Week 5: Protein Design II
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Part 1: Generate Binders with PepMLM
The human SOD1 sequence from UniProt (P00441) is:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
In the literature, the A4V mutation refers to the mature protein sequence where the initiator methionine is removed. In the full UniProt sequence (P00441), the mature form starts at position 2 (Ala). Therefore, the alanine at position 4 of the mature protein corresponds to position 5 in the full precursor sequence. So to introduce the A4V mutation in human SOD1, you change that alanine, the fifth residue, to valine.
After introducting the A4V mutation, the sequence becomes:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab, the following four peptides of length 12 aa conditioned on the mutant SOD1 sequence were generated and the known SOD1-binding peptide FLYRWLPSRRGG was added for comparison:
| Peptide | Perplexity |
|---|---|
| WLSGAQTGVLAG | 10.500361 |
| WIYAEVAVVHKA | 20.788801 |
| WRYSATGAKQAA | 10.341327 |
| WSYSVVAAEHLW | 18.361116 |
| FLYRWLPSRRGG | (known binder) |
Part 2: Evaluate Binders with AlphaFold3
| Peptide | ipTM | Binding Location Description |
|---|---|---|
| WLSGAQTGVLAG | 0.49 | Positioned near the β‑barrel region |
| WIYAEVAVVHKA | 0.38 | Located in the β‑barrel region |
| WRYSATGAKQAA | 0.44 | N‑terminal loop (ASP12) and the C‑terminal β‑barrel |
| WSYSVVAAEHLW | 0.29 | Spans the β‑barrel, the metal‑binding region, and the C‑terminal tail |
| FLYRWLPSRRGG | 0.30 | Near the active site, in a loop region, and in strand β7 of the β‑barrel |
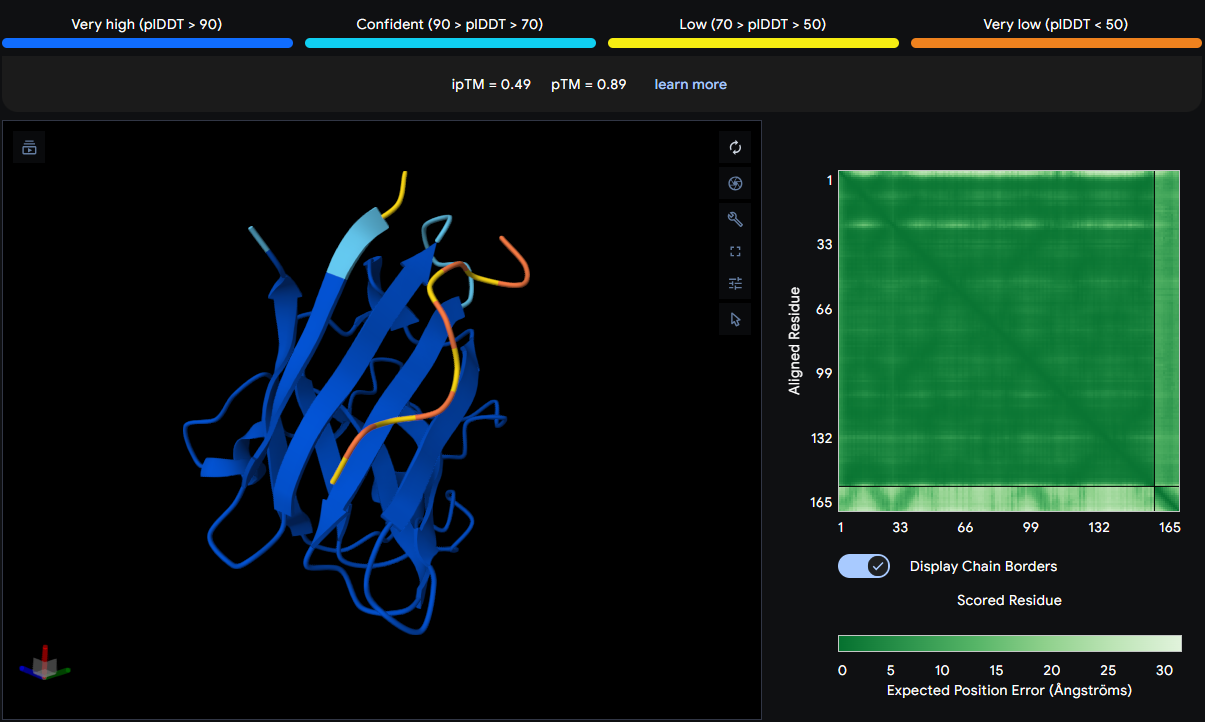
The peptide is positioned near the β‑barrel region of SOD1, specifically in the vicinity of residues 28–36 (a loop connecting β‑strands 2 and 3). It does not localize near the N‑terminus where the A4V mutation sits, nor does it approach the dimer interface. The peptide appears surface‑bound, sitting above the protein surface without forming stable contacts or burying into any pocket. The low ipTM score (0.49) indicates low confidence in this interaction, suggesting that the peptide may not bind stably to SOD1.
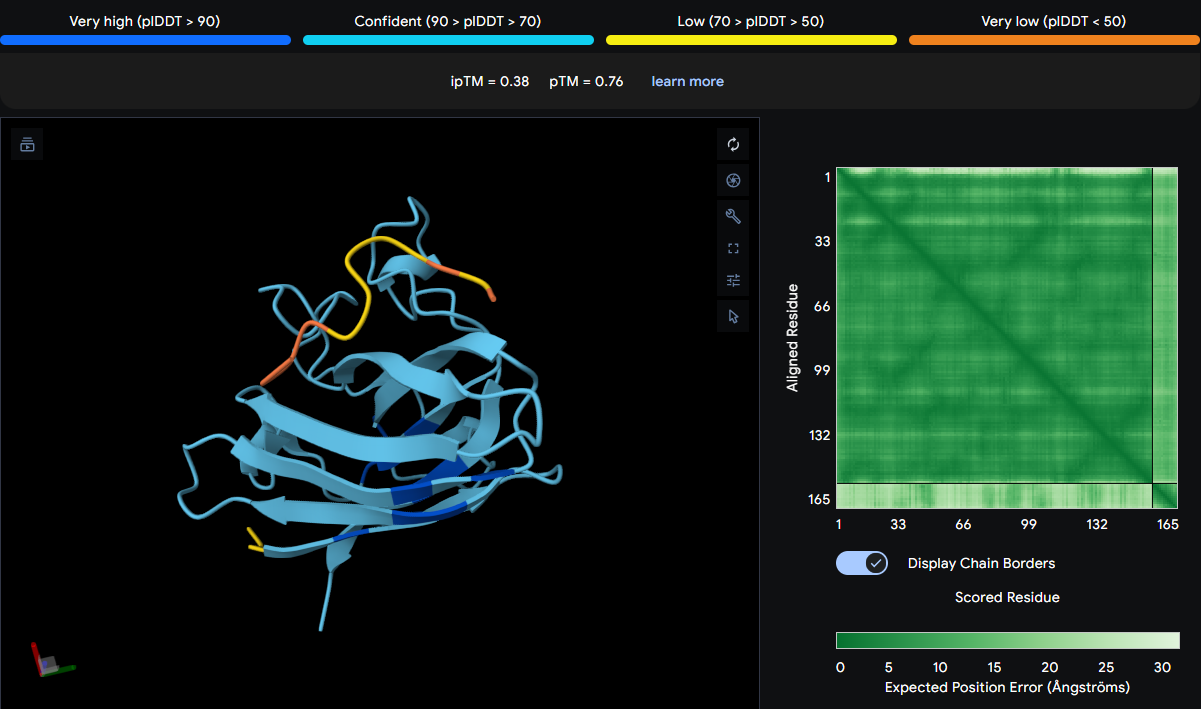
The peptide appears over GLY42 and ILE100, both of which are located in the β‑barrel region (GLY42 in strand β3, ILE100 in strand β6). It does not contact the N‑terminus or dimer interface. The peptide is surface‑bound, with yellow and orange sections suggesting low confidence in its exact placement. The low ipTM score reflects weak predicted binding.
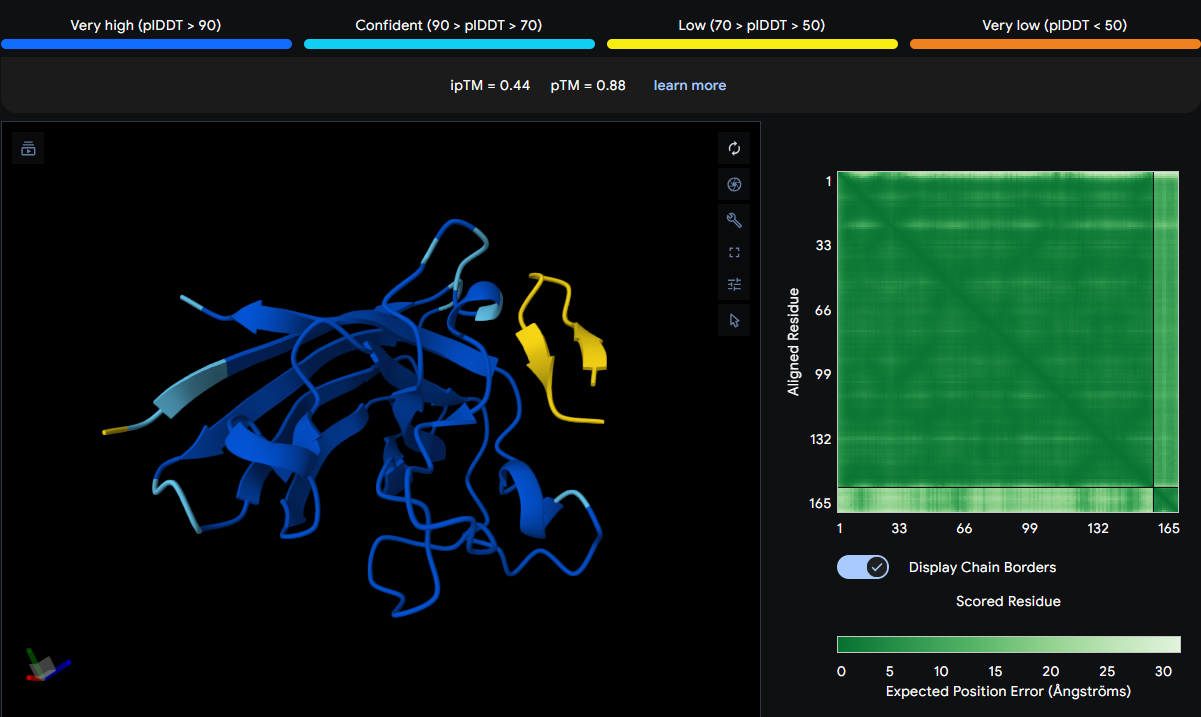
The peptide spans across multiple residues: ASN140, ASP12, THR138, VAL119, and HIS47. This places it in two distinct regions: the N‑terminal loop (ASP12) and the C‑terminal β‑barrel (residues 119–140). HIS47 is part of the metal‑binding loop. The peptide appears thicker and yellow in the viewer, indicating conformational flexibility or uncertainty. It is surface‑bound and does not engage a single defined site.
