Kate Carline is a fourth year undergraduate student studying Biology and Public Policy with a focus on synthetic biology and bacteriophage. She has published her research three times, including as first author for her work on William & Mary’s iGEM team, a globally-recognized group researching engineered bacteria and phage in soil, and in Dr. Margaret Saha’s SEA-PHAGES Lab, where she characterized a novel phage named Discoknowium. In addition to her work as a committed listner and a TA for the new HTGAA W&M node, Kate is currently participating in Honors research where she is investigating microbial community editing.
Class Assignment First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. I am interested in engineering bacteriophage (viruses which infect bacteria) in order to detect harmful bacterial pathogens by using the engineered viruses to deliver and express a certain reporter gene upon infection.
Class Assignment Waters Part I - Molecular Weight We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Class Assignment Part I. Example design performed in Benchling found here:
Design for lab found here:
Part II. Performed in lab. Resulting Gel Image here:
Class Assignment Python Script Uploaded to form and shared with lab.
Post-lab questions I investigated the paper “Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex” as I was interested in using GGA to assemble my engineered phage satellite for my final project and was curious as to how I could integrate an Opentrons workflow into the process. The paper tested two GGA kits, the MoClo Yeast Toolkit (YTK) and the SubtiToolKit (STK), with GFP as the reporter for both, then transformed the assembled plasmids into E. coli and Saccharomyces cerevisiae (yeast) or Bacilius subtilius based on the according kit. The authors found high assembly and transformation efficiencies using the opentrons to complete the protocols (17/17 colonies and 8/13 colonies contained the GGA construct with GFP using the YTK and STK kits respectively).
Class Assignment Part A. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) 500 grams is approximately 3.01e26 Daltons, which converts to 3.01e24 molecules of amino acids.
Class Assignment DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? We used NEB’s Q5 2X Hot Start Master Mix for our lab, but like the Phusion MM, it contains a DNA polymerase (for the extension step of PCR), as well as dNTPs and Mg2+ as polymerase cofactors and a buffer.
Class Assignment IANNs What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs can account for greater complexity, such as changing the level of output based on gradients or combinations of signals, which better reflects realistic biological systems.
Class Assignment General Qs Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Describe the main components of a cell-free expression system and explain the role of each component. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each. Kate Adamala Qs Design an example of a useful synthetic minimal cell as follows:
Subsections of Homework
Week 1: Principles and Practices
Class Assignment
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
I am interested in engineering bacteriophage (viruses which infect bacteria) in order to detect harmful bacterial pathogens by using the engineered viruses to deliver and express a certain reporter gene upon infection.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Safety of the use of these viruses is critical. Establishing good manufacturing practices and regulatory principles are complex, as phage replication is not as ‘consistent’ as a manufacturing a certain chemical drug would be, and combinations of certain phage and hosts may have implications for transduction of antibiotic resistance genes. Additionally, for therapeutic applications, accessibility and efficiency will be a priority. Questions on patenting phage, for example, would have to balance investment into this innovation (particularly given emerging phage therapies will be competing against thet traditional pharmaceutical drug market) with preventing artifically-high healthcare costs.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Policymakers (after consulting with members from the scientific community and industry) could mandate a set of good manufacturing practices specifically for the complexities of engineered phage, which would be required for FDA (or similar) approval and could be punished with fines or in extreme cases criminal liability. For example, regulatory organizations could adopt a modular approval process, such as the process for mRNA vaccines, where certain base manufacturing and delivery processes are reviewed but once approved minor swapping of phage ‘parts’ could be incorporated with a less lengthy review time. Currently, in the US, phage are generally regulated as ‘biological/medicinal products’ in the same category as drugs, although none has been approved with full FDA approval (their applications are just as an ‘Investigative New Drug’). The UK Medicines and Healthcare Regulatory Agency recently just released a set of guidelines specifically for phage therapy, to help clarify manufacturing expectations and the path to approval, yet did not address applications for engineered phage nor set any legally binding mandates.
The scientific community could share data on engineered phage reactions when co-cultured with other hosts and phage in order to better accumulate safety information about possible combinations. Currently, PhagesDB stores detailed information about sequenced phage found from various environments and the American Society for Microbiology’s Phage Therapy Coordination Network is analyzing clinical trial and manufacturing quality data, but no similar dataset exists for phage with engineered constructs. It is more difficult with engineered constructs as many labs and industries want to keep said constructs proprietary (whereas one cannot patent natural phage as the case for PhagesDB), and so the biggest hurdle would be to garner interest into voluntary information sharing.
