Week 2: DNA Read, Write, & Edit
Part 1. Benchling & In-silico Gel Art
This week’s lab followed the protocol detailed in “Gel Art: Restriction Digests and Gel Electrophoresis”. The first step was to make a free account at benchling.com and import the Lambda DNA as seen in [Figure 1] below.
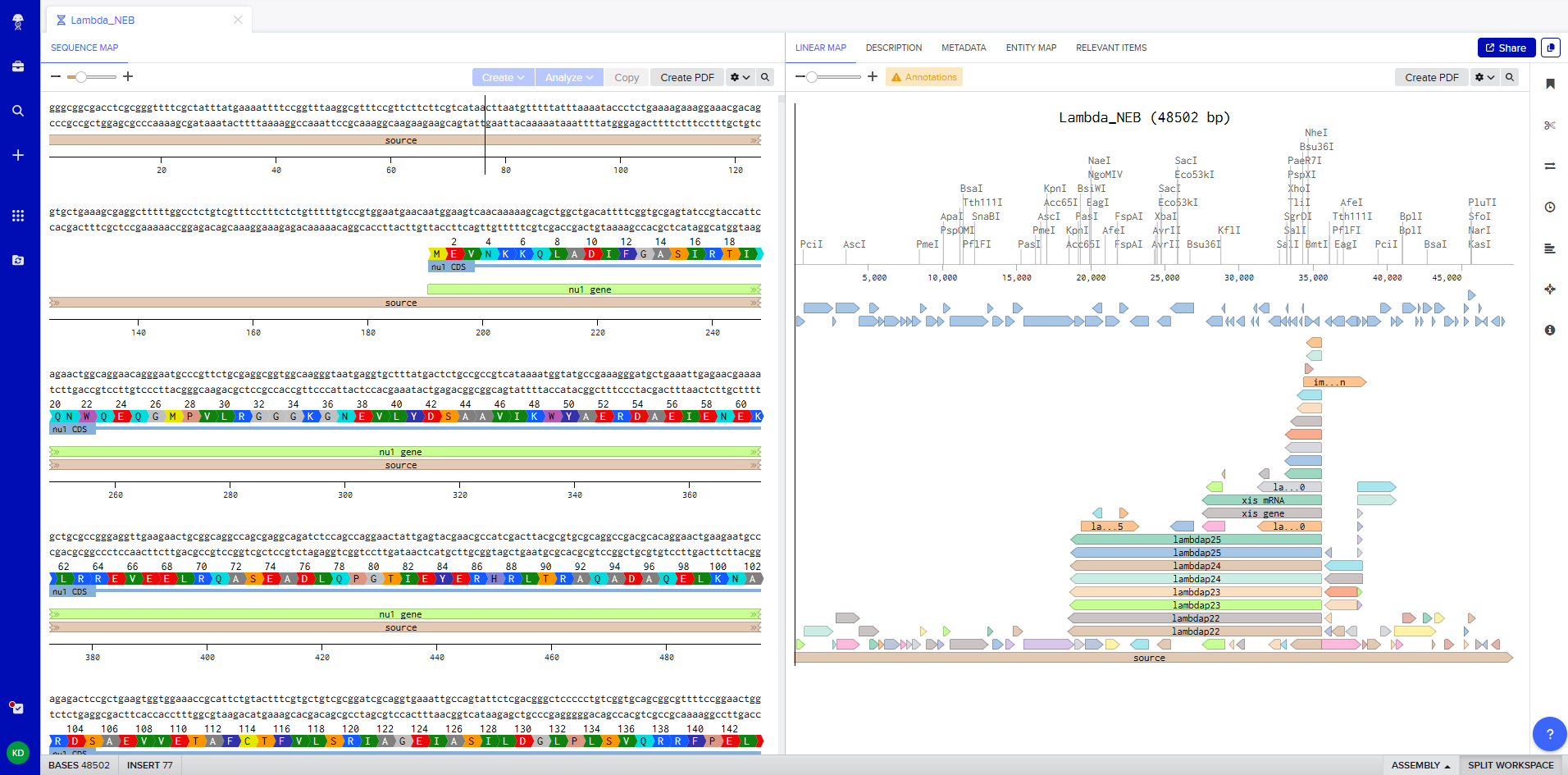 Figure 1. Imported the Lambda DNA.
Figure 1. Imported the Lambda DNA.
The next step was to simulate restriction enzyme digestion with the following enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
Figure 2 shows the virtual digest with EcoRI and Figure 3 shows the virtual digest with all the enzymes mentioned above.
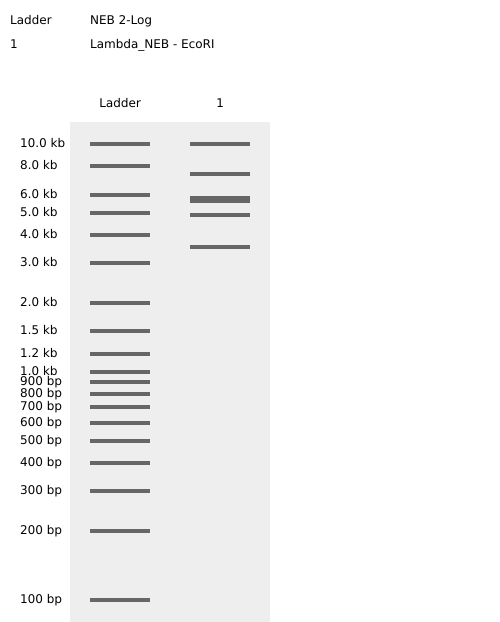 Figure 2. EcoRI virtual digest.
Figure 2. EcoRI virtual digest.
 Figure 3. Full virtual digest.