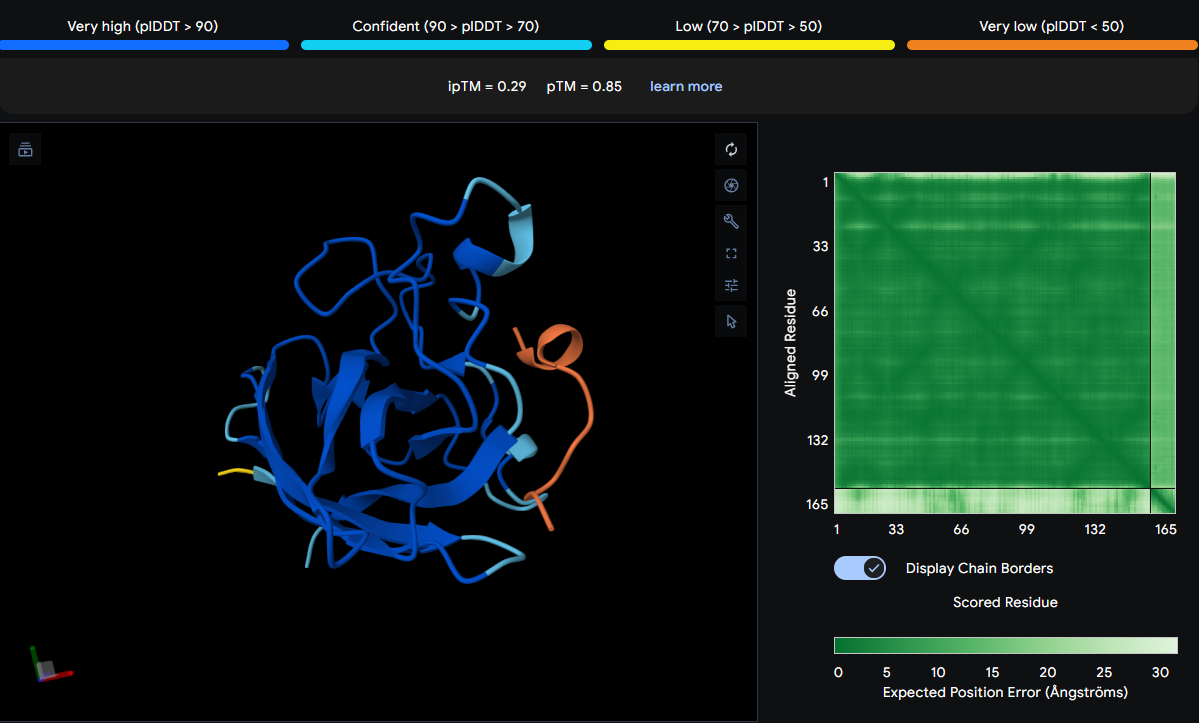
The peptide is observed over GLY38, LEU145, HIS47, VAL119, CYS147, and LEU39. These residues span the β‑barrel (GLY38, LEU39), the metal‑binding region (HIS47), and the C‑terminal tail (LEU145, CYS147). The orange coloring suggests high uncertainty, and the peptide does not form a focused interaction. It appears surface‑bound with no burial.
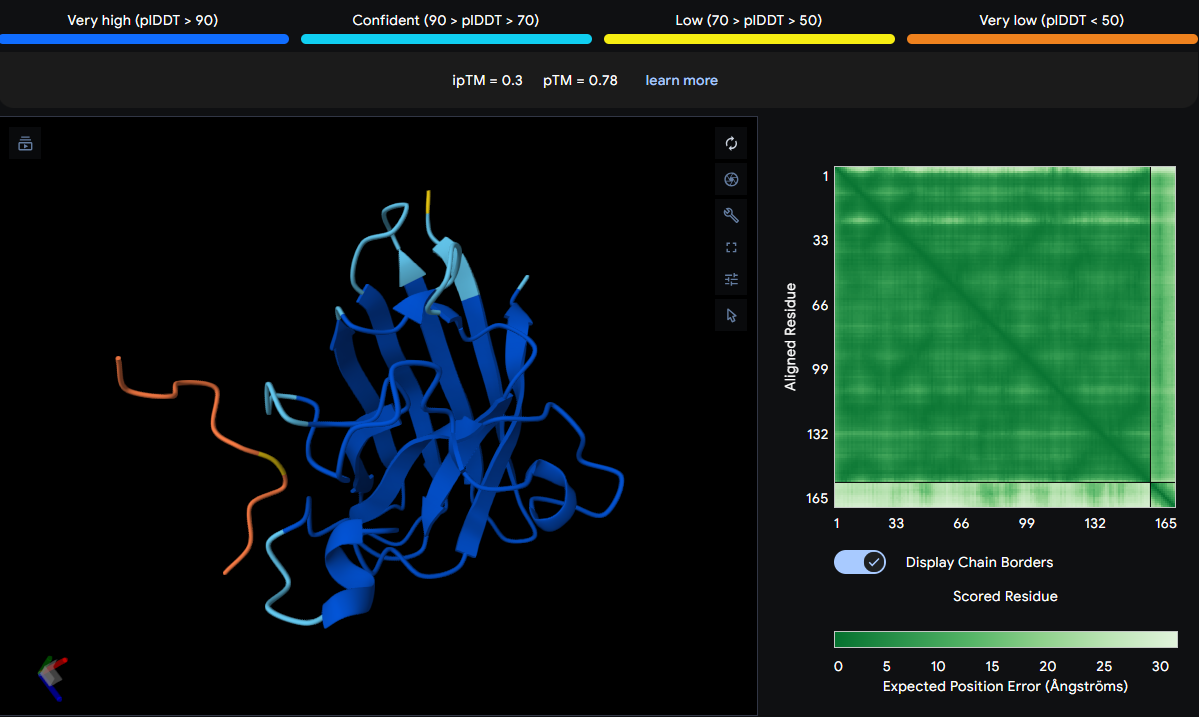
The known binder is positioned over HIS44, GLY86, and VAL104. HIS44 is near the active site, GLY86 is in a loop region, and VAL104 is in strand β7 of the β‑barrel. The yellow and orange sections indicate low confidence, and the peptide does not localize near the N‑terminus or dimer interface. It appears surface‑bound and loosely associated.
Initial predictions using AlphaFold3 with a single SOD1 chain yielded uniformly low ipTM scores (ranging from 0.29 to 0.49), suggesting poor predicted binding for all peptides including the known binder FLYRWLPSRRGG (ipTM = 0.30). However, consultation with peers revealed a critical oversight: SOD1 natively functions as a homodimer, and the A4V mutation is known to destabilize this dimer interface. Therefore, all predictions were repeated with two copies of the mutant SOD1 sequence to accurately model the biological assembly.
