Week 5 HW: Protein Design Part II
Week 5 HW: Protein Design Part II
(Carnaroli et al.,2025)
PART A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1. Then decide which ones are worth advancing toward therapy. You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling PeptiVerse: therapeutic property prediction moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
SEQUENCE OF SOD1 Protein with A4V MUTATION
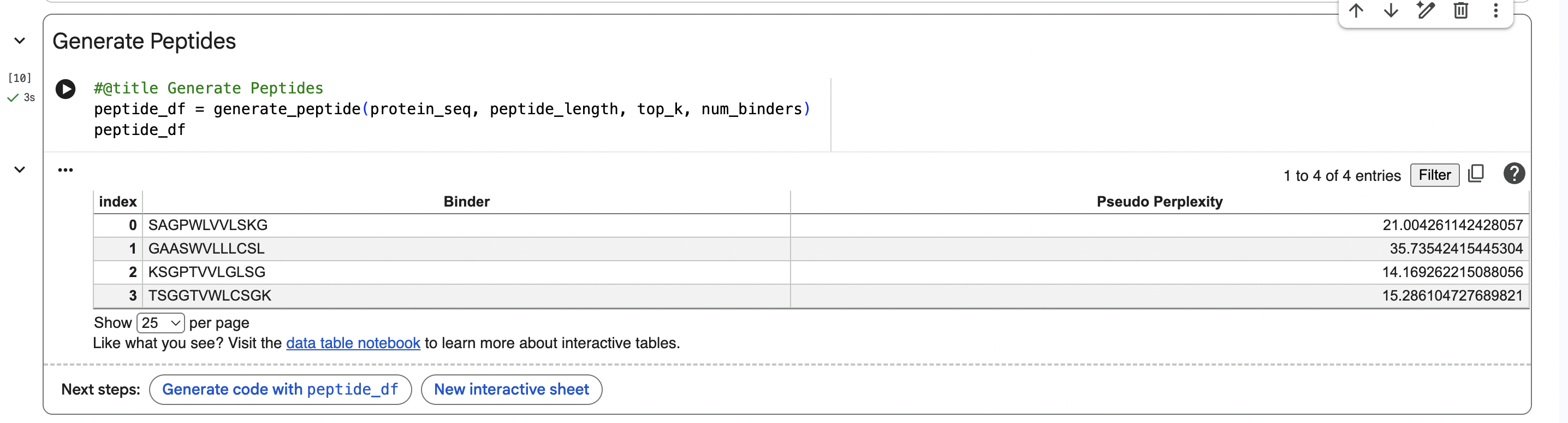