Figure 3. Full virtual digest.
The last step was to create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. For this I used Ronan’s website which was a helpful tool to iterate designs, especially since the physical lab experiment could not be carried out due to lab and equipment restrictions.
This was my original idea to make a penguin:
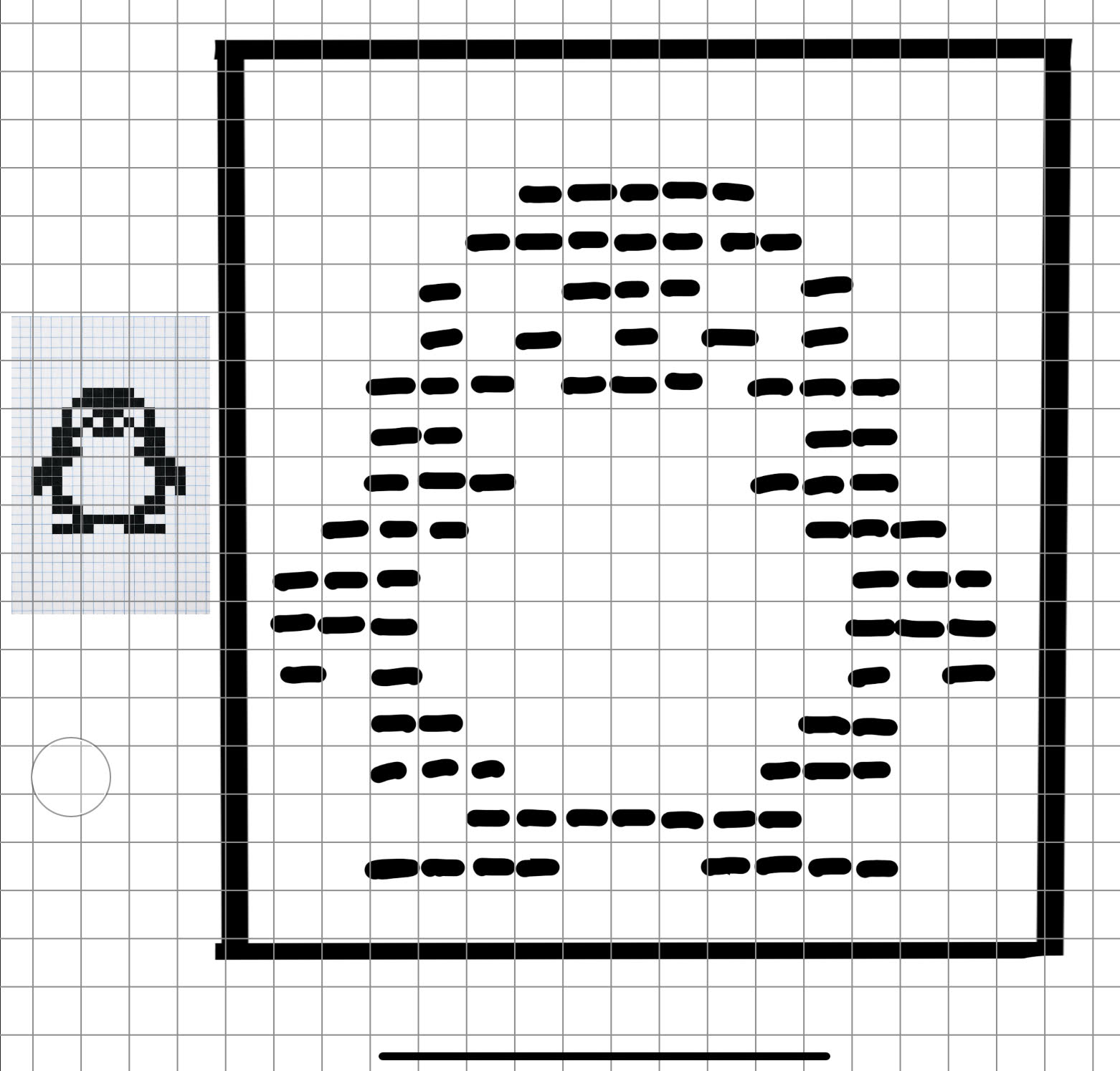 Figure 4. Pixel penguin sketch.
Figure 4. Pixel penguin sketch.
The result using Ronan’s website:
 Figure 5. Attempt at designing a penguin in the style of latent Figure Protocol.
Figure 5. Attempt at designing a penguin in the style of latent Figure Protocol.
These are the gel restriction digest per row:
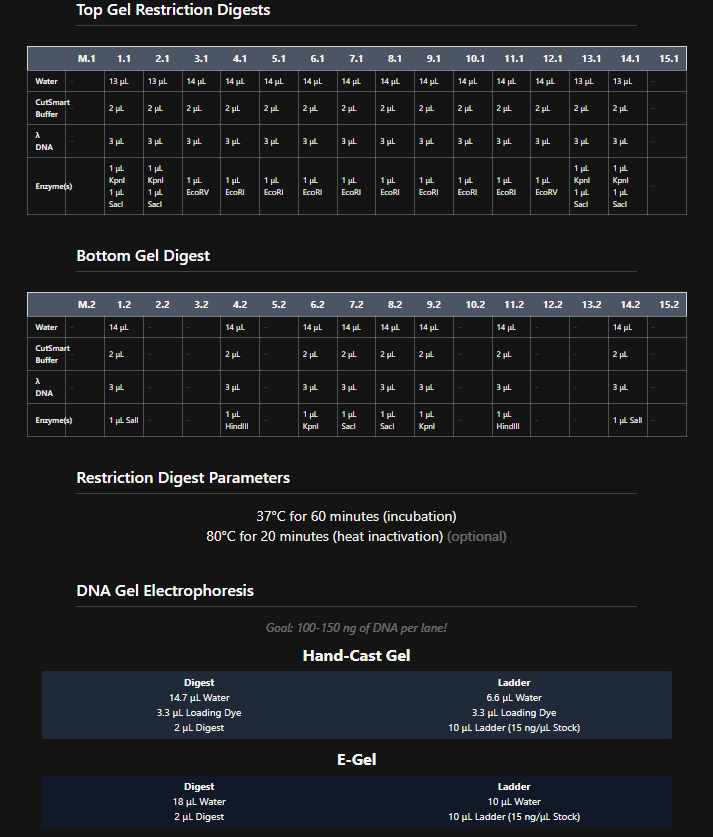 Figure 6. Tables of restriction enzymes used per row and per layer.