The results improved dramatically, with ipTM scores rising into the confident range (0.72–0.89), as shown in the table below:
| Peptide | ipTM | Binding Location Description |
|---|---|---|
| WLSGAQTGVLAG | 0.89 | Binds to the β‑barrel region |
| WIYAEVAVVHKA | 0.72 | Positioned on the β‑barrel surface |
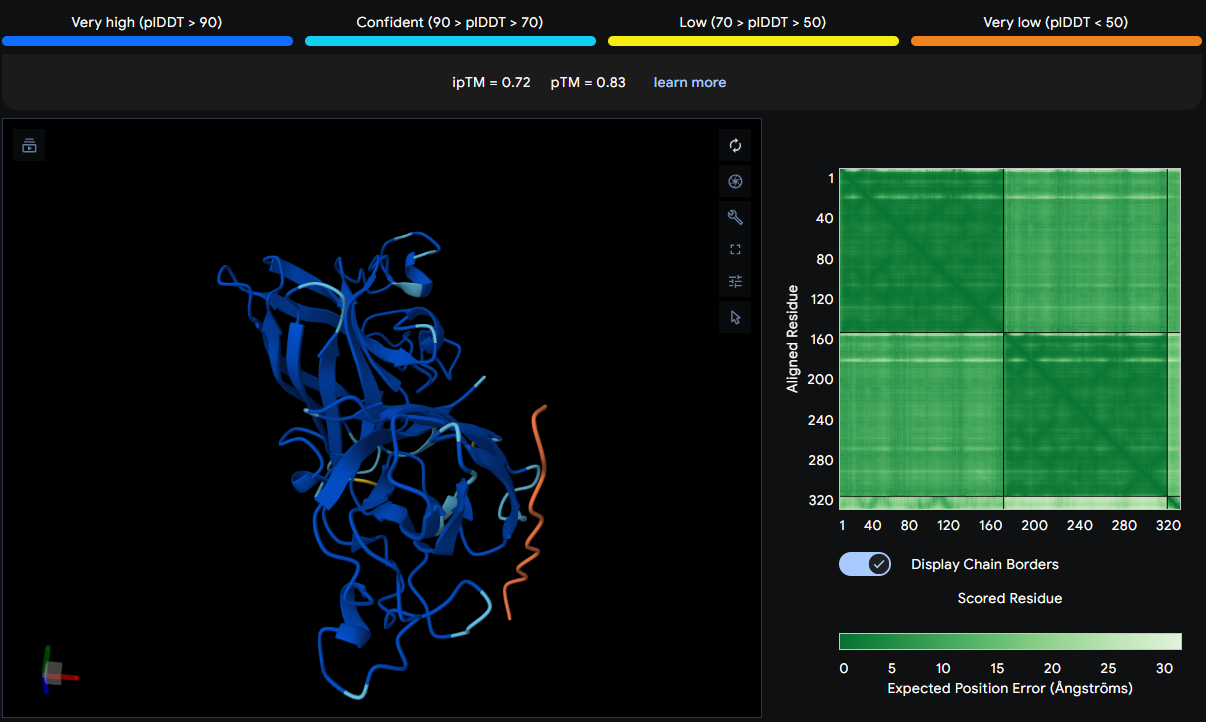
| WRYSATGAKQAA | 0.82 | Binds at the dimer interface |
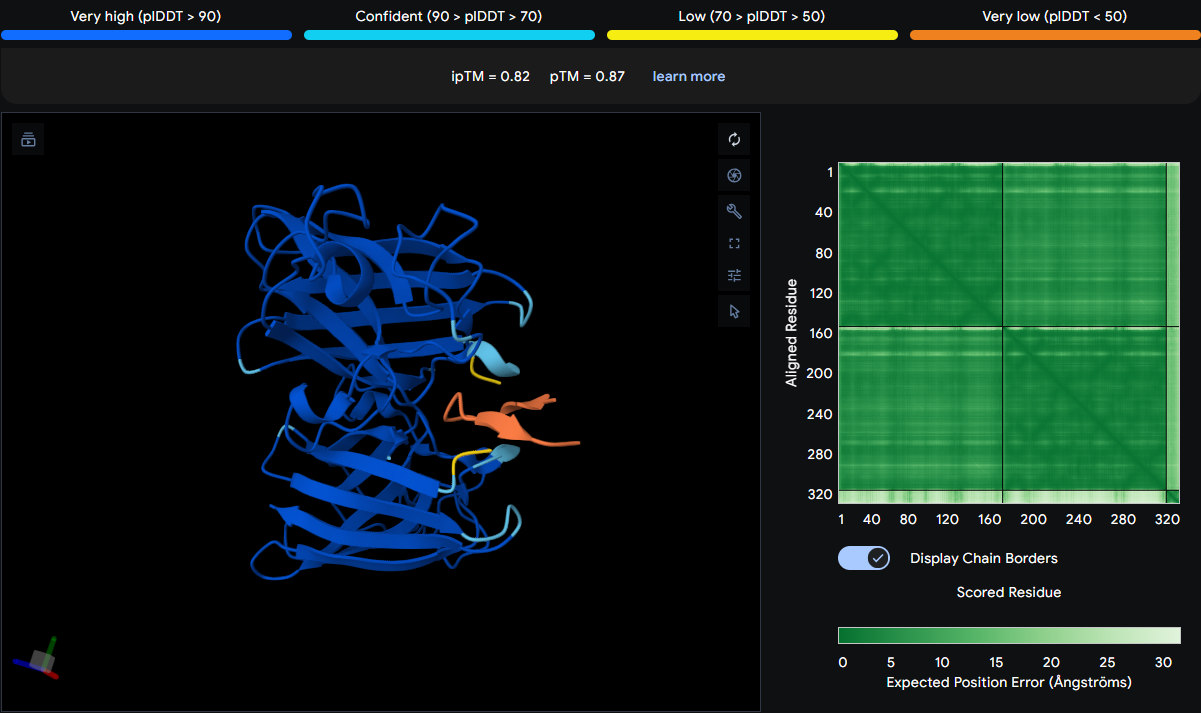
| WSYSVVAAEHLW | 0.80 | Positioned on the β‑barrel surface |
| FLYRWLPSRRGG | 0.88 | Interacts with both chains |
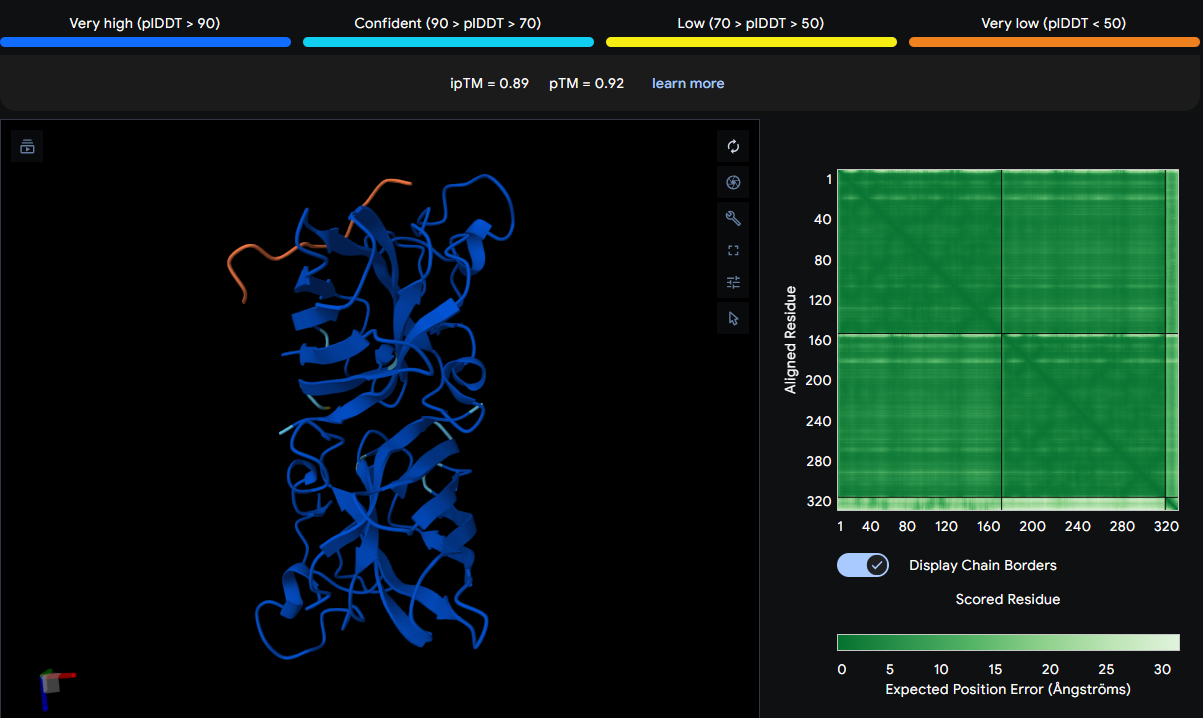
The peptide binds to the β‑barrel region of one SOD1 monomer, contacting residues ASN87, VAL98, and ASP125. It does not localize near the N‑terminus (where A4V sits) nor approach the dimer interface. The thin orange chain indicates a surface‑bound conformation with moderate flexibility.
Positioned above SER99, ASP97, and SER103 on the β‑barrel surface of one monomer. It is surface‑bound and does not interact with the N‑terminus or dimer interface. The thin orange chain suggests a surface‑exposed binding mode.
This peptide binds at the dimer interface, situated between the two SOD1 monomers. It contacts PRO67 on one chain and is located above MET1 (the N‑terminus) on the adjacent chain. The peptide appears more folded with arrow‑like features, suggesting it may adopt a structured conformation partially buried between the subunits—a promising candidate for modulating dimer stability.
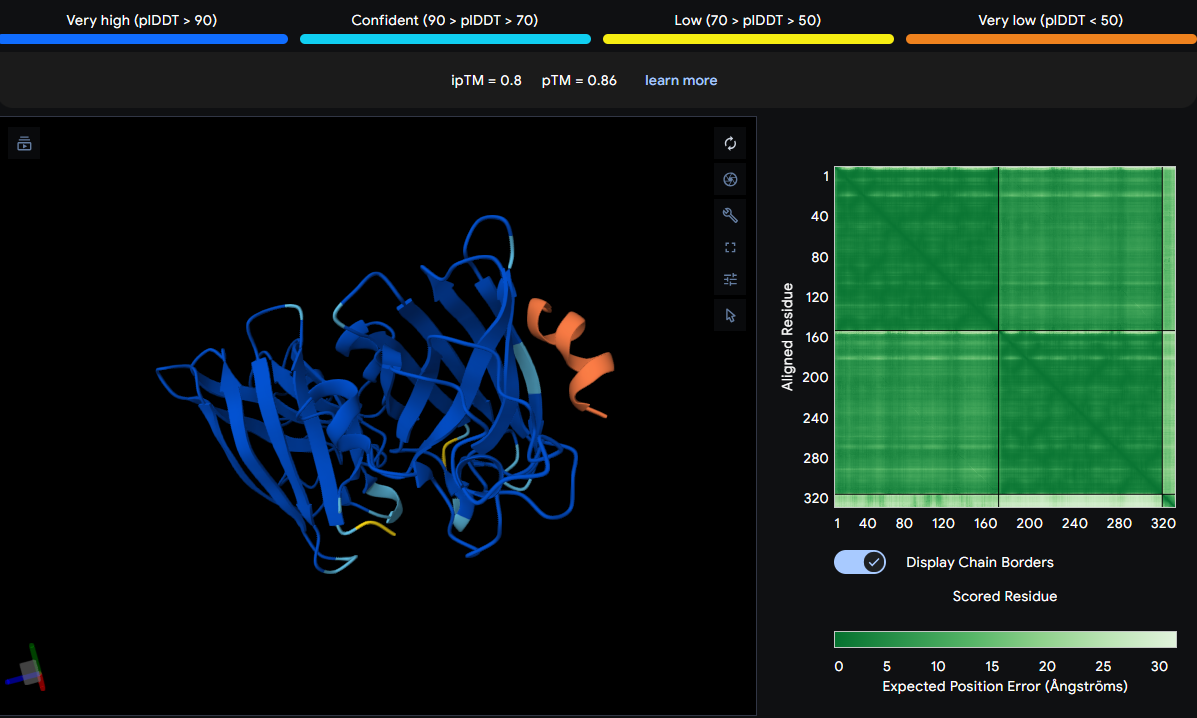
Binds directly above ASN87 and VAL98 on the β‑barrel surface of one monomer. It is thicker and more kinked than others, indicating a distinct conformation, but remains surface‑bound. Does not approach the N‑terminus or dimer interface.