PART 1: Generate Binders with PepMLM
PepMLM Sequences:
PART 2: Evaluate Binders with AlphaFold3
Alphafold outcome:
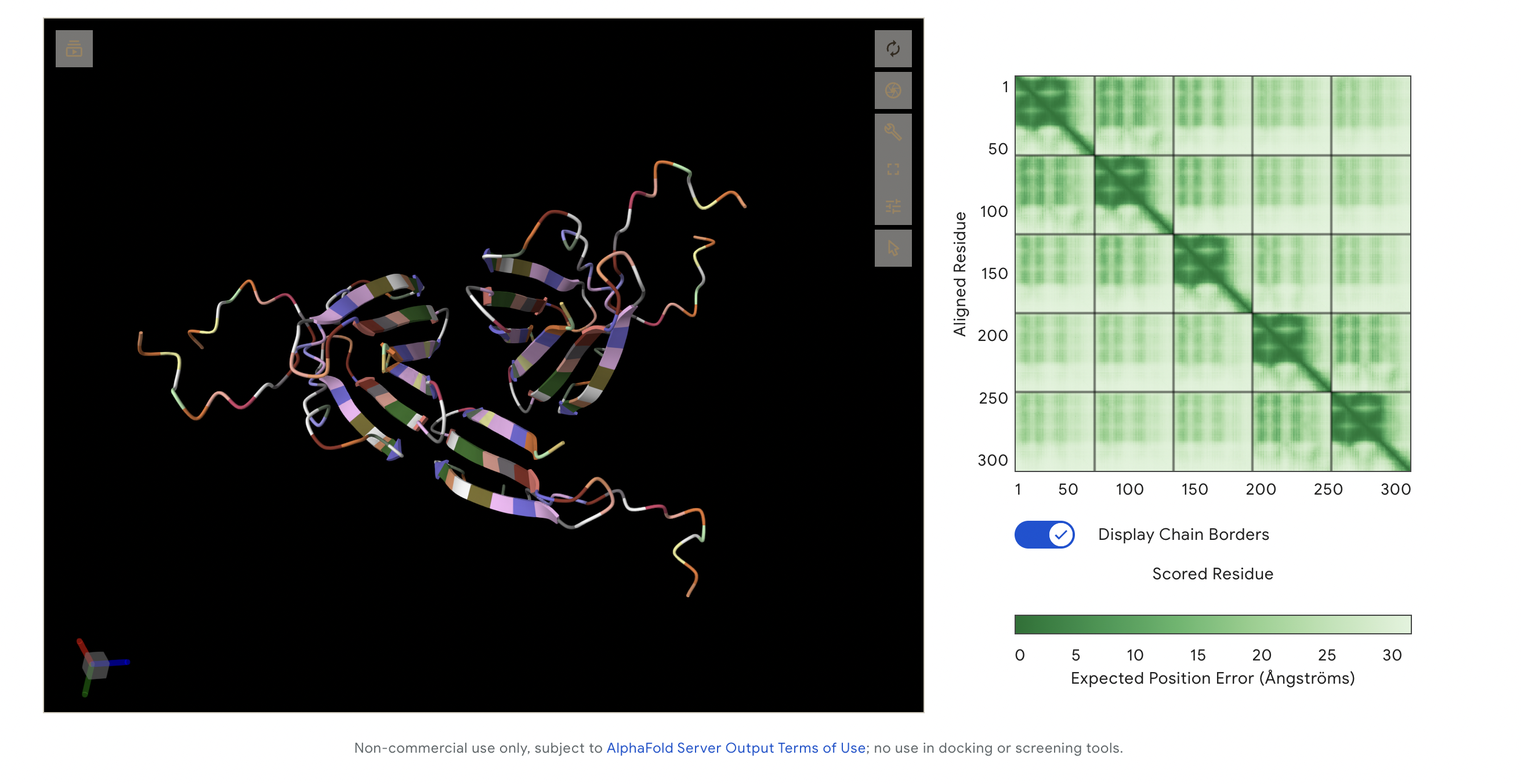
This image captures all peptides in one visualisation as I was meant to add them all together on alphafold. This made it hard to read, but I like the visualisation outcome, as it looks like a single protein made up of 3 large segments. The following are each of the peptide variants individually.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