Engineered phage companies could adopt post-treatment screening practices after applying engineered phage therapies. This data may be collected in certain clinical trials but there is no evidence of a norm at scale for all treatments to be monitored for a substantive period of time, particularly in non-therapeutic cases. Greater investment into diagnostic and sequencing labs will be necessary for this option, although costs could be offset with monetary incentives for screening (either government subsidies or even a market label that could attract consumers).
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Priotizing Patients
• By preventing incidents
1
2
3
• By helping respond
3
2
1
• By efficiently treating individuals
2
1
3
Accessibility
• By lowering costs
1
2
3
• By communicating safety information
2
3
1
Protect the environment
• By preventing incidents
1
2
3
• By helping respond
3
2
1
Economic considerations
• Minimizing costs and burdens to taxpayers
3
1
2
• Market competitiveness
1
3
2
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
I think Option 1 is the most feasible option, given the early precedent set by MHRA’s ‘regular’ phage therapy guidelines released last Summer, and has the widest impact at the national level. It paves the way for approved engineered phage therapy use, which is important to address accessibility and trust as well as introducing more safety review. By making the path more clear with a modular approval process, engineered phage therapy becomes an economically viable opportunity for the private sector to invest in as well, which will help advance research. The biggest challenge will be that any proposed regulatory system will be incomplete, as there a wide range of applications and cases for engineered phage therapy, as well as the financial burden for hiring more regulators to review these proposals. My proposed target would be to Senator Young, who is the Commissioner of the National Security Commission for Emerging Biotechnology and is already leading on introducing and implementing biotech-related legislation in Congress.
Instructor Questions
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
The error rate of eukaryotic DNA polymerase is 1 error every 106 bases. The human genome is approximately 3.2 x 109 base pairs long (6.4 billion is in most cells as they are diploid). This is problematic as this would introduce 3 errors for every two cells that replicate. There are thus other DNA repair polymerases that are dedicated to finding and fixing these errors.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Due to codon degeneracy (where a single codon could be encoded by several different DNA/RNA sequences), there are about 3 different sequences on average for each amino acid (64 possible codons for 20 amino acids). If an average human protein is 1036 bp, and each codon is 3 bp long, there are about 345 codons and thus about 3^345 ways the protein could be sequenced. mRNA regulations (different folding, silencers, etc) may be one reason as to why all these different codes don’t work for a protein of interest.
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite DNA Synthesis Cycle, a four step cycle where the existing strand is ‘deprotected’ with an acid or light to prepare for addition, a nucleotide base (often modified to include this protector) is added one at a time, the strand is capped (this step is sometimes included after each base in order to prevent errors), and then the phosphate is oxidized.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
The yield drops exponentially for long sequences as you are added bases one at a time, and shorter impurities become dominant.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Related to the answer above, the challenges in yield and purity for longer sequences makes directly synthesizing that long of a gene at once very difficult. But, you could synthesize it in smaller parts and use DNA assembly tools like Gibson to put the gene together!
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. The Lysine Contingency refers to a fictional genetic modification in Jurassic Park, where scientists knocked out the ability for the dinosaurs to produce their own lysine in order to prevent their successful survival in the wild if there were to escape the part (as they would be dependent on the park’s food supplements). As proven though with animals in real life lacking the ability to produce their own lysine, there are many ways to supplement these amino acids in the wild (other animals, dairy products, or soybeans are all lysine-rich foods, for example).
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine
for each adjacent pair of peaks
using:
Determine the MW of the protein using the relationship between
,
, and
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with $\frac{m}{z}$ values.
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with
values.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Homework: Waters Part II — Secondary/Tertiary structure
Assignees for this section
MIT/Harvard students Optional but highly recommended
Committed Listeners Optional but highly recommended
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800
? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $\frac{m}{z}$ on a mass spectrometer with 30,000 resolution.
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800
on a mass spectrometer with 30,000 resolution.
Homework: Waters Part III — Peptide Mapping - primary structure
Assignees for this section
MIT/Harvard students Required
Committed Listeners Required
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
Navigate to https://web.expasy.org/peptide_mass/
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Week 11 - Bioproduction & Cloud Labs
Class Assignment
Week 2: DNA Read, Write, Edit
Class Assignment
Part I.
Example design performed in Benchling found here:
Design for lab found here:
Part II.
Performed in lab. Resulting Gel Image here:
Part III.