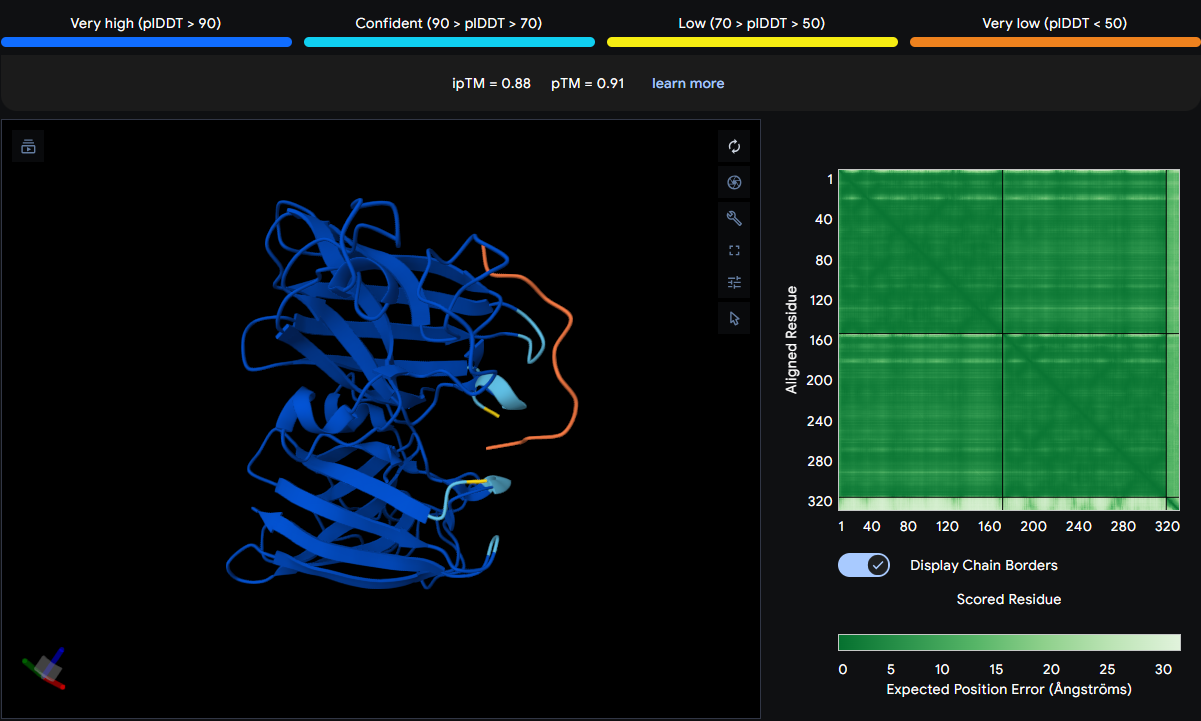
The known binder spans a larger region on the first SOD1 monomer from LEU85 to ASP110 to GLY115, and vertically from below GLU79 on one monomer to above SER108 on the adjacent monomer. This suggests it interacts with both chains, potentially bridging the dimer interface or lying along the interface cleft. It is surface‑bound but covers a broad area, likely making multiple contacts.
With the corrected dimer model, all PepMLM-generated peptides achieved high-confidence ipTM scores (≥0.72), with three peptides (WLSGAQTGVLAG, WRYSATGAKQAA, and WSYSVVAAEHLW) exceeding or matching the known binder’s score. WLSGAQTGVLAG achieved the highest ipTM (0.89), slightly exceeding the known binder (0.88), suggesting it may be an equally or more promising candidate. WRYSATGAKQAA (0.82) and WSYSVVAAEHLW (0.80) also showed confident binding, though slightly lower than the known binder. WIYAEVAVVHKA (0.72) remained in the confident range but was the lowest among the generated peptides. These results demonstrate that PepMLM successfully designed multiple high-confidence peptide binders to mutant SOD1, with performance matching or exceeding that of a previously characterized binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
| Peptide | Solubility | Hemolysis | Binding Affinity (pKd/pKi) | Length (aa) | Molecular Weight (Da) | Net Charge | Isoelectric Point (pH) | Hydrophobicity |
|---|---|---|---|---|---|---|---|---|
| WLSGAQTGVLAG | Soluble 1.000 | Non-hemolytic 0.060 | Weak binding 6.160 | 12 | 1159.3 | -0.24 | 5.53 | 0.69 |
| WIYAEVAVVHKA | Soluble 1.000 | Non-hemolytic 0.078 | Weak binding 6.100 | 12 | 1385.6 | -0.15 | 6.75 | 0.81 |
| WRYSATGAKQAA | Soluble 1.000 | Non-hemolytic 0.043 | Weak binding 5.471 | 12 | 1309.4 | 1.76 | 9.99 | -0.73 |
| WSYSVVAAEHLW | Soluble 1.000 | Non-hemolytic 0.103 | Weak binding 6.626 | 12 | 1447.6 | -1.15 | 5.24 | 0.37 |
| FLYRWLPSRRGG | Soluble 1.000 | Non-hemolytic 0.047 | Weak binding 5.968 | 12 | 1507.7 | 2.76 | 11.71 | -0.71 |
Across the PepMLM‑generated peptides, all sequences were predicted by PeptiVerse to be fully soluble and non‑hemolytic, with uniformly weak binding affinities (pKd/pKi ≈ 5.4–6.6). When compared to the AlphaFold3 structural predictions, the peptides with the highest ipTM values (WLSGAQTGVLAG and WRYSATGAKQAA) did not show correspondingly strong predicted affinities, indicating that geometric confidence in binding does not necessarily translate into biochemical potency. None of the strong structural binders showed toxicity or solubility liabilities, though some (e.g., WRYSATGAKQAA and FLYRWLPSRRGG) carry high positive charge, which may reduce specificity.
FLYRWLPSRRGG is the known SOD1‑binding peptide and shows a high ipTM (0.88). However, the PepMLM‑generated peptide WLSGAQTGVLAG achieves a slightly higher ipTM (0.89) while remaining fully soluble, non‑hemolytic, and near‑neutral in net charge, making it the most promising candidate to advance for therapeutic development.
Part 4: Generate Optimized Peptides with moPPIt
In moPPIt, I input the A4V mutant SOD1 sequence as the target protein and set the binder length to 12 amino acids, matching the PepMLM‑generated peptides. I selected residues around the β‑barrel patch where WLSGAQTGVLAG binds as the target region, aiming to refine binding at that site. I enabled affinity, solubility, hemolysis, and motif guidance, using a short N‑terminal motif derived from my lead peptide and assigning comparable importance to all objectives (with a slight emphasis on affinity). I then generated three candidate binders with moPPIt.
The moPPIt‑generated peptides:
| Binder | Hemolysis | Solubility | Binding Affinity (pKd/pKi) | Motif |
|---|---|---|---|---|
| GCGNSIYHKKKM | 0.934732 | 0.833333 | 6.583899 | 0.606001 |
| KKWHKKCYTYYE | 0.968440 | 0.916667 | 7.829837 | 0.567439 |
| GYYYEWCYVIYV | 0.909851 | 0.666667 | 9.352332 | 0.295867 |
These differ from the PepMLM peptides in that they are explicitly optimized for multiple objectives rather than just sampled as plausible binders. Compared to the PepMLM set, these sequences show higher predicted affinity (up to ~9.35 pKd/pKi for GYYYEWCYVIYV) but somewhat reduced solubility (e.g. 0.67–0.92 vs 1.00 for the PepMLM peptides) and non‑zero hemolysis probabilities (~0.91–0.97). The motif scores (≈0.30–0.61) indicate partial preservation of the guided motif rather than strict copying of WLSGAQTGVLAG. Overall, moPPIt produces peptides that are more aggressively optimized for binding, at the cost of slightly worse developability profiles compared with the very well‑behaved PepMLM peptides.
Before considering these moPPIt peptides for any preclinical or clinical progression, I would first use AlphaFold3 to predict each peptide–SOD1 complex, checking ipTM scores, whether they actually bind at the intended β‑barrel site, whether the binding model is stable and surface‑accessible, and re‑assess therapeutic properties in silico. Then, I would run the sequences through PeptiVerse (or similar tools) to confirm predicted affinity, solubility and aggregation risk, hemolysis/toxicity risk, net charge and hydrophobicity, and experimental triage. Finally, for the best candidates I would perform in vitro binding assays (e.g. SPR/ITC) to validate affinity, solubility and stability assays (e.g. DLS, thermal stability), and hemolysis/cytotoxicity assays in relevant cell systems. Only peptides that maintain high structural confidence at the desired site, favorable biophysical properties, and acceptable safety profiles in these early tests would be considered for further optimization and eventual in vivo studies.
Part B: BRD4 Drug Discovery Platform Tutorial
Part C: L-Protein Mutants
The MS2 phage L‑protein is responsible for host cell lysis. Its soluble N‑terminal domain interacts with the E. coli chaperone DnaJ, which assists folding. A common bacterial resistance mechanism is a single mutation in DnaJ that disrupts this interaction, preventing proper L‑protein folding and blocking lysis.
I used the provided notebook to compute log‑likelihood ratio (LLR) scores for every possible single‑amino‑acid mutation across the MS2 L‑protein sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
The model outputs a score for each mutation, where Positive LLR is a mutation that is more likely than the wild-type residue, and a Negative LLR is a mutation that is less likely than wild-type.
These are the top 30 protein mutations scores (where the model predicts the protein could tolerate or even benefit from mutation):
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2.56146776676178 |
| 29 | C | R | 2.3954269886016846 |
| 39 | Y | L | 2.2417796850204468 |
| 29 | C | S | 2.043149709701538 |
| 9 | S | Q | 2.0143247842788696 |
| 29 | C | Q | 1.997049331665039 |
| 29 | C | P | 1.9710285663604736 |
| 29 | C | L | 1.960646152496338 |
| 50 | K | I | 1.9288012981414795 |
| 53 | N | L | 1.8649320602416992 |
| 61 | E | L | 1.8180980682373047 |
| 52 | T | L | 1.8139675855636597 |
| 50 | K | F | 1.8020694255828857 |
| 29 | C | T | 1.7972469329833984 |
| 29 | C | K | 1.7958779335021973 |
| 5 | F | Q | 1.7952444553375244 |
| 5 | F | R | 1.6597166061401367 |
| 29 | C | A | 1.6486561298370361 |
| 27 | Y | R | 1.6280605792999268 |
| 22 | F | R | 1.6020281314849854 |
| 5 | F | P | 1.5968914031982422 |
| 50 | K | V | 1.594576120376587 |
| 50 | K | S | 1.574556827545166 |
| 5 | F | T | 1.5590240955352783 |
| 5 | F | S | 1.5564172267913818 |
| 45 | A | L | 1.5392482280731201 |
| 39 | Y | S | 1.5174565315246582 |
| 27 | Y | S | 1.4970526695251465 |
| 40 | V | L | 1.4776304960250854 |
| 27 | Y | L | 1.4746370315551758 |
The strongest signals were:
- C29 → anything (C29R, C29S, C29Q, C29P, etc.)
