Week 6: Genetic Circuits Part I - Assembly Technologies
DNA Assembly
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion HF PCR Master Mix is a 2X pre-formulated reagent containing several key components:
Phusion DNA Polymerase: A thermostable, high-fidelity polymerase derived from Pyrococcus-like archaeal enzymes, fused to a processivity-enhancing DNA-binding domain (Sso7d). It synthesizes new DNA strands from dNTPs.
3’→5’ proofreading exonuclease: Excises misincorporated nucleotides immediately after insertion, giving Phusion an error rate ~50× lower than Taq polymerase Molecular Biology International. This is critical for mutagenesis work where a single wrong base would ruin the experiment.
dNTPs (dATP, dCTP, dGTP, dTTP): The four deoxynucleotide building blocks incorporated into the growing DNA strand.
MgCl₂ (optimized concentration): Mg²⁺ is an essential cofactor for polymerase activity; it stabilizes the dNTP-polymerase complex and facilitates phosphodiester bond formation. Concentration affects both yield and fidelity.
Reaction buffer (pH ~8.0): Maintains optimal pH and ionic conditions for polymerase activity and primer annealing.
Stabilizers/additives: The HF buffer includes components that enhance specificity and reduce non-specific amplification.
The Sso7d DNA-binding domain is particularly important: it increases processivity (how far the polymerase travels before dissociating), which is why Phusion can amplify the ~3 kb backbone fragment efficiently. Phusion’s error rate is approximately 4.4 × 10⁻⁷ errors/bp/cycle, making it ideal for mutagenesis where sequence fidelity is paramount.
- What are some factors that determine primer annealing temperature during PCR?
The annealing temperature (T_a) is typically set 2–5°C below the lower of the two primer melting temperatures (T_m). Several factors govern T_m and therefore T_a:
Primer length -> Longer primers have more base pairs to stabilize, raising T_m. The protocol specifies 18–22 bp binding regions as a balance between specificity and manageable T_m.
GC content -> G:C base pairs form three hydrogen bonds vs. two for A:T pairs, making GC-rich primers more thermally stable. Higher GC% → higher T_m. The protocol targets 40–60% GC in the binding region.
DNA sequence -> context Nearest-neighbor thermodynamic interactions between adjacent base pairs affect stability. This is why tools like OligoCalc or Benchling give more accurate T_m estimates than simple formulas.
Salt/buffer concentration -> Higher Mg²⁺ and monovalent cation concentrations stabilize the DNA duplex by shielding the negatively charged phosphate backbone, raising T_m. The Phusion HF buffer is optimized for this.
Primer concentration -> Higher primer concentration slightly increases T_m (shifts equilibrium toward duplex formation), though this effect is minor in practice.
Presence of mismatches -> The Color Forward primers in this protocol contain intentional mismatches in the chromophore region. Mismatches destabilize the duplex and lower effective T_m — this is why the annealing temperature for the insert PCR (53°C) is lower than for the backbone PCR (57°C).
Overhang sequences -> The 5’ overhang portions (Gibson overlaps) do not contribute to annealing T_m during early PCR cycles, since they are not complementary to the template. Only the 3’ binding region determines T_m for primer design purposes.