3.1
I selected the Anti-CRISPR protein AcrIF7 in Pseudomonas aeruginosa prophage as I’m fascinated by the relationship between phages and their hosts and I believe Acrs could have an important role in biosafety for their potential to serve as off-switches for CRISPR editing.
The protein sequence (as identified from NCBI accession number WAX21977) is: mttftsivtt npdfggfefy veagqqfdds ayeeaygvsv ptavveemna kaaqlkdgew lnvshea
3.2
The reverse translated DNA sequence using the Bioinformatics.org online tool and the default E. coli codon table is: atgaccacctttaccagcattgtgaccaccaacccggattttggcggctttgaattttat
gtggaagcgggccagcagtttgatgatagcgcgtatgaagaagcgtatggcgtgagcgtg
ccgaccgcggtggtggaagaaatgaacgcgaaagcggcgcagctgaaagatggcgaatgg
ctgaacgtgagccatgaagcg
Electroporation of the DNA sequence on a plasmid (designed in Part IV) into P aeruginosa could enable production of the protein as the cell would utilize its own resources to transcribe and translate the sequence if an according promoter and RBS were on the plasmid with the sequence of interest. Electroporation is preferred to insert the plasmid into the cell as it is more efficient than other chemical techniques.
Part IV.
4.1
Twist account created.
4.2
Image of assembled sequence here:
Instructions followed to create vector in Twist for parts 4.3-4.6.
Part V.
5.1
I would be interested in sequencing a metagenomic analysis from a patient with an antibiotic resistant infection being treated with phage therapy. I would be curious to sequence DNA at several courses of time across the treatment to see how the entire microbial community in the environment (say, a gut microbiome) is affected by phage therapy. I would use Illumina sequencing as I don’t need full assembled genomes of each community species, but rather the high-throuput short-read approach would ensure I can capture a wide array of variants in the community.
5.2
I would be interested in synthesizing a construct using parts of a phage satellite than can integrate into a bacterial genome with a helpful anti-virulence circuit (like a CRISPR/Cas9 cassette targeting a virulence gene) to engineer bacterial hosts (particularly mycobacteria, which have key environmental and therapeutic applications and have challenges with developing antibiotic resistance). I would likely use PCR with primers designed to amplify parts of interest from a phage satellite (as I already know the genetic sequence) and could use Twist to synthesize the additional CRISPR/Cas genetic insert. PCR includes the denaturation of the template DNA, annealing of primers, extension of the amplicon, and the repetition of many cycles to produce many copies: one advantage is this high quantity of DNA of the part produced, although it is limited in the length of amplicons it can be effectively used. Twist synthesis is highly accurate and can synthesize longer segments, but does not have as many copies.
5.3
Since we have already identified a mutation in a singular, specific gene that causes Huntington’s Disease, I would be fascinated to edit out the trinucleotide repeat mutation in that gene that causes the disorder. Already, microRNA treatments have been employed to control expression (so not a DNA editing approach), and base editing through CRISPR/Cas9 has proven success in mice at a permanent, one-time treatment. The base editing CRISPR system uses prime editors, and so uses a gRNA designed for this trinucleotide mutation in the huntingtin gene alongside a mutated Cas9 enzyme that only cuts one strand and a catalytic enzyme capable of inducing single base pair mutations. To test the realistic constrains on delivery, the input would likely by Huntington affected cells and the editing system would need to be injected and localize to the nucleus to make the DNA edits.
Week 3: Automation
Class Assignment
Python Script
Uploaded to form and shared with lab.
Post-lab questions
I investigated the paper “Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex” as I was interested in using GGA to assemble my engineered phage satellite for my final project and was curious as to how I could integrate an Opentrons workflow into the process. The paper tested two GGA kits, the MoClo Yeast Toolkit (YTK) and the SubtiToolKit (STK), with GFP as the reporter for both, then transformed the assembled plasmids into E. coli and Saccharomyces cerevisiae (yeast) or Bacilius subtilius based on the according kit. The authors found high assembly and transformation efficiencies using the opentrons to complete the protocols (17/17 colonies and 8/13 colonies contained the GGA construct with GFP using the YTK and STK kits respectively).
This paper shows promise for using the opentrons to run the protocols of other GGA kits for my final project. I also plan to use opentrons for phage plating, particularly for spot tests covering the plate in a webbed lysis form that can be flooded to increase the titer of phage and phage satellites, and have started working on a Python script for such applications.
Week 4: Protein Design 1
Class Assignment
Part A.