Figure 6. Tables of restriction enzymes used per row and per layer.
Part 3: DNA Design Challenge
1. Choose your protein.
For the Week 1 HW I focused on engineering a biodegradable pod that would control the release of localized beneficial bacteria that could help corals survive heat stress. For this week’s HW, I’ve selected the protein manganese superoxide dismutase (MnSOD) which is the enzyme that helps corals neutralize harmful reactive oxygen species (ROS). When ocean temperatures rise during bleaching events, corals experience oxidative stress, and their cells produce dangerous levels of superoxide radicals (O₂⁻). MnSOD is part of the coral’s natural antioxidant defense system, converting these damaging radicals into hydrogen peroxide (H₂O₂) and oxygen (O₂) [1], which are then further neutralized by other enzymes like catalase.
Protein Selected: Manganese Superoxide Dismutase (MnSOD) from Stylophora pistillata
Accession Numbers:
- UniProt: A3KLM5 (A3KLM5_STYPI)[2]
- NCBI GenBank: AAX99423.1[3]
Organism: Stylophora pistillata (Smooth cauliflower coral)
 Figure 1. A photo of Stylophora pistillata [4].
Figure 1. A photo of Stylophora pistillata [4].
Length: 156 amino acids (partial sequence)
Protein Existence: Evidence at transcript level
Protein Sequence:
AAX99423.1 manganese superoxide dismutase, partial [Stylophora pistillata] YDYDALQPAISAEIMQLHHQKHHATYVNNLNVAAEEKFSEAQAKGDTSAMISLQPALKFNGGGHINHSIFWTNLSPNGGGEPT GALMEAIKEDFGSFENFKERFNAATVAVQGSGWGWLGYSKADKGLVITTCANQDPLQATTGLVPLLGMDVWEHA
2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Using the Central Dogma framework, I worked backwards from the MnSOD protein sequence to find its corresponding DNA sequence. The protein sequence I selected (AAX99423.1) was originally derived from the mRNA record AY916505.1 [2,5]. Because this record includes a /codon_start=3 annotation, I removed the first two nucleotides (which likely represent 5’ UTR sequence) to obtain the actual coding sequence. The resulting 468-nucleotide sequence (156 codons) exactly matches the partial MnSOD protein.
DNA sequence (from NCBI AY916505.1):
MnSOD DNA sequence (partial cds) tacgatgctttacaaccagcaatcagtgcagaaattatgcaacttcatcaccagaaacatcatgcaacatatgtgaacaacttgaatgtagccgaagaaaagttttctgaggcgcaagctaaaggagataccagtgctatgatatcactccagccagccttgaaattcaatggaggaggacatattaatcactcaatattttggacaaatctctctcctaatggtggaggtgaaccaacaggagccttgatggaagctatcaaggaagactttggttcatttgaaaactttaaggaaaggttcaatgcagcaactgtagctgtgcagggctcaggatggggttggctgggttatagcaaggctgacaagggcctggtgatcaccacatgtgccaatcaagaccctctccaggccaccacaggactggtgccacttcttggaatggatgtctgggaacacgca
3. Codon optimization.
Codon optimization is necessary because of codon usage bias. Most amino acids are encoded by multiple codons, however, different organisms have evolved preferences for specific codons based on the availability of matching tRNA molecules in their cells [6,7]. For example, attempting to express the coral MnSOD gene in E. coli could result in low protein yield, truncated proteins, or no expression at all because there would be codons that are rare in bacteria [7]. By codon-optimizing the sequence, the coral gene is essentially translated for the host organism to replace those rare codons with preferred ones but still maintaining the same amino acid sequence. This maximizes translational efficiency and protein yield.
I’ve chosen Escherichia coli (E. coli) as the host organism for codon optimization as it is the most well-characterized expression system, it has well-documented codon usage tables, optimization tools freely available, and it is fast, cheap, and scalable [8]. I used the GenScript GenSmart™ Codon Optimization Tool [9] with the following parameters:
- Host organism: E. coli
- Excluded restriction sites: BsaI, BsmBI, BbsI (to eliminate Type IIs enzyme recognition sequences
- Optimize for high protein expression
Original MnSOD DNA sequence (from 3.2):
tacgatgctttacaaccagcaatcagtgcagaaattatgcaacttcatcaccagaaacatcatgcaacatatgtgaacaacttgaatgtagccgaagaaaagttttctgaggcgcaagctaaaggagataccagtgctatgatatcactccagccagccttgaaattcaatggaggaggacatattaatcactcaatattttggacaaatctctctcctaatggtggaggtgaaccaacaggagccttgatggaagctatcaaggaagactttggttcatttgaaaactttaaggaaaggttcaatgcagcaactgtagctgtgcagggctcaggatggggttggctgggttatagcaaggctgacaagggcctggtgatcaccacatgtgccaatcaagaccctctccaggccaccacaggactggtgccacttcttggaatggatgtctgggaacacgca
Codon-optimized MnSOD sequence for E. coli:
TATGATGCACTACAACCCGCTATATCAGCGGAGATCATGCAACTGCATCACCAGAAGCACCACGCCACGTACGTGAATAACTTAAATGTTGCGGAAGAGAAGTTCAGCGAAGCGCAGGCGAAAGGTGACACCAGCGCAATGATCTCGCTCCAACCGGCTTTGAAATTCAACGGCGGCGGCCATATCAACCACAGCATTTTTTGGACCAACTTGTCCCCGAATGGTGGCGGAGAACCGACTGGTGCACTGATGGAAGCGATTAAAGAGGACTTCGGCTCCTTCGAGAACTTTAAAGAGCGTTTTAACGCCGCTACCGTTGCGGTCCAGGGTTCTGGTTGGGGTTGGCTGGGCTATAGCAAGGCCGATAAGGGCCTGGTTATTACCACGTGCGCTAATCAGGATCCACTGCAAGCGACCACCGGTCTGGTGCCGTTGCTGGGTATGGACGTGTGGGAACATGCG