- There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
| Feature | PCR | Restriction Enzyme Digest |
|---|---|---|
| Mechanism | Exponential amplification using primers and thermostable polymerase | Sequence-specific endonuclease cuts at recognition sites |
| Input material | Any DNA template (plasmid, genomic, cDNA) | Purified plasmid or genomic DNA |
| Fragment definition | Defined entirely by primer placement — any region, any size | Defined by restriction site locations in the sequence |
| Overhang type | Blunt ends (Phusion) or custom overhangs via primer design | Sticky ends (4–6 bp overhangs) or blunt ends depending on enzyme |
| Sequence modification | Can introduce mutations, add sequences, change ends | Cannot alter sequence; cuts only at recognition sites |
| Cleanup | Requires PCR cleanup + DpnI digest (to remove template) | Requires gel purification or column cleanup |
| Time | ~1.5–2 hours (thermocycler + cleanup) | ~1–2 hours (digest + cleanup) |
| Error risk | Polymerase errors possible (mitigated by Phusion) | No sequence errors introduced |
| Scalability | Amplifies from nanogram quantities of template | Requires sufficient starting plasmid |
When to Prefer PCR:
- to introduce mutations (as in this lab — chromophore mutagenesis)
- the desired fragment lacks convenient restriction sites flanking it
- to add specific overhangs for Gibson/HiFi assembly
- working from low-abundance templates (genomic DNA, cDNA)
- building synthetic constructs from scratch
When to Prefer Restriction Enzyme Digest:
- working with well-characterized vectors with mapped restriction sites
- need directional cloning with defined sticky ends (ensures correct orientation)
- fragment fidelity is paramount and you want to avoid polymerase errors
- performing diagnostic digests to verify plasmid identity
- for large fragments (>10 kb) that are difficult to amplify by PCR
In this lab, PCR is the only viable choice because the goal is to introduce specific point mutations in the chromophore region, something restriction enzymes cannot do.
- How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembly requires that adjacent fragments share 20–40 bp of identical sequence at their junctions (overlaps). To ensure compatibility:
- Design overlaps into primers -> The 5’ overhangs of your primers must be complementary to the end of the adjacent fragment. In this protocol:
- The Backbone Reverse primer’s 3’ end overlaps with the Color Forward primer’s binding region
- The Color Reverse primer’s 3’ end overlaps with the Backbone Forward primer’s binding region Verify this in Benchling by aligning the primer sequences to the mUAV plasmid map.
Verify fragment sizes by gel electrophoresis -> Run a diagnostic agarose gel after PCR. Compare band sizes to predicted sizes calculated from your primer positions on the mUAV map. Unexpected bands indicate mispriming or contamination.
Confirm concentration by Nanodrop/Qubit -> Fragments should be ≥30 ng/µL. Insufficient DNA leads to failed assembly. The 260/280 ratio should be ~1.8 (pure DNA); lower values indicate protein contamination.
Check for correct orientation -> Ensure both fragments are in the same 5’→3’ orientation relative to the final circular plasmid. In Benchling, simulate the assembly to confirm the final construct is correct before running the reaction.
Remove template with DpnI -> Residual methylated mUAV template would compete with your PCR fragments in transformation, yielding colonies with the original (unmutated) plasmid. DpnI selectively digests the methylated template, leaving only your unmethylated PCR products.
Confirm overlap sequences are free of secondary structure -> Use NUPack or Benchling to check that the overlap regions don’t form strong hairpins (ΔG > −10 kcal/mol), which would prevent efficient annealing during assembly.
Calculate molar ratios -> Use the NEBioCalculator or similar tool to ensure a 2:1 insert:vector molar ratio. Since molar amount = (mass in ng / fragment length in bp) × (1/660), a longer backbone requires proportionally more mass to achieve the same molar amount as a shorter insert.
- How does the plasmid DNA enter the E. coli cells during transformation?
In this protocol, heat-shock transformation is used with chemically competent DH5α cells. The mechanism involves two phases:
Preparation of competent cells: Cells are treated with divalent cations (typically CaCl₂) during their preparation. Ca²⁺ ions neutralize the negative charges on both the bacterial outer membrane (lipopolysaccharide) and the DNA phosphate backbone, reducing electrostatic repulsion and allowing DNA to associate with the cell surface.
Ice incubation (30 minutes): The plasmid-cell mixture is kept on ice. At low temperature, the membrane is in a more ordered (gel) phase, and DNA-Ca²⁺ complexes associate with the outer membrane. The 30-minute incubation allows this association to occur.