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
500 grams is approximately 3.01e26 Daltons, which converts to 3.01e24 molecules of amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The fish/cow DNA introduced by the food you consume does not transform into your cells (and furthermore their nuclei), and so cannot even be read by human transcription/translation machinery to produce fish/cow proteins. Furthermore, specialization of promoters and ribosomes mean that the human machinery cannot ‘read’ this foreign DNA as if it were human DNA.
Why are there only 20 natural amino acids?
The leading hypothesis is that so far evolution has optimized for this number to introduce enough variation for protein diversity but not so much that amino acids are not stable.
Can you make other non-natural amino acids? Design some new amino acids.
Absolutely! There is a lot of existing research to produce non-natural amino acids for applications in drug therapeutics and molecular imaging (such as photo-reactive amino acids).
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids were first formed from random interactions in the ‘primordial soup’ of water, inorganic gases, and energy sources such as heat.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
We would expect left handedness.
Why are most molecular helices right-handed?
In nature, amino acids exist in L confirmation which results in righ-handed helices due to sterical constraints for stability, but there are growing efforts to make D-amino acids (mirror images). This ‘mirror life’ efforts have been controversial, however, with many scientists signing a statement about the risks of mirror life in December 2024.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The backbone of the Beta sheet wants to form hydrogen bonds with other Beta sheet backbones, leading to aggregation. Stacked Beta sheets have greater thermodynamic stability (lower energy state) and so this aggregation is preferred.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Based on how proteins misfold in amyloid diseases, the beta-sheet stacking structure is preferred to create the large aggregates of amyloid fibers. These amyloid beta sheet structures are very strong, and so have been employed as materials for tissue engineering or even environmental filtering!
Part B.
Briefly describe the protein you selected and why you selected it.
I selected the mCherry protein as I plan to use it as a ‘proof of concept’ reporter gene to see whether my integrative phage satellite can be used to engineer M. aichiense.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
It is 236 amino acids long and the most frequent amino acid is Glycine (25 residues).
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Uniprot reports two similar proteins (both uncharacterized, from Pseudotamlana agarivorans and Purpureocillium lilacinum (Paecilomyces lilacinus)) at 100% identity match, but no official homologs.
Does your protein belong to any protein family?
Yes, the pFam PF01353, which also includes many green fluorescent proteins.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better.
The crystal structure I found was published in 2006 with a 1.36 Å resolution, so a high resolution and thus a good quality structure.
Are there any other molecules in the solved structure apart from protein?
There are some water molecules and one small ’non-standard’ molecule CH6 at the center of the protein, but otherwise the solved structure is just the protein.
Does your protein belong to any structure classification family?
Its immediate family is the mFruits family of other red fluorescent proteins.
Open the structure of your protein in any 3D molecule visualization software. Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
I used the RCSB visualization software.
Cartoon visualization
Ribbon visualization
(In RCSB, this is called ‘Backbone’ representation type)
Ball and stick visualization
Color the protein by secondary structure. Does it have more helices or sheets?