4. You have a sequence! Now what?
There are two main approaches I could take to produce the MnSOD protein:
- using living organisms as protein factories (cell-dependent methods)
- using extracted cellular machinery in a test tube (cell-free methods)
The cell-dependent method, which uses living bacteria, would first require cloning the MnSOD gene into an expression vector [10]. To do that the gene would have to be inserted into a plasmid that contains all the elements needed for expression in E. coli. That would include: a promoter to initiate transcription, a ribosome binding site to start translation, a terminator to stop transcription, and an antibiotic resistance gene to select for bacteria that took up the plasmid. Then, the plasmid would be introduced into competent E. coli cells through heat shock or electroporation. The bacteria that take up the plasmid become antibiotic-resistant, so they can be grown on plates containing that antibiotic. Once there’s a colony of transformed bacteria, they can be grown in liquid culture. When the culture reaches the right density, an inducer like IPTG for a lac promoter would be added to turn on transcription of the MnSOD gene. The bacteria’s RNA polymerase reads the DNA, makes mRNA, and the ribosomes translate that mRNA into protein. Finally, after a few hours of expression, spin down the bacteria, lyse them, and purify the MnSOD protein using techniques like affinity chromatography.
The cell-free methods are essentially protein synthesis in a tube and skips the living organism [11]. For the MnSOD gene, begin by taking E. coli cells, lysing them, and spinning out all the cell debris and genomic DNA. What’s left are all the components needed for transcription and translation, such as ribosomes, tRNAs, amino acids, RNA polymerase, energy regeneration systems, etc. Then, add the purified MnSOD gene with a strong promoter like T7 directly to this extract. The cell-free system does the transcribing and translating of the gene in a few hours. Since there’s no cell wall to break open, purification is simpler. Spin down any precipitates and collect the protein from the supernatant. The cell-free methos is faster, works for toxic proteins that might kill living cells, and allows for easy modification of reaction conditions [12]. The trade-off is cost since it’s more expensive per milligram of protein than living cultures [11].
Whether in cells or in a tube, the fundamental processes of transcription and translation are the same. For MnSOD, the ribosome would link 156 amino acids in exactly the order specified by the optimized codons, and the newly made protein would fold into its functional 3D shape.
In general, the process is the following: Transcription (DNA → RNA) [13] -
- An enzyme called RNA polymerase binds to the promoter region of the gene
- It unwinds the DNA and reads the template strand
- It builds a complementary mRNA molecule, replacing every T with a U
- When it hits the terminator, it releases the finished mRNA
Translation (RNA → Protein) [13]-
- The mRNA binds to a ribosome
- tRNAs bring specific amino acids (each tRNA has an anticodon that matches a codon on the mRNA)
- The ribosome moves along the mRNA, three nucleotides at a time
- Amino acids are linked together in a growing chain
- When the ribosome hits a stop codon (UAA, UAG, or UGA), it releases the finished protein
5. How does it work in nature/biological systems?
In nature, organisms can produce multiple different proteins from a single gene through several mechanisms. The two most common are alternative splicing in eukaryotes, and alternative translation initiation in both prokaryotes and eukaryotes. Alternative splicing happens after transcription but before translation. When a gene is transcribed, the initial RNA (pre-mRNA) contains both exons and introns. The spliceosome removes the introns and joins the exons together. By choosing different combinations of exons, a single gene can produce multiple different mRNA variants, each encoding a different protein isoform [14]. This is very common in humans, with over 95% of our genes undergoing alternative splicing, which is part of why we can have around 20,000 genes but make thousands of different proteins.
Alternative translation initiation happens when a single mRNA has multiple start codons (AUG) in different reading frames or at different positions. Ribosomes can start translation at these different sites, producing proteins with different N-termini from the same transcript. This is more common in viruses and bacteria but happens in eukaryotes too. For the MnSOD gene, it is just one protein, but the central dogma still applies, information flows from DNA to RNA to protein.
To see how this work, I’ve taken a small section of the MnSOD gene and traced it through transcription and translation. I used the first 15 amino acids of the protein to create the alignment. This is the DNA sequence segment from the optimized gene:
TATGATGCACTACAACCCGCTATATCAGCGGAGATCATGCAACTGCATCACCAGAAG
During transcription, an enzyme called RNA polymerase reads the DNA template strand and builds a complementary mRNA copy. Every T in the DNA becomes a U in the RNA. Here’s what the RNA looks likes:
AUACUACGUGAUGUUGGGCGAUAUAGUCGCCUCUAGUACGUUGACGUAGUGGUCUUC
The ribosome reads the mRNA in groups of three nucleotides and matches each codon to an amino acid. Transfer RNAs (tRNAs) bring the right amino acids based on the codon sequence. This is where the genetic code gets translated into protein.