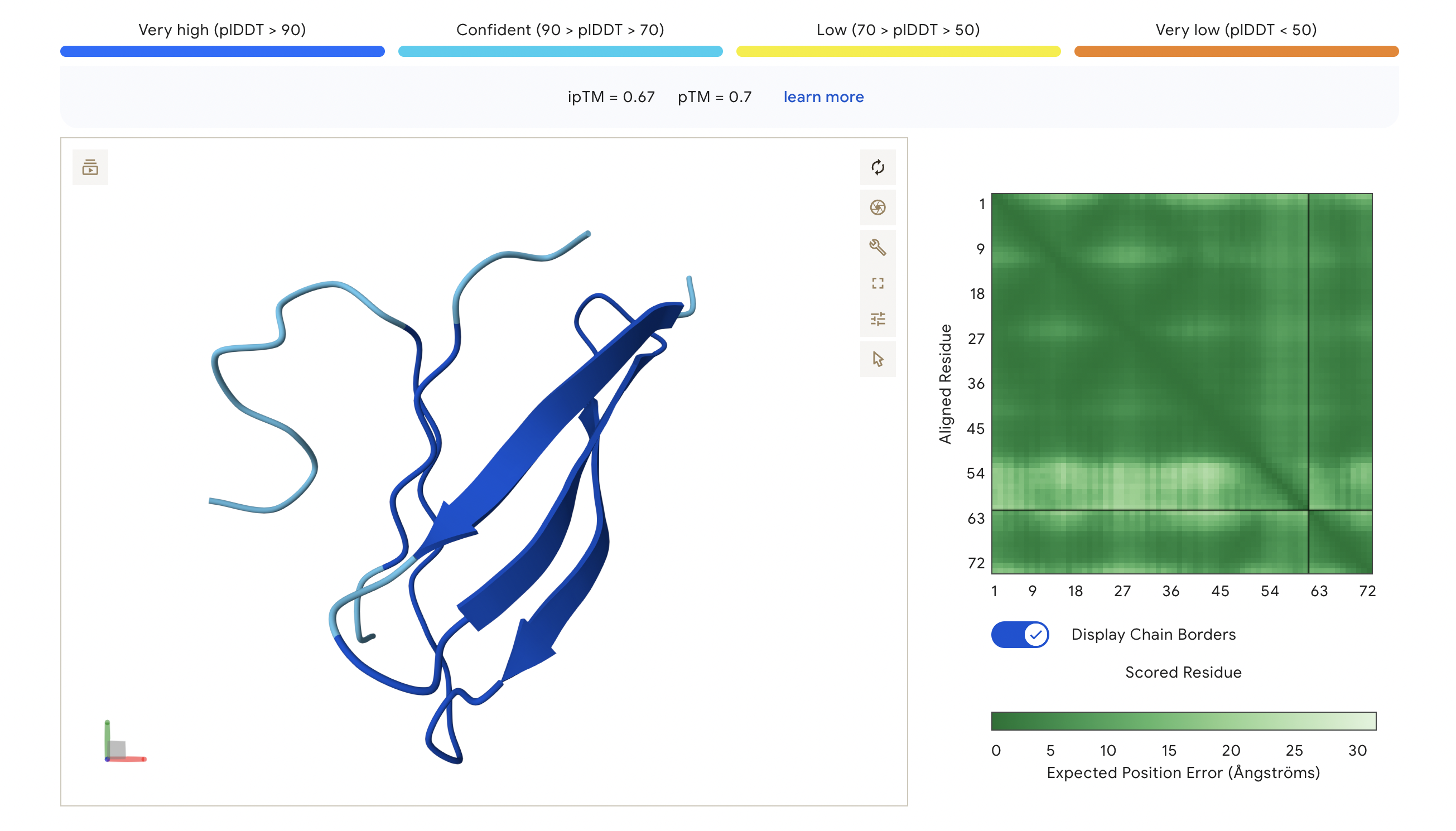 Figure 1
Figure 1
Peptide 1: SAGPWLVVLSKG
The ipTM score of this peptide is 0.67. This is the highest ipTM of all the peptides, indicading the most confident interaction between the peptide and A4V mutation. As seen in (Figure 1), the peptide seems to bind at the surface of the β-barrel but tucked stretching from one end of the protein chain to the other.