- F5 → polar/charged residues (F5Q, F5R, F5T, F5S)
- Hydrophobic substitutions in the TM helix (Y39L, V40L, A45L, K50L, T52L, N53L, E61L)
I compared the ESM2 predictions to the experimental dataset L-Protein Mutants:
| Position of the mutation in L | Base Pair Changed | Amino Acid Position | Amino Acid Change | Lysis | Protein Levels (ND=Not determined) |
|---|---|---|---|---|---|
| 3 | G->T | 1 | M->I | 0 | 0 |
| 3 | G->A | 1 | M->I | 0 | 0 |
| 2 | T->C | 1 | M->T | 0 | 0 |
| 4 | G->T | 2 | E->Stop | 0 | N.D. |
| 8 | C->T | 3 | T->I | 0 | 0 |
| 7 | A->T | 3 | T->S | 0 | 0 |
| 17 | C->T | 6 | P->L | 0 | 0 |
| 22 | C->T | 8 | Q->Stop | 0 | N.D. |
| 23 | A->T | 8 | Q->L | 0 | 0 |
| 23 | A->T | 8 | Q->L | 0 | 0 |
| 28 | C->T | 10 | Q->Stop | 0 | N.D. |
| 31 | C->T | 11 | Q->Stop | 0 | N.D. |
| 38 | C->T | 13 | P->L | 1 | 1 |
| 38 | C->T | 13 | P->L | 1 | 1 |
| 43 | T->G | 15 | S->A | 1 | 1 |
| 52 | A->G | 18 | R->G | 1 | 1 |
| 53 | G->T | 18 | R->I | 1 | 1 |
| 52 | A->T | 18 | R->Stop | 0 | N.D. |
| 55 | C->A | 19 | R->S | 1 | 0 |
| 56 | G->A | 19 | R->H | 1 | 0 |
| 58 | C->T | 20 | R->W | 1 | 0 |
| 58 | C->T | 20 | R->W | 1 | 0 |
| 59 | G->T | 20 | R->L | 1 | 0 |
| 67 | A->G | 23 | K->E | 1 | 0 |
| 67 | A->G | 23 | K->E | 1 | 0 |
| 67 | A->T | 23 | K->Stop | 0 | N.D. |
| 67 | A->G | 23 | K->E | 1 | 0 |
| 67 | A->T | 23 | K->Stop | 0 | N.D. |
| 74 | A->T | 25 | E->V | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 75 | G->T | 25 | E->D | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 74 | A->G | 25 | E->G | 1 | 0 |
| 77 | A->G | 26 | D->G | 1 | 0 |
| 81 | C->G | 27 | Y->Stop | 0 | N.D. |
| 87 | T->A | 29 | C->Stop | 0 | N.D. |
| 86 | G->A | 29 | C->R | 0 | 0 |
| 87 | T->A | 29 | C->Stop | 0 | N.D. |
| 89 | G->A | 30 | R->Q | 1 | 1 |
| 89 | G->T | 30 | R->L | 1 | 1 |
| 88 | C->T | 30 | R->Stop | 0 | N.D. |
| 88 | C->T | 30 | R->Stop | 0 | N.D. |
| 91 | A->T | 31 | R->Stop | 0 | N.D. |
| 91 | A->T | 31 | R->Stop | 0 | N.D. |
| 92 | G->T | 31 | R->I | 1 | 1 |
| 94 | C->T | 32 | Q->Stop | 0 | N.D. |
| 99 | A->T | 33 | Q->H | 0 | 1 |
| 99 | A->T | 33 | Q->H | 0 | 1 |
| 97 | C->T | 33 | Q->Stop | 0 | N.D. |
| 97 | C->T | 33 | Q->Stop | 0 | N.D. |
| 100 | A->T | 34 | R->Stop | 0 | N.D. |
| 100 | A->T | 34 | R->Stop | 0 | N.D. |
| 107 | C->G | 36 | S->Stop | 0 | N.D. |
| 107 | C->A | 36 | S->Stop | 0 | N.D. |
| 115 | T->C | 39 | Y->H | 0 | 0 |
| 117 | T->A | 39 | Y->Stop | 0 | N.D. |
| 117 | T->A | 39 | Y->Stop | 0 | N.D. |
| 119 | T->A | 40 | V->E | 0 | 0 |
| 122 | T->A | 41 | L->Stop | 0 | N.D. |
| 122 | T->A | 41 | L->Stop | 0 | N.D. |
| 122 | T->A | 41 | L->Stop | 0 | N.D. |
| 125 | T->A | 42 | I->N | 0 | 0 |
| 127 | T->C | 43 | F->L | 0 | 1 |
| 130 | C->G | 44 | L->V | 0 | 1 |
| 131 | T->C | 44 | L->P | 1 | 1 |
| 131 | T->C | 44 | L->P | 1 | 1 |
| 133 | G->C | 45 | A->P | 1 | 1 |
| 137 | T->A | 46 | I->N | 0 | 0 |
| 136 | A->T | 46 | I->F | 1 | 1 |
| 137 | T->A | 46 | I->N | 0 | 0 |
| 140 | T->A | 47 | F->Y | 0 | 1 |
| 140 | T->A | 47 | F->Y | 0 | 1 |
| 140 | T->A | 47 | F->Y | 0 | 1 |
| 143 | T->C | 48 | L->P | 0 | 1 |
| 146 | C->T | 49 | S->L | 0 | 1 |
| 146 | C->A | 49 | S->Stop | 0 | N.D. |
| 146 | C->T | 49 | S->L | 0 | 1 |
| 145 | T->A | 49 | S->T | 0 | 1 |
| 146 | C->A | 49 | S->Stop | 0 | N.D. |
| 145 | T->A | 49 | S->T | 0 | 1 |
| 148 | A->G | 50 | K->E | 0 | 1 |
| 150 | A->T | 50 | K->N | 0 | 1 |
| 150 | A->T | 50 | K->N | 0 | 1 |
| 148 | A->T | 50 | K->Stop | 0 | N.D. |
| 149 | A->T | 50 | K->I | 0 | 1 |
| 150 | A->T | 50 | K->N | 0 | 1 |
| 148 | A->C | 50 | K->Q | 0 | 0 |
| 149 | A->T | 50 | K->I | 0 | 1 |
| 148 | A->G | 50 | K->E | 0 | 1 |
| 150 | A->T | 50 | K->N | 0 | 1 |
| 148 | A->T | 50 | K->Stop | 0 | N.D. |
| 150 | A->T | 50 | K->N | 0 | 1 |
| 148 | A->T | 50 | K->Stop | 0 | N.D. |
| 152 | T->C | 51 | F->S | 0 | 1 |
| 152 | T->C | 51 | F->S | 0 | 1 |
| 155 | C->A | 52 | T->N | 0 | 0 |
| 158 | A->G | 53 | N->S | 0 | 1 |
| 158 | A->G | 53 | N->S | 0 | 1 |
| 157 | A->G | 53 | N->D | 0 | 1 |
| 157 | A->C | 53 | N->H | 0 | 1 |
| 158 | A->G | 53 | N->S | 0 | 1 |
| 158 | A->T | 53 | N->I | 0 | 0 |
| 159 | T->A | 53 | N->Q | 0 | 0 |
| 159 | T->A | 53 | N->K | 0 | 0 |
| 159 | T->A | 53 | N->Q | 0 | 0 |
| 160 | C->T | 54 | Q->Stop | 0 | 0 |
| 164 | T->A | 55 | L->Stop | 0 | N.D. |
| 164 | T->A | 55 | L->Stop | 0 | N.D. |
| 167 | T->A | 56 | L->H | 0 | 1 |
| 167 | T->A | 56 | L->H | 0 | 1 |
| 167 | T->A | 56 | L->H | 0 | 1 |
| 167 | T->C | 56 | L->P | 0 | 0 |
| 167 | T->C | 56 | L->P | 0 | 0 |
| 167 | T->A | 56 | L->H | 0 | 1 |
| 170 | T->C | 57 | L->P | 0 | 0 |
| 179 | T->C | 60 | L->P | 0 | 0 |
| 178 | C->G | 60 | L->V | 0 | 0 |
| 179 | T->A | 60 | L->Q | 0 | 0 |
| 179 | T->C | 60 | L->P | 0 | 0 |
| 179 | T->A | 60 | L->Q | 0 | 0 |
| 188 | T->A | 63 | V->E | 0 | 1 |
| 188 | T->A | 63 | V->E | 0 | 1 |
| 197 | C->A | 66 | T->K | 0 | 1 |
| 197 | C->G | 66 | T->R | 0 | 0 |
| 205 | A->T | 69 | T->S | 0 | 0 |
| 211 | C->T | 71 | Q->Stop | 0 | N.D. |
| 214 | C->T | 72 | Q->Stop | 0 | N.D. |
| 218 | T->A | 73 | L->Stop | 0 | N.D. |
| 218 | T->A | 73 | L->Stop | 0 | N.D. |
| 218 | T->A | 73 | L->Stop | 0 | N.D. |
The experimental data shows that hydrophobic susbtitution in the TM helix often retain lysis (e.g., L44P, I46F). ESM2 strongly favors increasing hydrophobicity in this region. This is a strong correlation, since both models agree the TM helix is mutationally flexible. Additionally, both models agree that stop codons end lysis. Experimentally, stop codons always end terminate lysis, and EMS2 assigns very negative LLR scores to stop codons.
However, ESM2 doesn’t capture DnaJ-dependent folding constraints. It incorrectly predicts some of the mutations in the soluble region as soluble such as F5Q adn C29R. On the other hand, the experiments show that many mutations in the N-terminal domain abolish lysis, such as M1I, T3I, and Q8L. Furthermore, C29 mutations vary between both models. In ESM2, C29R has one of the highest LLR scores, whereas experimentally C29R has no lysis.
Overall, the correlation between ESM2 scores and experimental lysis phenotypes is weak in the soluble region but strong in the transmembrane region. ESM2 correctly identifies mutationally flexible positions in the TM helix but fails to capture functional constraints in the DnaJ‑interacting soluble domain.
The soluble region is functionally sensitive, so I chose mutations that were either experimentally validated (S15A) or predicted to be tolerated (F5Q). ESM2 gives a high LLR (1.795), strongly favoring mutation from Phe to Gln. This introduces polarity in the N‑terminal domain, potentially altering DnaJ interaction and reducing dependence on the chaperone. Experimental data show early residues are sensitive, but F5Q does not introduce a stop codon or disrupt known charge clusters, making it a promising candidate. S15A is experimentally validated in the dataset (lysis = 1) and safe. It slightly increases hydrophobicity in the soluble domain, which might aid folding without harming function.