The protein appears to have more sheets (colored in yellow).
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophilic residues are colored in orange and hydrophobic residues are colored in green. The protein appears to have a fairly even distrubition of residues that tend to alternate.
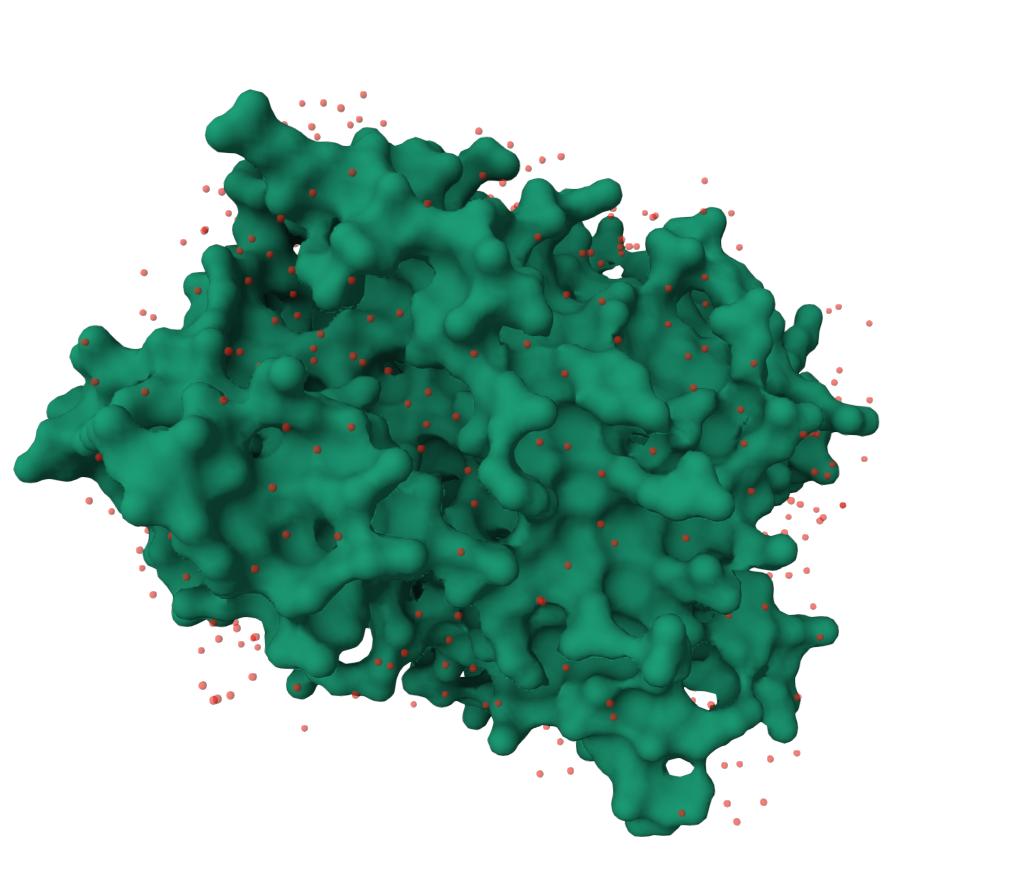
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
The image has been rotated to highlight the deepest hole observed in the surface of the protein, but I would not say there is an clearly apparent large binding pockets.
Part C.
Week 5: Protein Design II
Class Assignment
Week 6: Genetic Circuits I
Class Assignment
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
We used NEB’s Q5 2X Hot Start Master Mix for our lab, but like the Phusion MM, it contains a DNA polymerase (for the extension step of PCR), as well as dNTPs and Mg2+ as polymerase cofactors and a buffer.
What are some factors that determine primer annealing temperature during PCR?
The make up of nucleotides in the primer sequence (more GC content means a higher annealing temperature as it takes more energy to form the three hydrogen bonds between the base pairs) as well as the overall primer length.
There are two methods from this class that create linear fragments of DNA: PCR and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR denatures DNA to pull the two strands apart, uses primers designed to target regions to anneal and extend certain regions, and amplifies the target product. Restriction Digests cut the template DNA at specific recognition sites based on the employed enzyme. PCR is preferred to produce high-quantity outputs of DNA for a particular region, whereas restriction digests can be used to produce cut DNA with ends compatible for assembling with another piece of DNA not from the template (such as a plasmid with complementary cut ends).
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
PCR cleanups and assessing through Nanodrop scores can be used to make sure there is not additional junk (such as from the polymerases in the PCR solution reaction) in the product and that there is sufficient concentration of DNA for an efficient cloning reaction.
How does the plasmid DNA enter the E. coli cells during transformation?
Either electroporation or chemical transformation can work with E. coli - for our lab, we used chemical transformation with the NEB PCR Cloning kit.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly uses Type II Restriction Enzymes that cut outside of their recognition sites to create 4 bp overhangs. Fragments are designed so that the resulting base pair overhangs after a restriction digest are complementary to the overhangs of the neighboring fragments so that the DNA parts can assemble like puzzle pieces (using a ligase). This assembly tool is modeled with my final project.
Asimov Kernel
Work documented in Asimov Kernel notebook.
Week 7: Genetic Circuits II
Class Assignment
IANNs
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs can account for greater complexity, such as changing the level of output based on gradients or combinations of signals, which better reflects realistic biological systems.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs could be used in therapeutic applications in a gut microbiome, such as to produce an anti-inflammatory cytokine inhibitor at low, medium, or high levels based on the intensity of several signals for inflammation (tetrathionate, nitric oxide, etc). This would however have high metabolic burden, and the engineered bacteria may struggle in the competitive gut microbiome.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials have been used for everything from fashion (as a leather-like material) to shipping (biodegradable packaging) to construction (insulation). They are biodegradable, which is a huge advantage over the counterparts they replace (like styrofoam) but are not are strong (more susceptible to moisture changes, can’t hold as much weight).