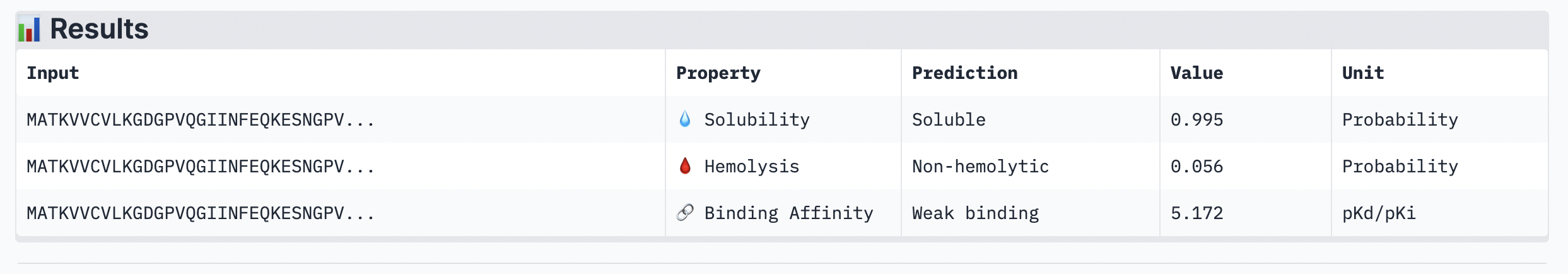
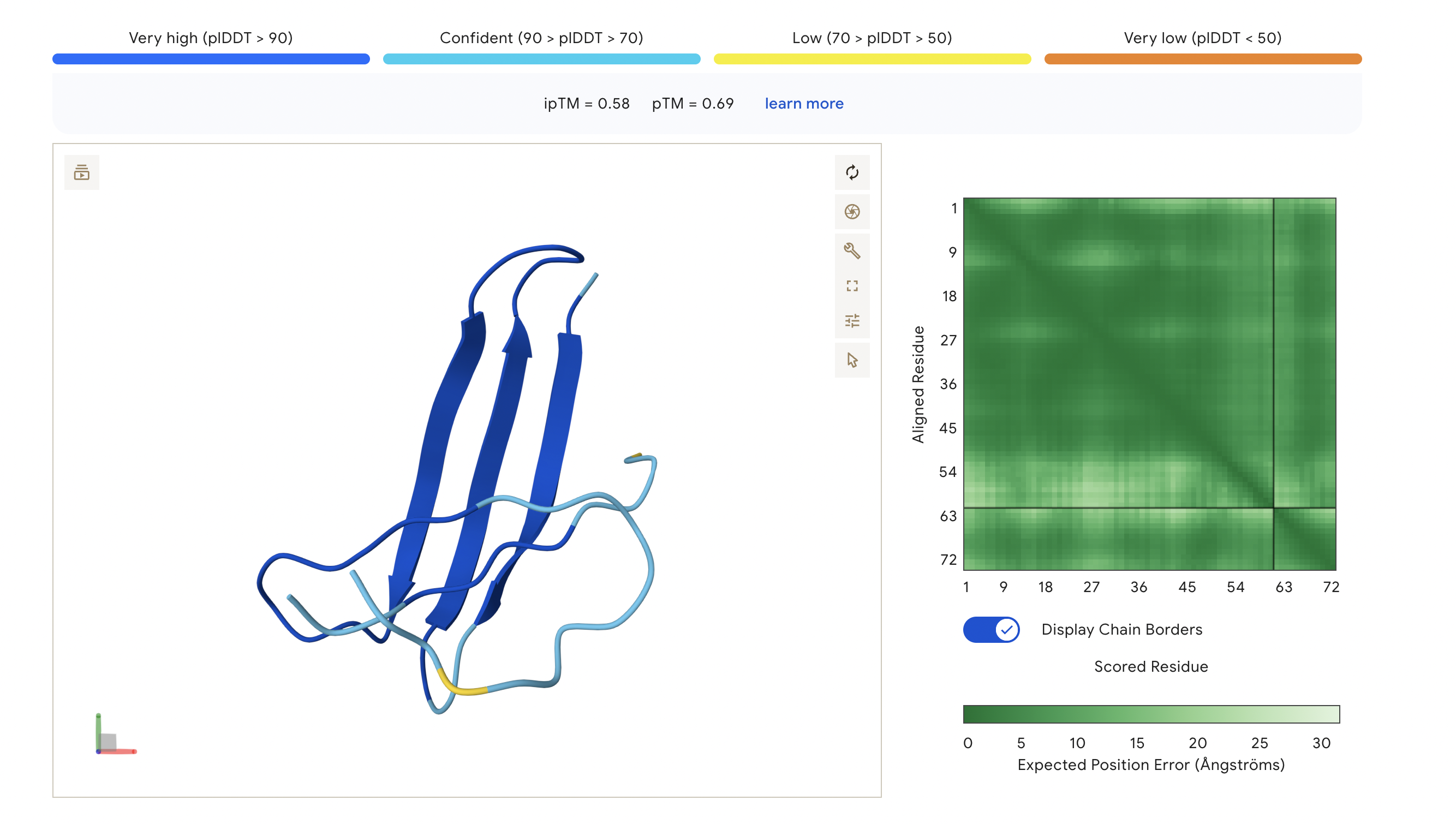 Figure 2
Figure 2
Peptide 2: GAASWVLLLCSL
The ipTM score of this peptide is 0.58 which is the second most confident, a moderately stable interaction. The peptide is also at the surface and more tucked in near the C terminus, stabalizing a different interaction rather than the mutation located at the 4th amino acid position of the protein sequence.
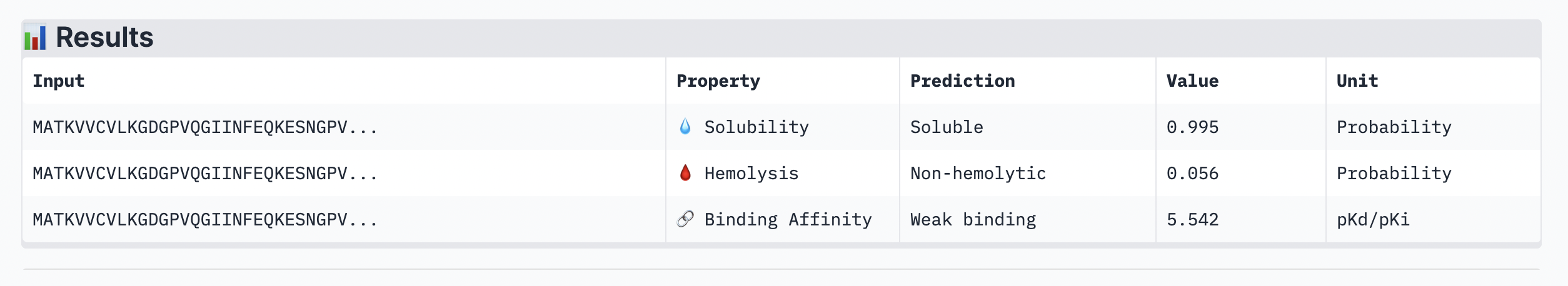