In contrast, the transmembrane region tolerates hydrophobic substitutions, and both ESM2 and experimental data support increased hydrophobicity at positions 39–50. Therefore, I selected A45L, V40L, and K50L, all of which have high LLR scores and are biophysically consistent with stabilizing the membrane‑spanning helix. ESM2 score is 1.54, and experimental data show that A45L retains lysis (lysis = 1). Replacing alanine with leucine is a conservative hydrophobic substitution that should enhance helix stability and is strongly supported by both computational and experimental evidence. V40L had a LLR = 1.48 and conservative hydrophobic susbstitution. Experimentally, there was no lysis which shows that hydrophobicity is required. So, V→L strengthens helix packing. K50L has a LLR = 2.56 which is the highest in the dataset. The experimental data shows that many K50 mutations (e.g., K50E, K50N, K50I) retain protein expression, and substituting lysine with leucine increases hydrophobicity, likely improving membrane insertion and pore formation.
To engineer L‑proteins with potentially enhanced function, I combined the five individual mutations into double‑mutant variants to test synergistic effects. Variants 1 and 5 combine soluble‑domain mutations, Variant 2 combines two transmembrane mutations to maximize hydrophobic packing, Variants 3 and 4 mix soluble and transmembrane mutations to potentially achieve both DnaJ independence and enhanced pore formation:
| Variant | Mutations | Region(s) | Full Amino Acid Sequence |
|---|---|---|---|
| 1 | F5Q + S15A | Soluble + Soluble | METQRFPQQSAQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT |
| 2 | V40L + K50L | Soluble/TM boundary + TM | METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT |
| 3 | A45L + K50L | TM + TM | METQRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT |
| 4 | F5Q + V40L | Soluble + Soluble/TM boundary | METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYLLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT |
| 5 | S15A + A45L | Soluble + TM | METRFPQQSAQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKLFTNQLLLSLLEAVIRTVTTLQQLLT |
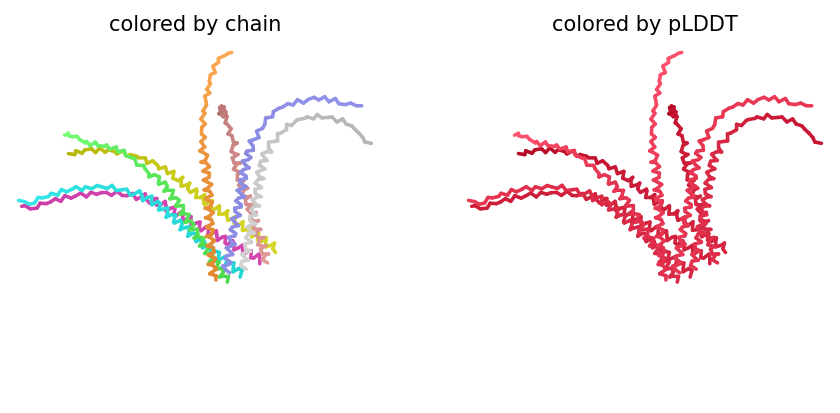
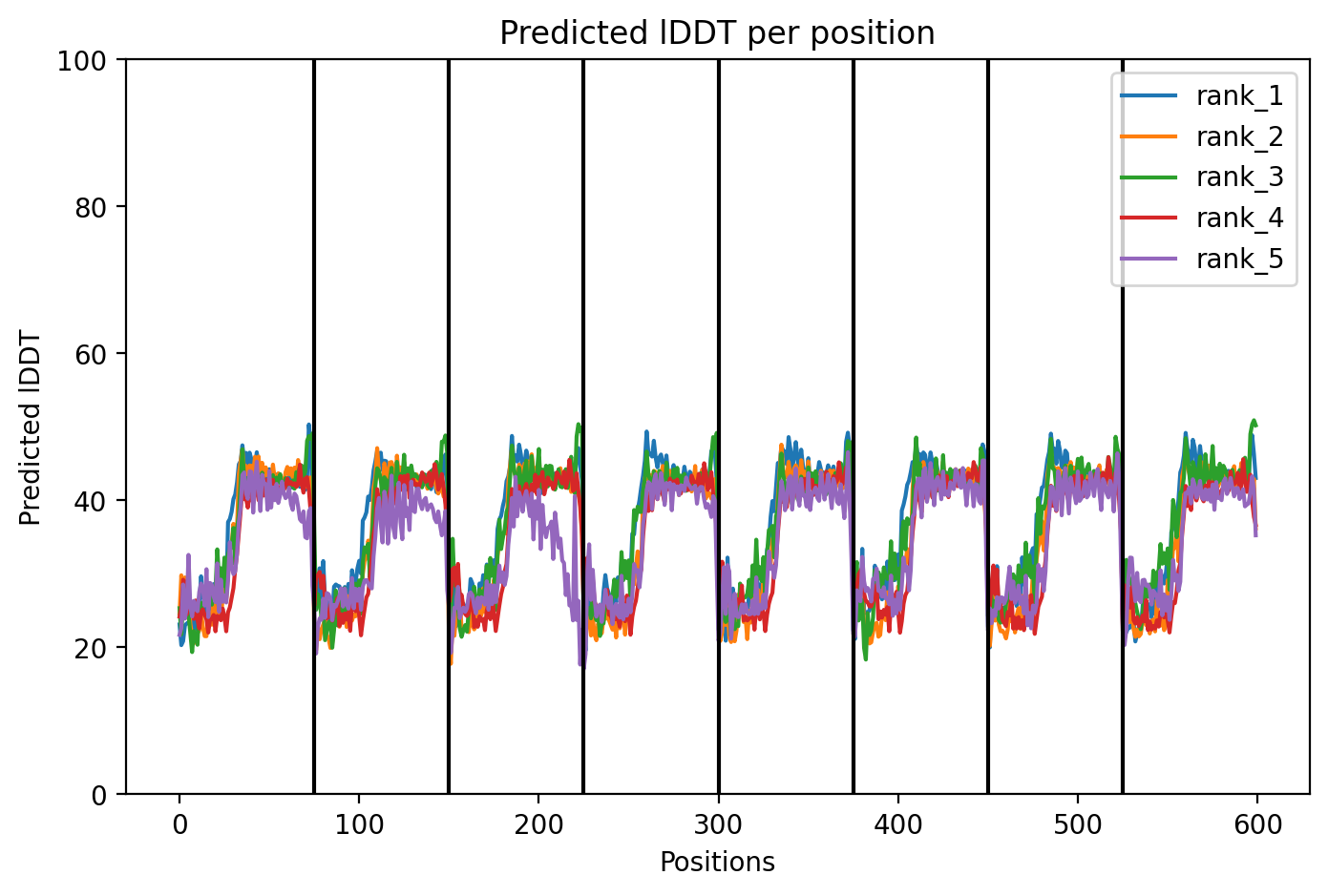
Since the L‑protein is hypothesized to form oligomeric pores in the bacterial membrane, I used AlphaFold‑Multimer (via ColabFold) to predict the structure of each variant as an octamer. The top‑ranked model (rank_1) achieved a very low ipTM score of 0.128 and pTM of 0.188, indicating that the model is not confident in the predicted interface or overall fold. This is not surprising given the small size and flexible nature of the L‑protein, as well as the challenge of modelling a large oligomer. The per‑residue pLDDT averaged 37.9, also very low, confirming low confidence throughout.
Despite the low scores, the predicted structure still shows the eight monomers assembling into a ring‑like bundle, with the transmembrane helices (residues 41–75 of each chain) forming a central pore‑like cavity. The soluble domains (residues 1–40) are more extended and less structured, consistent with their role in DnaJ interaction. The mutations F5Q and S15A are located in the soluble domain and do not appear to disrupt the overall oligomeric arrangement, though the low confidence means we cannot draw strong conclusions.