Heat shock (42°C, 45 seconds): The abrupt temperature increase causes a rapid phase transition in the lipid bilayer — from ordered to disordered (fluid) phase. This creates transient hydrophilic pores or channels in the membrane. The thermal expansion also creates a pressure differential that drives DNA into the cell by passive diffusion down a concentration gradient (extracellular DNA concentration » intracellular).
Return to ice (5 minutes): The membrane rapidly re-seals as it returns to the ordered phase, trapping internalized DNA inside the cell.
SOC recovery (37°C, 60 minutes): Cells recover, repair membrane damage, and begin expressing the antibiotic resistance gene (chloramphenicol acetyltransferase in this case). This expression window is critical — without it, cells plated directly onto selective media would die before resistance is established.
Only cells that successfully incorporated the plasmid express chloramphenicol acetyltransferase, which inactivates the antibiotic by acetylation, allowing those cells to survive and form colonies.
Note: The efficiency of heat-shock transformation is typically 10⁵–10⁸ CFU/µg DNA — lower than electroporation (10⁸–10¹⁰ CFU/µg), but sufficient for this application since Gibson assembly produces circular, supercoiled-like plasmids that transform efficiently.
- Describe another assembly method in detail (such as Golden Gate Assembly)
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a one-pot, scarless DNA assembly method that uses Type IIS restriction enzymes (most commonly BsaI or BsmBI) to generate user-defined 4-bp sticky ends on DNA fragments, which are then ligated in a defined order. Unlike conventional Type IIP restriction enzymes (e.g., EcoRI, BamHI) that cut within their recognition sequence, Type IIS enzymes cut at a fixed distance outside their recognition site, meaning the recognition sequence is removed from the final product, leaving no scar. This is the key innovation: by placing the Type IIS recognition site in the primer overhang (outside the desired insert), the enzyme cuts away its own recognition sequence after digestion, leaving only the custom 4-bp overhang you designed. The reaction is run as