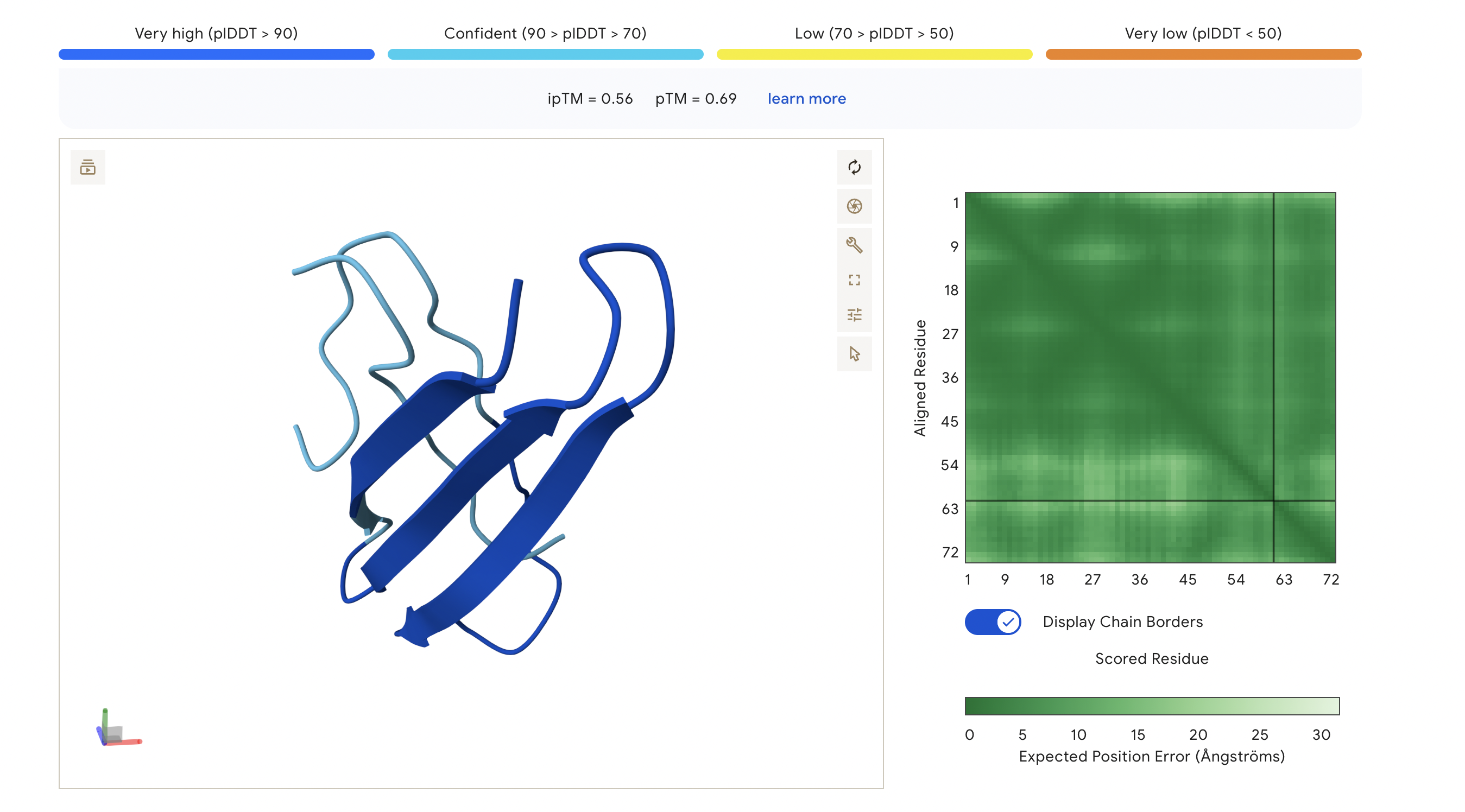 Figure 3
Figure 3
Peptide 3: KSGPTVVLGLSG
The ipTM score for this peptide is 0.56 which is the third most confident interaction. Even though it’s lower than the peptide 2 in (Figure 2), this peptide shows a more structural and stable composition, indicated by the blue colours in the regions, compared to the other, with some more unstable yellow regions. The peptide is also tucked and surfacing near the C terminus.