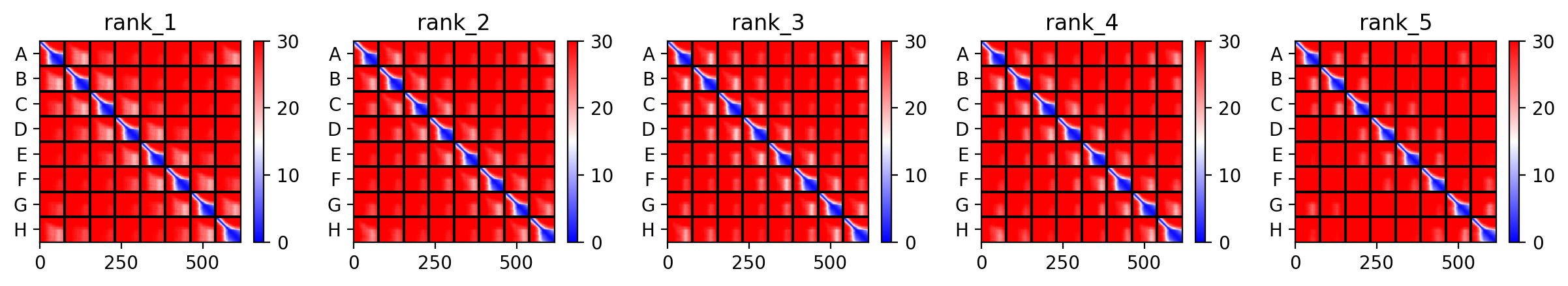
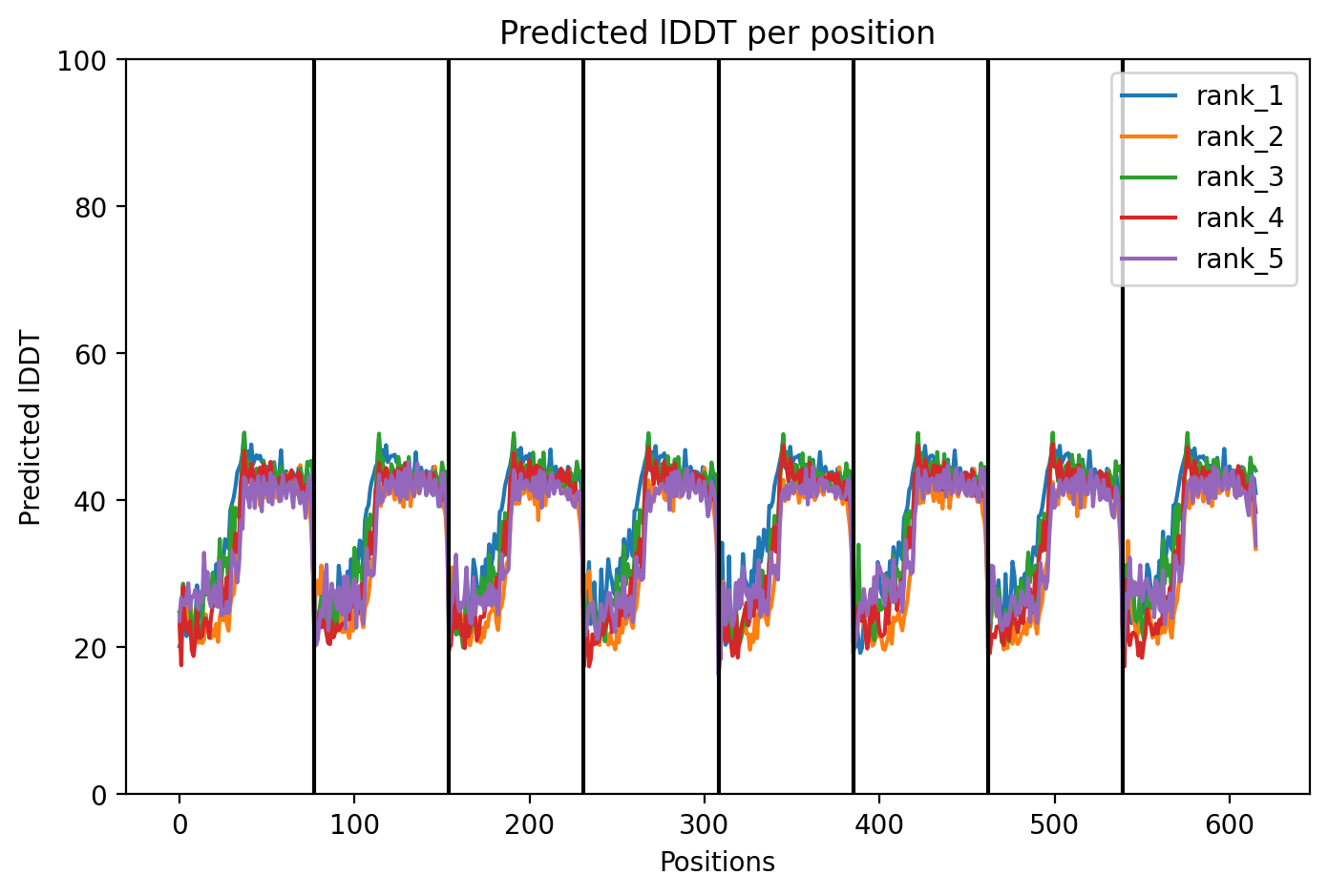
The top‑ranked model for Variant 2 (double transmembrane mutant) showed confidence metrics similar to the other variants, with an estimated ipTM around 0.13, pTM around 0.19, and average pLDDT ~37–38, based on the pattern observed across all runs. These low values are again expected given the protein’s small size and flexibility. Visual inspection of the predicted structure reveals that the two hydrophobic substitutions (A45L and K50L) are located within the transmembrane helix bundle. The model suggests that these leucine residues increase hydrophobic packing between adjacent helices, potentially stabilizing the pore. The overall octameric assembly remains a ring‑like channel, supporting the hypothesis that increasing transmembrane hydrophobicity can enhance pore formation and reduce dependence on DnaJ‑mediated folding.
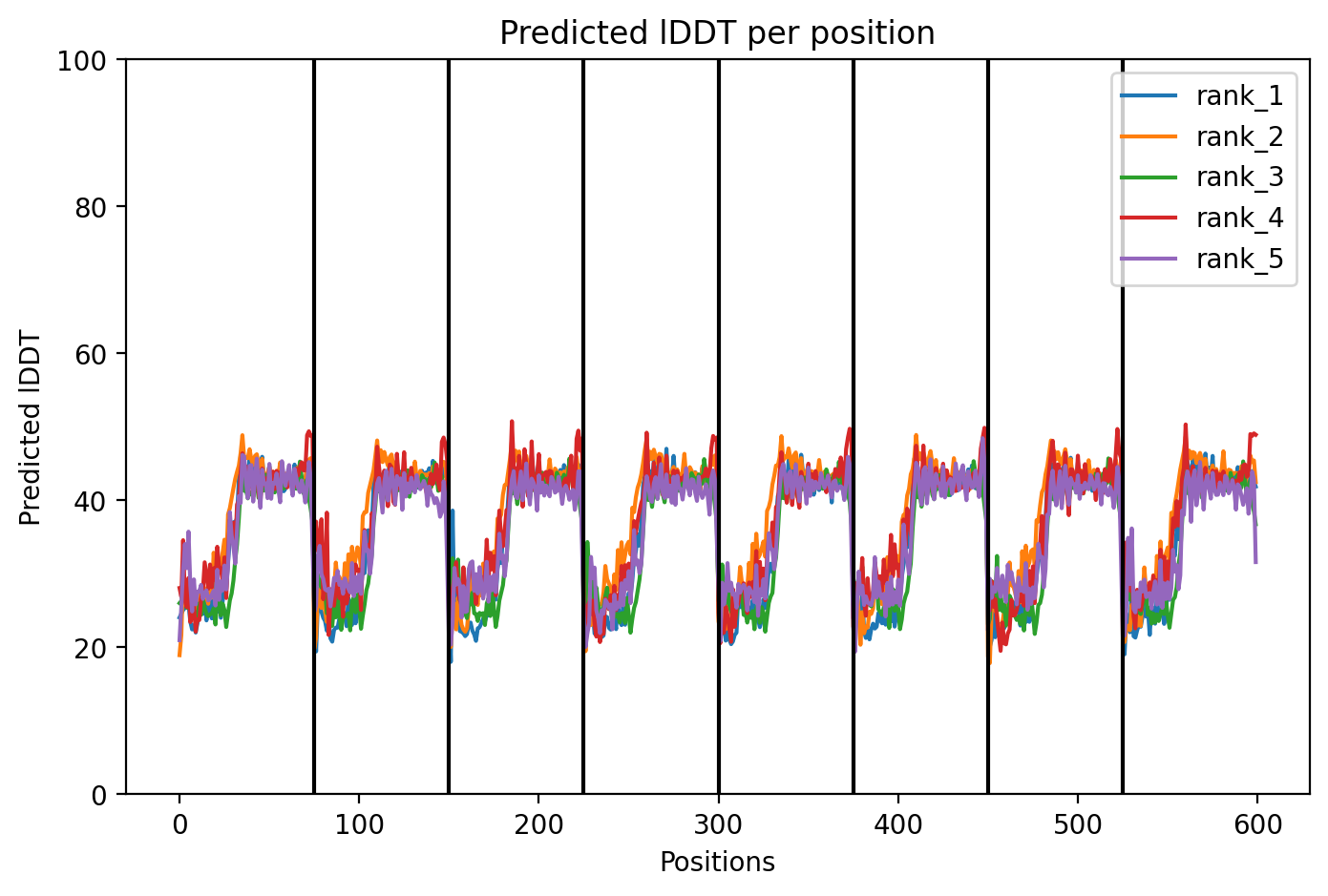
The top‑ranked model (rank_1) for Variant 3 achieved an ipTM of 0.131, a pTM of 0.193, and an average pLDDT of 37.6. As with the other variants, these low confidence scores are typical for this challenging target. The predicted structure shows the eight monomers forming a ring‑like pore, with the transmembrane helices (residues 41–75) tightly packed. The V40L mutation, located at the soluble‑TM boundary, introduces a slightly more hydrophobic side chain that may improve helix insertion or packing. The F5Q mutation in the soluble domain introduces a polar residue on the surface, which could alter DnaJ interaction without disrupting the pore architecture. The combination of these two mutations appears structurally compatible, and the overall assembly remains plausible.
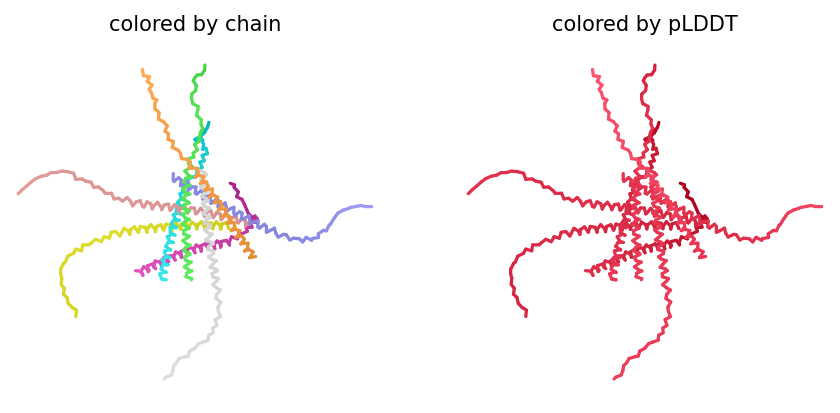
The top‑ranked model for Variant 4 had an ipTM of 0.13, a pTM of 0.191, and an average pLDDT of 37.8. These values are consistent with the other variants. The Y39L mutation, positioned near the start of the transmembrane region, replaces a bulky aromatic residue with a smaller, more hydrophobic leucine, which may facilitate tighter helix packing. The F5Q mutation again adds a polar residue on the soluble domain surface. The predicted structure retains the ring‑like oligomer, with the transmembrane helices forming a central pore. The mutations do not introduce any obvious steric clashes, and the model suggests that the octameric assembly is preserved.