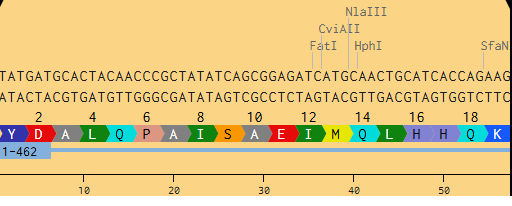 Figure 2. Snapshot from Benchling of MnSOD from DNA to Protein.
Figure 2. Snapshot from Benchling of MnSOD from DNA to Protein.
References
- Demicheli V, Moreno DM, Radi R. Human Mn-superoxide dismutase inactivation by peroxynitrite: a paradigm of metal-catalyzed tyrosine nitration in vitro and in vivo. Metallomics. 2018 May 23;10(5):679-695. doi: 10.1039/c7mt00348j.
- National Center for Biotechnology Information. Manganese superoxide dismutase, partial [Stylophora pistillata]. GenBank: AAX99423.1 [Internet]. 2005 [cited 2026 Feb 17]. Available from: https://www.ncbi.nlm.nih.gov/protein/AAX99423.1.
- UniProt Consortium. Superoxide dismutase (Fragment) OS=Stylophora pistillata OX=50429 GN=AWC38_SpisGene15259 PE=4 SV=1. UniProtKB A3KLM5 [Internet]. 2007 [cited 2026 Feb 17]. Available from: https://www.uniprot.org/uniprotkb/A3KLM5/entry.
- Rusconi G. Stylophora pistillata [image]. In: Sealifebase [Internet]. 2005 [cited 2026 Feb 17]. Available from: https://www.sealifebase.ca/summary/Stylophora-pistillata.html.
- Furla P, Richier S, Allemand D. Stylophora pistillata manganese superoxide dismutase mRNA, partial cds [Internet]. GenBank: AY916505.1; 2005 [cited 2026 Feb 17]. Available from: https://www.ncbi.nlm.nih.gov/nuccore/AY916505.1.
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011 Jan;12(1):32-42. doi: 10.1038/nrg2899.
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004 Jul;22(7):346-53. doi: 10.1016/j.tibtech.2004.04.006.
- Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014 Apr 17;5:172. doi: 10.3389/fmicb.2014.00172.
- GenScript. GenSmart™ Codon Optimization [Internet]. Piscataway (NJ): GenScript; 2026 [cited 2026 Feb 17]. Available from: https://www.genscript.com/gensmart-free-gene-codon-optimization.html.
- Green MR, Sambrook J. Molecular Cloning: A Laboratory Manual. 4th ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2012.
- Gregorio NE, Levine MZ, Oza JP. A User’s Guide to Cell-Free Protein Synthesis. Methods Protoc. 2019 Mar 20;2(1):24. doi: 10.3390/mps2010024.
- Silverman AD, Karim AS, Jewett MC. Cell-free gene expression: an expanded repertoire of applications. Nat Rev Genet. 2020 Mar;21(3):151-170. doi: 10.1038/s41576-019-0186-3.
- Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 6th ed. New York: Garland Science; 2014. Chapter 6: How Cells Read the Genome: From DNA to Protein.
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291-336. doi: 10.1146/annurev.biochem.72.121801.161720.
Part 4: Prepare a Twist DNA Synthesis Order
After creating my Twist and Benchling account, I built my DNA insert sequence to make MnSOD (see [Figure 7,8]). I went through each piece of the DNA sequence and annotated the parts and finally got a Linear Map of the entire sequence [Figure 9]. Then on Twist, I imported the sequence by uploading the FASTA file from Benchling. Since the order is for a clonal gene, I had to then select a cloning vector like pTwist Amp High Copy. I then proceeded to download construct (GenBank) to get the full plasmid sequence and imported this to Benchling to see the plasmid with the expression cassette [Figure 10].
 Figure 7. Annotations for MnSOD.
Figure 7. Annotations for MnSOD.
 Figure 8. MnSOD annotations continued.
Figure 8. MnSOD annotations continued.
 Figure 9. MnSOD Linear Map.
Figure 9. MnSOD Linear Map.
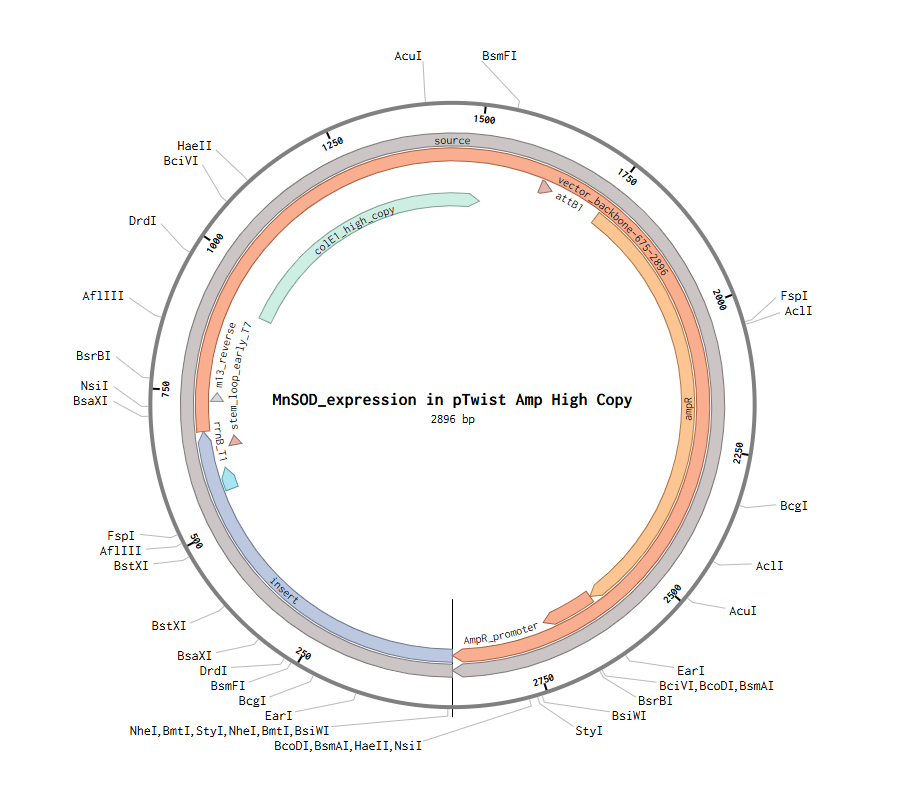 Figure 10. MnSOD plasmid with expression cassette.