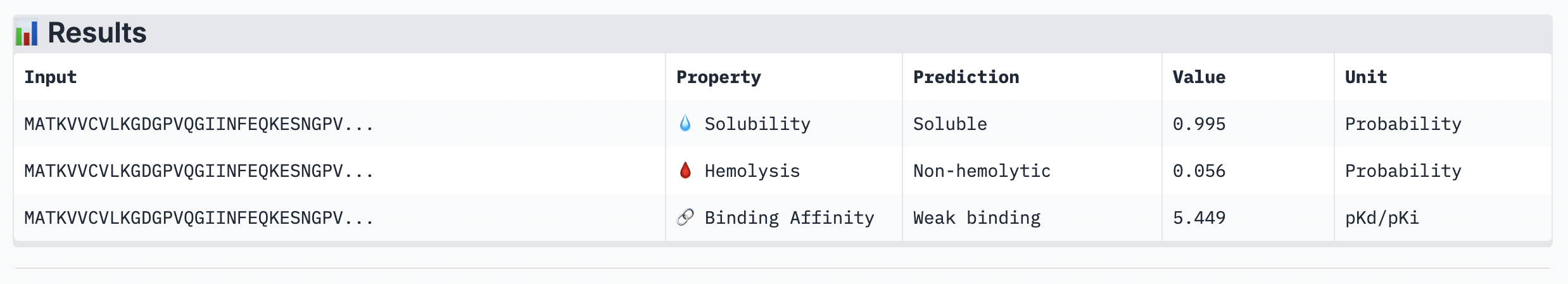
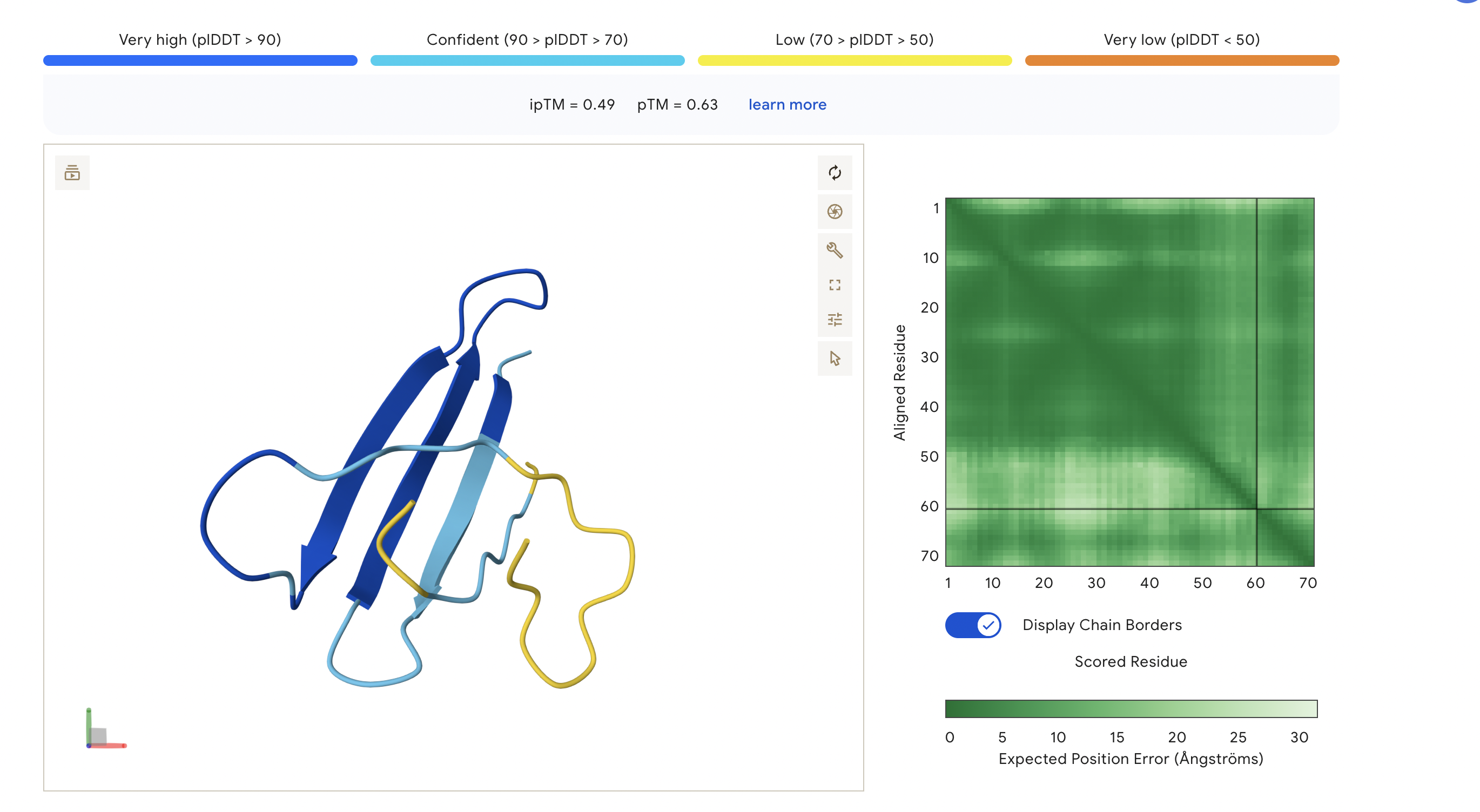 Figure 4
Figure 4
Peptide 4: TSGGTWLCSGK
The ipTM score for this peptide is 0.49 and is the second least confident. The peptide seems to stretch tucked from the N terminus to the C terminus, which has low structural stability.