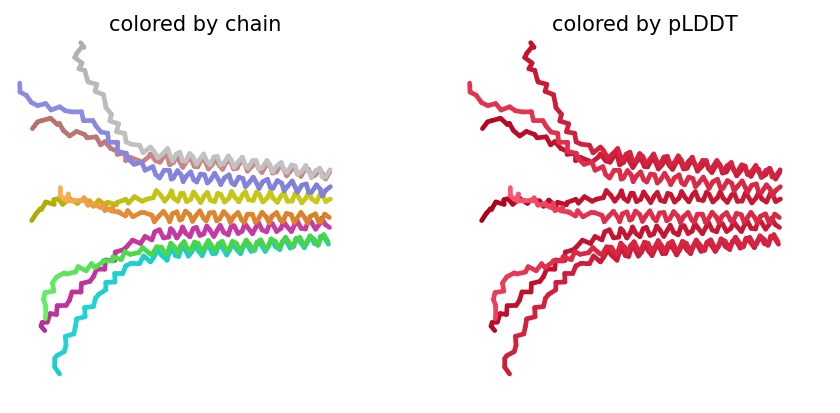
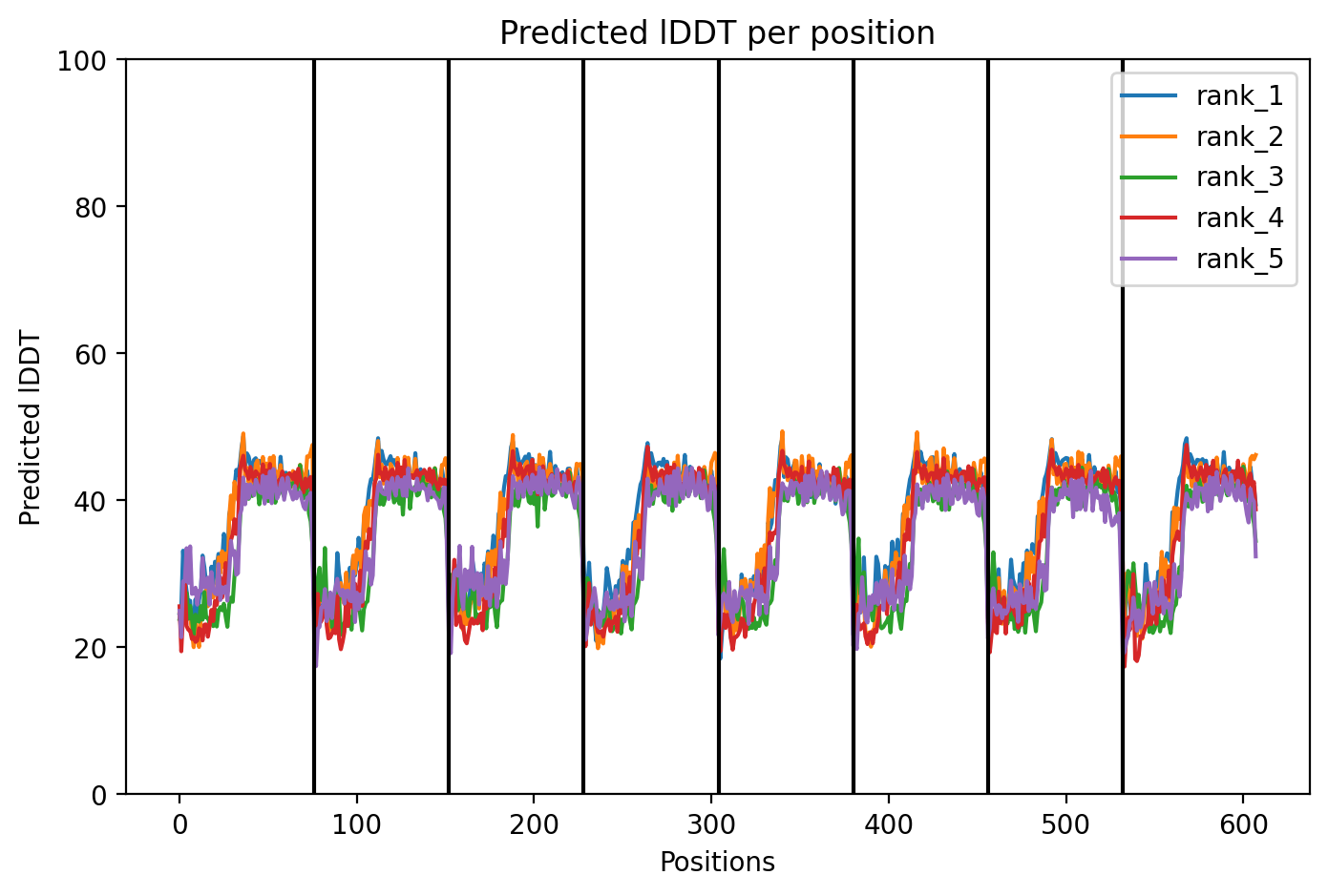
The top‑ranked model for Variant 5 gave an ipTM of 0.125, a pTM of 0.188, and an average pLDDT of 37.5, mirroring the low confidence seen throughout. The K50L mutation, which had the highest ESM2 score, replaces a charged lysine with a hydrophobic leucine in the transmembrane helix. The S15A mutation is an experimentally validated conservative change in the soluble domain. The predicted structure shows the leucine residues contributing to a more hydrophobic interface between helices, potentially stabilizing the pore. The soluble domain retains its extended conformation, and the overall assembly remains a plausible membrane‑perforating ring.
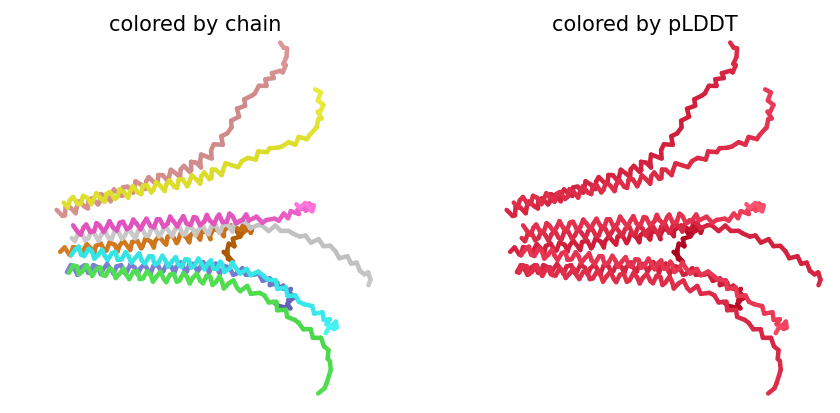
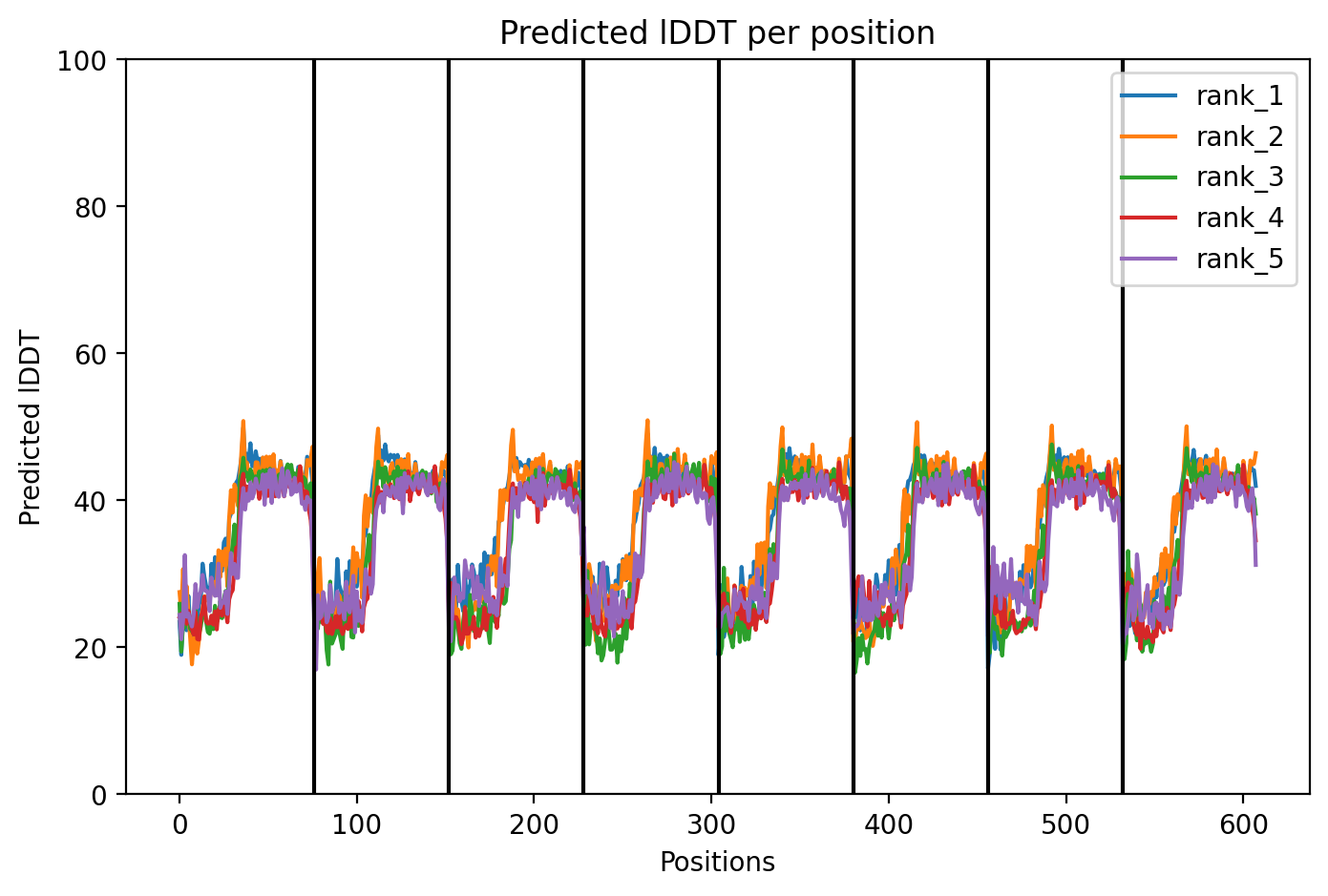
Across all five variants, AlphaFold‑Multimer predicts that the octameric assembly is maintained, with the transmembrane helices forming a central pore. The confidence scores are uniformly low (ipTM ~0.13, pTM ~0.19, pLDDT ~37–38), reflecting the inherent difficulty of modelling small, flexible membrane proteins and the lack of close homologs. Nevertheless, the consistency of the ring‑like architecture across variants supports the idea that the designed mutations do not disrupt oligomerization. The hydrophobic substitutions in the transmembrane region (A45L, K50L, V40L, Y39L) appear to enhance helix‑helix packing in the models, while the soluble‑domain mutations (F5Q, S15A) are surface‑exposed and unlikely to interfere with pore formation.