Figure 10. MnSOD plasmid with expression cassette.
Part 5: DNA Read/Write/Edit
DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence environmental DNA (eDNA) from coral reef water samples around deployment sites of the biodegradable pods. If the MnSOD-producing bacteria where deployed on a reef, various factors would have to be monitored. The first factor would be whether the strain persists in the environment which eDNA sequencing from water samples could detect using the MnSOD gene itself [1]. Water samples collected at various distances from deployment sites would tell if containment is working by showing whether or not it has spread beyond the target area [1]. Additionally, coral-associated samples would reveal if introducing the probiotic is disrupting the natural microbial community [2].
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina next-generation sequencing (NGS). [3,4] For eDNA metagenomics, the high throughput is essential for capturing the full diversity of microbial communities. Illumina’s short reads have very low error rates (around 0.1-1%) [5], which matters when confirming the gene sequence. Illumina also offers the best balance of data quality and price considering the scale of monitoring required in this specific case. This is a second-generation sequencing technology, and it’s defined by massively parallel sequencing, i.e. reading millions of short DNA fragments simultaneously instead of one at a time like Sanger sequencing [3,4]. The main difference between Sanger, which is first-generation, and Illumina, is the number of fragments read at one time. Sanger reads one fragment at a time [6,7], whilst Illumina can read millions of fragments in parallel [3,4]. Other distinctions include long reads up to 900 bp, slow and expensive but accurate for each base for Sanger [7] vs short reads of up to 300 bp, high accuracy and lower cost per base for Illumina [4]. Third generation methods like PacBio and Nanopore can achieve very long reads up to 100 kb or more but they have higher error rates [8,9].
The input for Illumina NGS would be the DNA extracted from filtered seawater samples or coral mucus swabs. The following are the preparation steps [1,10]:
- Filter large volumes of seawater to capture microbial biomass
- Extract total DNA (including human, fish, microbial, and free DNA)
- Quantify and check quality
- Fragmentation (mechanical or enzymatic)
- Adapter ligation with barcodes (to multiplex many samples)
- PCR amplification
- Library pooling
The first essential step of Illumina sequencing is the library preparation during which DNA is fragmented, and adapters are ligated to both ends [3,4]. Then, the library fragments must be attached to a flow cell surface. Bridge amplification creates thousands of identical copies of each fragment in tight clusters. Fluorescently labeled nucleotides would then be added one at a time. As each nucleotide incorporates, a camera takes an image of the flow cell. The color tells you which base (A, T, C, or G) was added. Then, a software analyzes the images, identifying which base was added to each cluster in each cycle. The sequence of colors across cycles gives you the DNA sequence for each fragment [3,4]. Finally, the millions of short reads are assembled and annotated. The output is then millions of short sequencing reads in FASTQ format, which is a text file containing a read identifier, the nucleotide sequence (A, T, C, G), and quality scores for each base (Phred scores) indicating the confidence in each call [11]. This would provide the community composition data which tells you which microbial species are present and in what relative abundances.
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I want to synthesize a heat-sensing genetic circuit that could eventually be used to monitor coral reef health. The idea is to create bacteria that glow when the water gets too warm providing an early warning sign before corals start to bleach. The DNA I’d synthesize is a single construct containing two parts: a heat-shock promoter from a coral or its symbiotic algae, and a fluorescent protein gene (mCherry) that makes bacteria turn red. When the temperature rises, the promoter turns on, the bacteria produce mCherry, and they start to glow. If it’s possible to make bacteria that glow when the water changes temperature, then it would be possible to swap the mCherry gene for the MnSOD gene later and have bacteria that actually protect corals instead of just reporting on them [2].