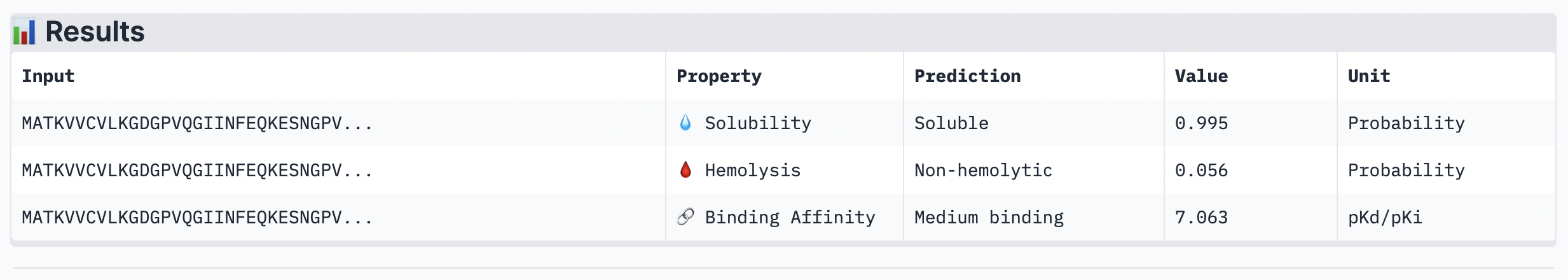
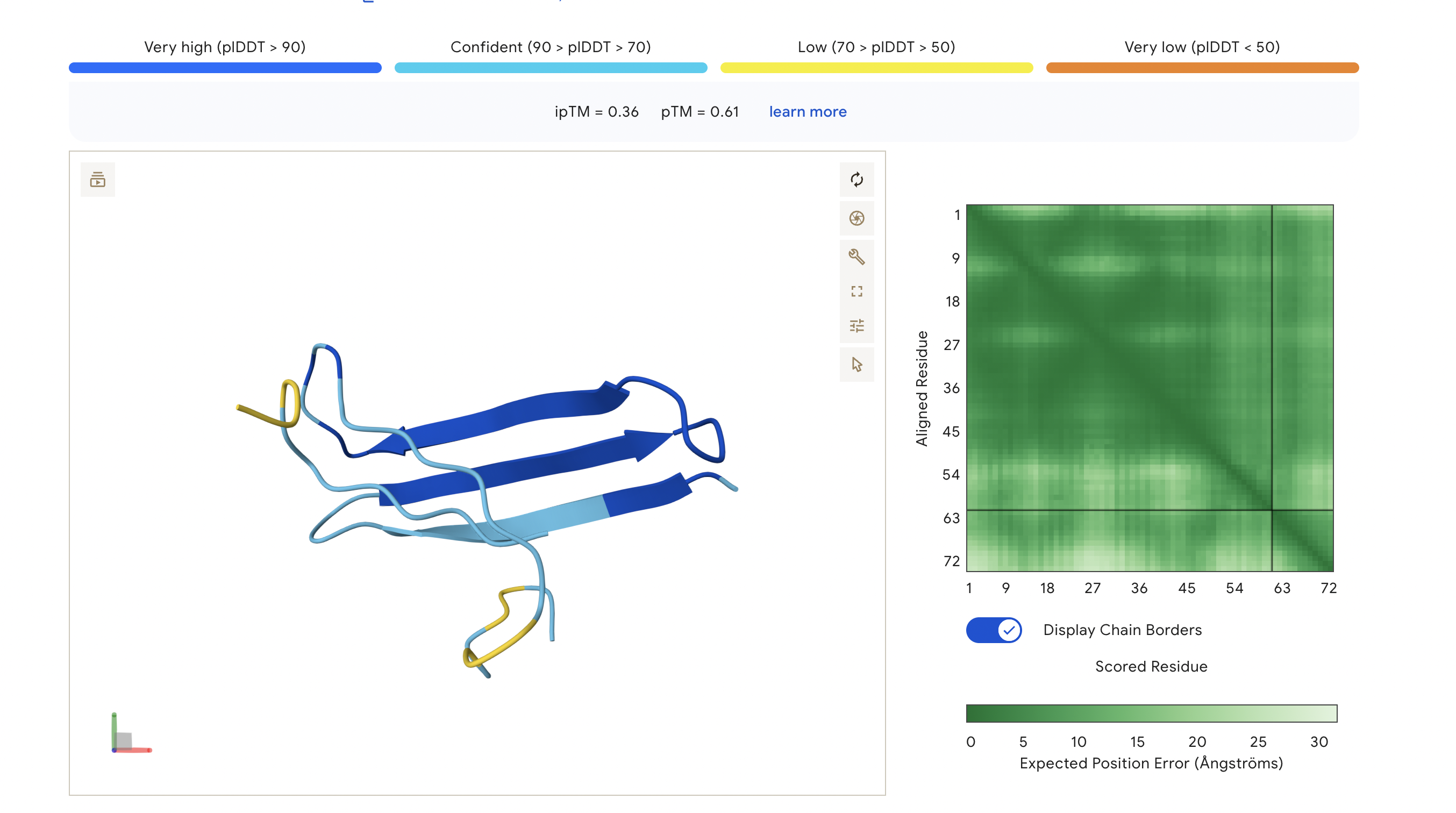 Figure 5
Figure 5
Peptide 5: FLYRWLPSRRGG
The ipTM score for this peptide is 0.36, and is the least confident bind. The peptide is surfacing near the C terminus, and shows little structural stability, whereas the β-barrel is structurally confident.
