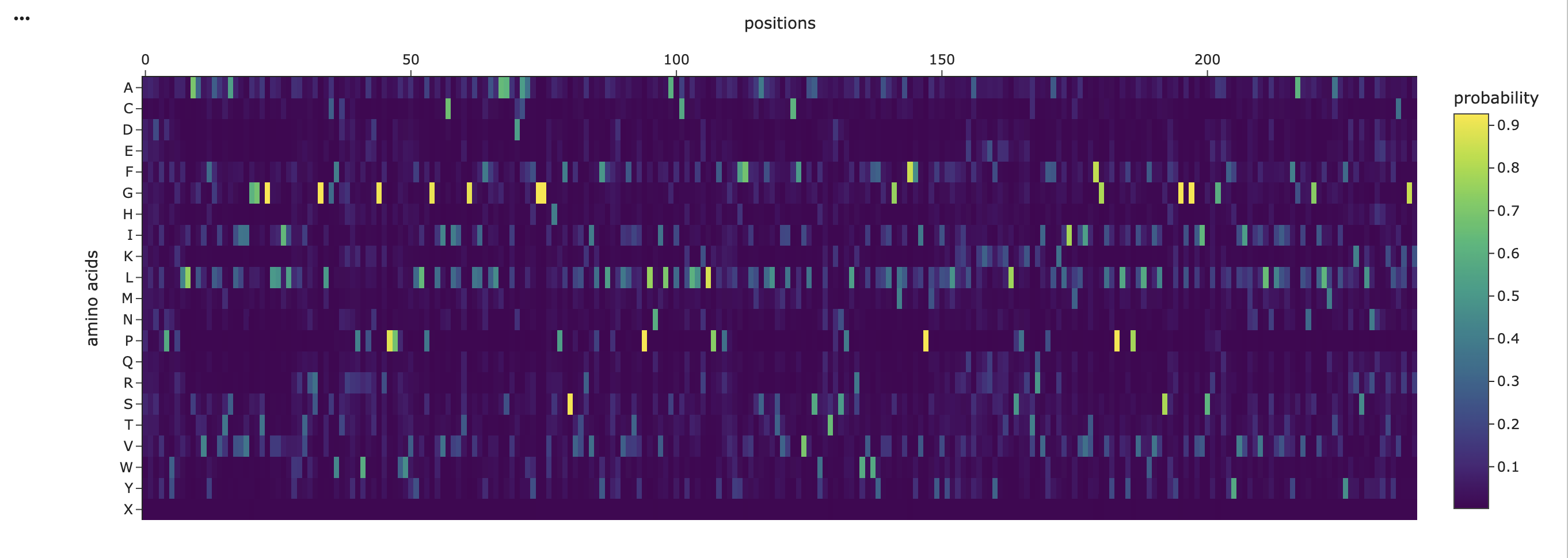
Figure 5
The amino acid probability heatmap shows that most positions in the protein exhibit a strong preference for a single amino acid, indicated by sharp high-probability peaks. This suggests that many residues are structurally or functionally constrained, which is important for maintaining the protein’s fold. In contrast, some of the positions display more distributed probabilities across multiple amino acids, indicating regions of greater flexibility that may tolerate mutation. Hydrophobic residues such as leucine (L), isoleucine (I), and valine (V) are highly present across many positions, and are consistent with the transmembrane nature of the protein. Overall, the probability distribution highlights a balance between conserved structural regions and more flexible positions, which can guide rational mutation design.
RESOURCES:
American Chemical Society, 2023. Unpicking nature’s invisibility cloak [online image]. Available at: https://axial.acs.org/biology-and-biological-chemistry/unpicking-natures-invisibility-cloak [Accessed 29 Mar. 2026].
RCSB PDB (2023) 8GI8: Kalium channelrhodopsin 1 from Hyphochytrium catenoides (HcKCR1) embedded in peptidisc. Available at: https://www.rcsb.org/structure/8GI8 (Accessed: 29 March 2026).
USE OF AI
“Help me analyze this output from collab and take me step by step on what is happening and why, take me step by step”
“Why is the prediction of a 3d fold so different from one from PDB? What is happening?”
“Edit this paragraph analysis”