The mCherry sequence I’m referring to is the standard version developed by Shaner et al. [12] and codon-optimized for E. coli expression based on the iGEM Parts Registry [13]. I have to find the exact promoter sequence from a coral database separately but the reporter part, the mCherry sequence, is the following:
ATGGTGAGCAAGGGCGAGGAGGATAACATGGCCATCATCAAGGAGTTCATGCGCTTCAAGGTGCACATGGAG GGCTCCGTGAACGGCCACGAGTTCGAGATCGAGGGCGAGGGCGAGGGCCGCCCCTACGAGGGCACCCAGACC GCCAAGCTGAAGGTGACCAAGGGTGGCCCCCTGCCCTTCGCCTGGGACATCCTGTCCCCTCAGTTCATGTAC GGCTCCAAGGCCTACGTGAAGCACCCCGCCGACATCCCCGACTACTTGAAGCTGTCCTTCCCCGAGGGCTTC AAGTGGGAGCGCGTGATGAACTTCGAGGACGGCGGCGTGGTGACCGTGACCCAGGACTCCTCCCTGCAGGAC GGCGAGTTCATCTACAAGGTGAAGCTGCGCGGCACCAACTTCCCCTCCGACGGCCCCGTAATGCAGAAGAAG ACCATGGGCTGGGAGGCCTCCTCCGAGCGGATGTACCCCGAGGACGGCGCCCTGAAGGGCGAGATCAAGCAG AGGCTGAAGCTGAAGGACGGCGGCCACTACGACGCTGAGGTCAAGACCACCTACAAGGCCAAGAAGCCCGTG CAGCTGCCCGGCGCCTACAACGTCAACATCAAGTTGGACATCACCTCCCACAACGAGGACTACACCATCGTG GAACAGTACGAACGCGCCGAGGGCCGCCACTCCACCGGCGGCATGGACGAGCTGTACAAGTAA
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use Twist Bioscience’s silicon-based DNA synthesis platform [14]. Twist synthesizes DNA by building short fragments on silicon chips and then assembling them into longer pieces. Their method is accurate, reliable, and perfect for a project like this where I only need one construct to start testing. First, I would design my sequence and upload it to Twist’s portal. Inside their machine, they synthesize short overlapping oligonucleotides adding one nucleotide at a time in a series of chemical cycles. Then they assemble those fragments into the full-length gene using PCR assembly, where the overlapping ends act like puzzle pieces that only fit together one way. Once the full gene is made, they insert it into a plasmid and transform it into E. coli. They grow up the bacteria, sequence the DNA to make sure it’s 100% correct, and then send me either the purified DNA or a tube of bacteria with my gene inside [14].
In terms of speed, it takes about 5-10 business days to get the DNA [14]. That’s fine for planning experiments but not something you’d use in an emergency. For accuracy, the chemistry isn’t perfect since each coupling step is about 99.5% efficient, meaning that for a 1000 bp gene, only about 1% of full-length products might be error-free. That’s why Twist sequences everything before shipping, so they only send out perfect clones. For scalability, Twist can make thousands of genes at once on their silicon chips but for a single gene like in my case, that doesn’t really matter. The main limitation for me would be cost.
DNA Edit
(i) What DNA would you want to edit and why?
Building on my project to engineer beneficial bacteria for coral reefs, I would want to edit two different targets: the MnSOD gene itself to create improved versions, and the genome of my engineered E. coli chassis to add biosafety features.
First, I would edit the MnSOD gene to test variants that might have higher activity or better stability. By introducing targeted mutations at specific amino acid positions, I could potentially create a superoxide dismutase enzyme that works more efficiently at the higher temperatures corals experience during bleaching events. This kind of protein engineering through gene editing is a common approach to improve enzyme function. Second, and more importantly for my project’s real-world application, I would edit the genome of my engineered E. coli strain to add a kill switch. In my Week 1 governance section, I discussed the importance of containment and preventing engineered bacteria from spreading uncontrollably in the environment. A kill switch is a genetic circuit designed to cause cell death under specific conditions, for example, if the bacteria escape the coral reef environment or if a certain time period has passed. This would address the ethical concerns I raised about disrupting native microbiomes and would make the whole approach much safer.
The specific edit would involve inserting a synthetic gene circuit into a neutral site in the E. coli chromosome. This circuit could be designed so that the bacteria require a synthetic amino acid or a specific chemical signal to survive. Alternatively, it could be a temperature-sensitive switch that kills the bacteria if they leave the warm reef waters. These kinds of biocontainment strategies are actively being developed in synthetic biology to address exactly the safety concerns I outlined in my earlier work [15].
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-Cas9 for both types of edits [16]. CRISPR is the most versatile and precise genome editing tool available, and it works well in E. coli which is my chassis organism. For editing the MnSOD gene on a plasmid, CRISPR-Cas9 would allow me to introduce specific point mutations efficiently. For the more complex task of inserting a kill switch into the bacterial chromosome, CRISPR is ideal because it can target a specific genomic location with high precision [17]. CRISPR-Cas9 works through a simple two-component system. The Cas9 protein is a nuclease that cuts DNA, and a guide RNA (gRNA) directs it to the right location [16].
There are five essential steps to CRISPR-Cas9. First, there needs to be a guide RNA with a 20-nucleotide sequence that matches my target site in the E. coli genome. The target site must be next to a PAM sequence (NGG for the standard Cas9 from Streptococcus pyogenes) which is required for Cas9 to bind. For the kill switch insertion, there would also need to be a donor DNA template which is the kill switch gene sequence flanked by homology arms that match the genomic target site [18]. Then, three components would be introduced into the E. coli cells: a plasmid expressing Cas9, a plasmid expressing the guide RNA, and the donor DNA template for insertion edits. This is typically done through electroporation, which uses an electric field to make cells temporarily permeable to DNA. Inside the cell, the guide RNA binds to Cas9 and directs it to the matching genomic sequence. Cas9 checks for the PAM sequence, unwinds the DNA, and if the match is good, it makes a double-strand break about three nucleotides upstream of the PAM [17]. Next, the cell’s natural repair systems kick in. Without a donor template, the break is repaired by non-homologous end joining (NHEJ), which is error-prone and often disrupts the gene. With a donor template present, the cell can use homology-directed repair (HDR) to copy the donor sequence into the genome, which is how I would insert my kill switch [18]. After editing, I would sequence the target region to confirm the edit worked correctly and that there were no off-target mutations.