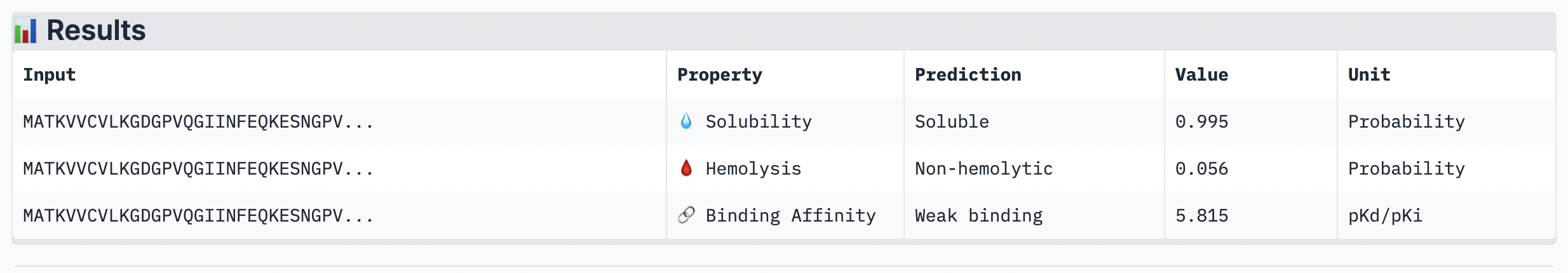
Analysis
While ipTM often reflects how well the peptide fits structurally, it doesn’t always guarantee stronger binding affinity. In the analysis, peptide 4 had a lower ipTM score (0.49), indicating a less stable structural fit. Yet, it showed the highest predicted binding affinity (7.063), suggesting it forms key interactions despite imperfect alignment. This highlights that structural confidence and binding predictions can diverge. In choosing a peptide to advance, one can balance both structural fit and therapeutic properties. Peptide 4 stands out as a strong candidate due to its higher affinity, making it worth advancing despite the structural uncertainty.
PART 4: Generate Optimized Peptides with moPPIt
Final peptides produced
AGKKKEKEKKLN Final score: [0.9788, 0.9779, 0.8333]
KEKKKNTFEKKN Final score: [0.9860, 0.9842, 0.9167]
KKKKGDTKESQE Final score: [0.9886, 0.9891, 1.0000]
BEST —> WORST KKKKGDTKESQE, KEKKKNTFEKKN, AGKKKEKEKKLN
The mmplot/moPPIt optimisation appears to have been successful, as all three candidate peptides showed clear improvements in score over the course of sampling. Among the generated sequences, KKKKGDTKESQE was the strongest overall candidate, achieving the best final multi-objective profile (0.9886, 0.9891, 1.0000). KEKKKNTFEKKN also performed very well and would be a strong secondary candidate, while AGKKKEKEKKLN was somewhat weaker due to a lower third-objective score. Overall, the results suggest successful convergence toward high-scoring peptides, though the strong lysine enrichment across all candidates may indicate limited sequence diversity.
RESOURCES
Carnaroli, M.; Deriu, M.A.; Tuszynski, J.A. Computational Search for Inhibitors of SOD1 Mutant Infectivity as Potential Therapeutics for ALS Disease. Int. J. Mol. Sci. 2025, 26, 4660. https://doi.org/10.3390/ijms26104660
USE OF AI
“Why is my protein so symmetrical? Have I done it wrong? Walk me step by step on using AlphaFold with 5 different peptides to analyze seperatley (key word)”
“Help me understand the nature of this protein and walk me through how one would approach analysis on alphafold”
“Explain to me each part of the protein and what exactly I should be looking for when analysing its structure and confidence as a successful protein peptide.”
“What am I looking for on mmplot, walk be step by step”
“Edit this paragraph analysis”
Part C: Final Project: L-Protein Mutants
{{PhageLysisProteinDesignChallenge.pdf}} !()[PhageLysisProteinDesignChallenge.pdf]