a thermocyclic digest-ligation: alternating between ~37°C (restriction enzyme active) and ~16°C (ligase active) for 25–30 cycles, driving the equilibrium toward correctly assembled, ligated products. Because each junction has a unique 4-bp overhang, fragments can only assemble in one specific order and orientation, which enables simultaneous, directional assembly of up to 35+ fragments in a single tube. The assembled product cannot be re-cut by the enzyme (since the recognition site is gone), which drives the reaction to completion and gives Golden Gate its characteristically high efficiency. This makes Golden Gate especially powerful for combinatorial library construction and modular cloning (MoClo) systems in synthetic biology, where standardized parts (promoters, RBS, CDS, terminators) are assembled hierarchically into complex multi-gene constructs.
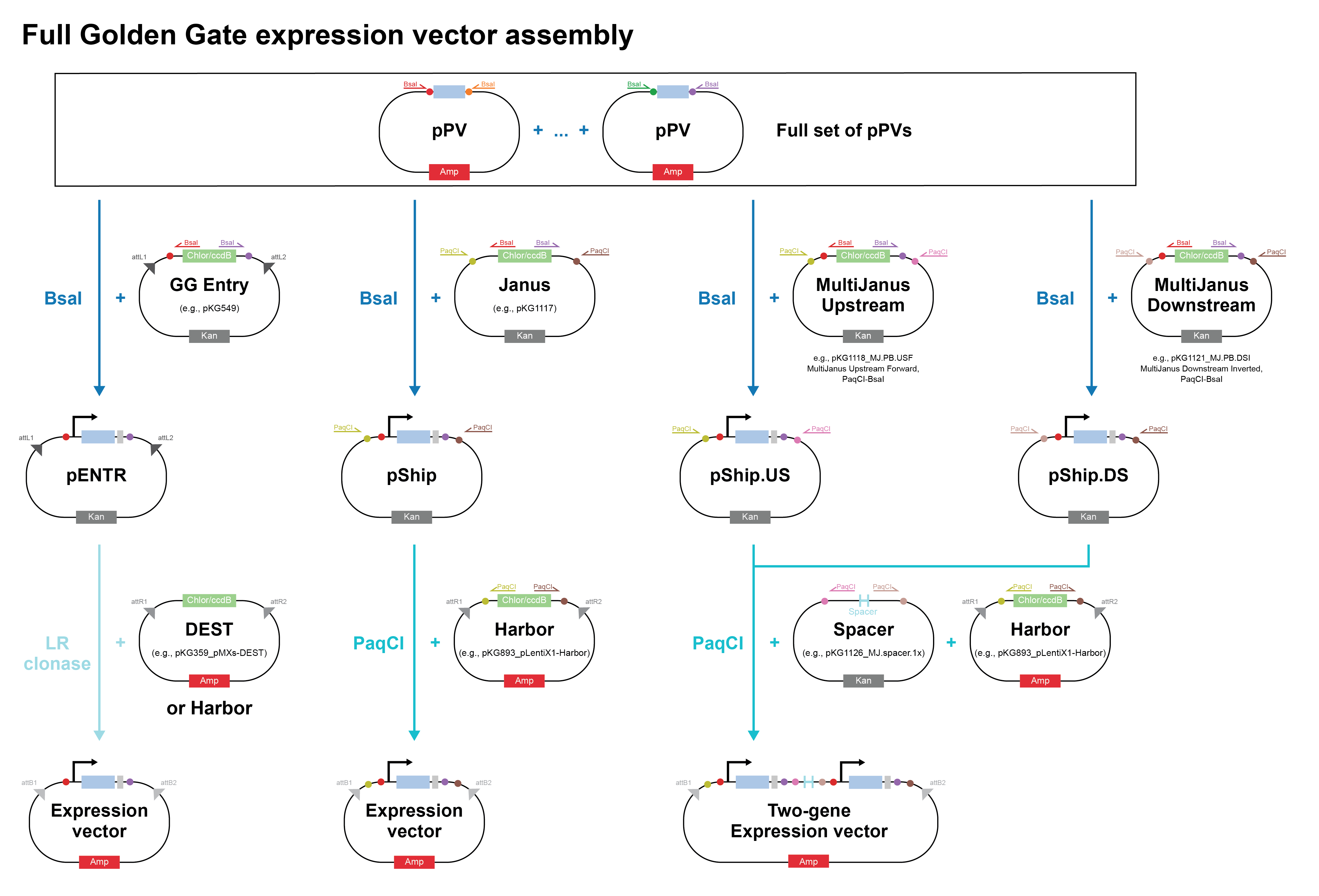
| Feature | Golden Gate | Gibson Assembly |
|---|---|---|
| Key enzymes | Type IIS restriction enzyme + ligase | T5 exonuclease + polymerase + ligase |
| Overhang type | 4-bp sticky ends (enzyme-generated) | ~20–40 bp ssDNA overhangs (exonuclease-generated) |
| Scar sequence | None (recognition site removed) | None (seamless) |
| Fragment number | Up to 35+ in one pot | Typically 2–6 (efficiency drops with more) |
| Reaction | Thermocyclic (37°C ↔ 16°C) | Isothermal (50°C, 15–60 min) |
| Directionality | Enforced by unique 4-bp overhangs | Enforced by overlap sequence design |
| Reusability of parts | High — standardized parts in MoClo libraries | Lower — overlaps are construct-specific |
| Internal restriction sites | Must avoid internal BsaI/BsmBI sites | No restriction site constraints |
| Best for | Combinatorial assembly, large multi-part constructs, standardized libraries | Mutagenesis, 2–4 fragment assemblies, adding overhangs to existing fragments |
For this lab specifically, Gibson Assembly is the better choice because you only have 2 fragments, you need to introduce specific point mutations via primer design, and the mUAV plasmid likely contains internal BsaI sites that would complicate Golden Gate design.
b. Model this assembly method with Benchling or Asimov Kernel!