For the kill switch insertion, the preparation steps would include:
- Designing the kill switch genetic circuit (promoter, toxin gene, regulator elements)
- Designing guide RNAs targeting a neutral integration site in the E. coli genome (sites like attTn7 that are known to tolerate insertions without disrupting essential genes)
- Designing and ordering the donor DNA template (the kill switch flanked by 500-1000 bp homology arms)
- Cloning the guide RNA into a suitable expression plasmid
- Preparing the Cas9 expression plasmid (or using a strain that already expresses Cas9)
- Preparing competent E. coli cells for transformation
The input materials would be: the two plasmids (Cas9 and gRNA), the donor DNA template (either as linear DNA or on a separate plasmid), and competent cells of my target strain [18].
CRISPR-Cas9 is powerful but has limitations. In terms of efficiency, homology-directed repair (HDR) is much less efficient than non-homologous end joining (NHEJ), especially in bacteria. For inserting a kill switch, only a small percentage of cells that take up the DNA will undergo the correct HDR event. This means I would need a good screening method to find the correct clones [17]. In terms of precision, CRISPR can sometimes cut at off-target sites that are similar but not identical to the intended sequence. This could introduce unwanted mutations elsewhere in the genome. Using high-fidelity Cas9 variants and carefully designing guide RNAs with unique targets can minimize this risk [16]. Another limitation is delivery. Getting all the components into cells efficiently can be challenging, especially for non-model organisms. For E. coli, electroporation works well, but for editing coral symbionts directly, delivery would be much harder. Finally, there’s the PAM requirement. Cas9 can only target sequences next to a PAM, which limits where I can edit. For my kill switch integration site, I would need to find a safe genomic location that also has a suitable PAM sequence nearby. Despite these limitations, CRISPR-Cas9 is still the best choice because it is precise, programmable, and has a huge research community developing improved versions and methods.
References
- Liles MR, Williamson LL, Rodbumrer J, Torsvik V, Goodman RM, Handelsman J. Isolation and Cloning of High-Molecular-Weight Metagenomic DNA from Soil Microorganisms. Cold Spring Harb Protoc. 2009;2009(8):pdb.prot5271.
- Gao B, Ruiz D, Case H, Jinkerson RE, Sun Q. Engineering bacterial warriors: harnessing microbes to modulate animal physiology. Curr Opin Biotechnol. 2024;87:103113.
- Illumina Inc. An Introduction to Next-Generation Sequencing Technology [Internet]. San Diego: Illumina; 2021 [cited 2026 Feb 17]. Available from: https://www.illumina.com/technology/next-generation-sequencing.html.
- Illumina Inc. HiSeq 4000 Sequencing System [Internet]. San Diego: Illumina; 2024 [cited 2026 Feb 17]. Available from: https://www.biocompare.com/23967-Next-Generation-Sequencers/11182181-HiSeq-4000-Sequencing-System/.
- De-Kloet RE, Jansen HJ, Groenen MAM, Megens HJ. Comparison of Pacific Biosciences (PacBio), Oxford Nanopore Technologies (ONT), Illumina (IL), 10× Genomics linked-read sequencing on the Illumina platform (10×), RNA sequencing on the Illumina platform (RNA-seq), BioNano Genomics (BNG) and the genome-wide chromatin conformation capture protocol Hi–C (Hi–C). [Table 3] In: Comprehensive comparison of long-read and short-read sequencing technologies for de novo genome assembly. 2020 [cited 2026 Feb 17]. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC7925608/table/Tab3/.
- Thermo Fisher Scientific. How to Conduct Sanger Sequencing [Internet]. Waltham (MA): Thermo Fisher Scientific; 2025 [cited 2026 Feb 17]. Available from: https://www.thermofisher.com/hk/en/home/life-science/sequencing/sequencing-learning-center/capillary-electrophoresis-information/how-conduct-fragment-analysis0.html.
- University of British Columbia Sequencing and Bioinformatics Consortium. Sanger FAQ [Internet]. Vancouver: UBC; 2025 [cited 2026 Feb 17]. Available from: https://sequencing.ubc.ca/our-services-equipment/sanger-sequencing/sanger-faq.
- Yale School of Medicine. PacBio Single Molecule Real-Time (SMRT) Sequencing System [Internet]. New Haven: Yale University; 2024 [cited 2026 Feb 17]. Available from: https://dev.medicine.yale.edu/keck/microarray/services/long-reads-sequencing/pacbio/.
- Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021 Nov 8;39(11):1348-1365. doi: 10.1038/s41587-021-01108-x.
- Peabody MA, Hullahalli K, Sistu H, Pritchard JR, Walker S. Preparation of functional metagenomic libraries from low biomass samples using METa assembly and their application to capture antibiotic resistance genes. mSystems. 2025 Oct 21;10(10):e01039-25. doi: 10.1128/msystems.01039-25.
- Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010 Apr;38(6):1767-71. doi: 10.1093/nar/gkp1137.
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004 Dec;22(12):1567-72.
- iGEM Foundation. mCherry coding sequence (BBa_J06504) [Internet]. 2022 [cited 2026 Feb 17]. Available from: http://parts.igem.org/Part:BBa_J06504.
- Twist Bioscience. DNA Synthesis Technology [Internet]. San Francisco: Twist Bioscience; 2026 [cited 2026 Feb 17]. Available from: https://www.twistbioscience.com/.
- Chan CTY, Lee JW, Cameron DE, Bashor CJ, Collins JJ. ‘Deadman’ and ‘Passcode’ microbial kill switches for bacterial containment. Nat Chem Biol. 2016 Feb;12(2):82-6. doi:10.1038/nchembio.1979.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21. doi: 10.1126/science.1225829.
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013 Mar;31(3):233-9. doi: 10.1038/nbt.2508.
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013 Nov;8(11):2281-2308. doi: 10.1038/nprot.2013.143.