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
You could engineer fungi to have greater fiber strength for the above applications to address this weakness. Many fungi are eukaryotic and so are better at producing certain eukaryotic proteins than bacterial chassis and their multicellular capability expands potential applications (such as with fungal materials).
Twist Order
Insert sequence recorded in Benchling.
Week 8 - Cell Free Systems
Class Assignment
General Qs
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Kate Adamala Qs
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it. What would your synthetic cell do? What is the input and what is the output? Could this function be realized by cell-free Tx/Tl alone, without encapsulation? Could this function be realized by genetically modified natural cell? Describe the desired outcome of your synthetic cell operation.
Design all components that would need to be part of your synthetic cell. What would be the membrane made of? What would you encapsulate inside? Enzymes, small molecules. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian) How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.) How will you measure the function of your system?
Peter Nguyen Qs
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail?
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Ally Huang Qs
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Final Project
Updates to final project can be found on the Final Project page.
Abstract Phage satellites–mobile genetic elements which hijack a helper phage to propagate–have great potential to revolutionize synthetic biology applications. In particular, phage satellites have also been employed to deliver genetic circuits to the bacterial chromosome, including CRISPR/Cas9 cassettes that can target virulence genes to weaken pathogens (Ram et al 2018). A novel phage satellite class, Extracellular Prophage-Inducing Particles (EPIPs), can target Mycobacteriaceae and thus have particularly relevant applications for engineering native soil microbial communities to improve environmental health as well as treating common mammalian pathogens causing tuberculosis and leprosy (Qian et al., 2025). Mycobactericae can be particularly tricky to engineer using traditional plasmid techniques due to their mycolic acid walls preventing easy uptake of foreign DNA (Dao et al 2025). Already, mycobacteriophage-based DNA delivery has been found to increase transduction efficiency, including to deliver resolvases that knockout hygromycin antibiotic resistance in M. tuberculosis (Jain et al 2014), and other phage satellite classes infecting different bacterial hosts (such as SaPIs) can deliver circuits in a similar infection and integration manner (Ram et al 2018). These phage and phage-satellite methods are also advantageous in that modifications do not require antibiotic selection to persist, which is important given the rising biosafety concerns of horizontal gene transfer of antibiotic resistance (de Lorenzo and Martinez-Garcia 2025) particularly given a common antibiotic resistance cassette used in plasmid engineering, kanamycin, also confers resistance to amino glycides, a drug class used to treat tuberculosis infections (Yang et al 2015). I thus propose to modify one such EPIP capable of integrating into the M. aichiense genome, the EPIP named Bernie, to investigate its potential use as a microbial community editor. Specificially, I will use Golden Gate Assembly to re-assemble Bernie with the red fluorescence protein mCherry and the strong psmyc promoter optimized for myco, so that engineered Bernie DNA could be electroporated into the M. aichiense host, the phage satellite could reboot and infect, and the bacterial host would then express the red fluorescence gene newly integrated into its genome (as assessed using plate reader techniques).
Phage satellites–mobile genetic elements which hijack a helper phage to propagate–have great potential to revolutionize synthetic biology applications. In particular, phage satellites have also been employed to deliver genetic circuits to the bacterial chromosome, including CRISPR/Cas9 cassettes that can target virulence genes to weaken pathogens (Ram et al 2018). A novel phage satellite class, Extracellular Prophage-Inducing Particles (EPIPs), can target Mycobacteriaceae and thus have particularly relevant applications for engineering native soil microbial communities to improve environmental health as well as treating common mammalian pathogens causing tuberculosis and leprosy (Qian et al., 2025). Mycobactericae can be particularly tricky to engineer using traditional plasmid techniques due to their mycolic acid walls preventing easy uptake of foreign DNA (Dao et al 2025). Already, mycobacteriophage-based DNA delivery has been found to increase transduction efficiency, including to deliver resolvases that knockout hygromycin antibiotic resistance in M. tuberculosis (Jain et al 2014), and other phage satellite classes infecting different bacterial hosts (such as SaPIs) can deliver circuits in a similar infection and integration manner (Ram et al 2018). These phage and phage-satellite methods are also advantageous in that modifications do not require antibiotic selection to persist, which is important given the rising biosafety concerns of horizontal gene transfer of antibiotic resistance (de Lorenzo and Martinez-Garcia 2025) particularly given a common antibiotic resistance cassette used in plasmid engineering, kanamycin, also confers resistance to amino glycides, a drug class used to treat tuberculosis infections (Yang et al 2015). I thus propose to modify one such EPIP capable of integrating into the M. aichiense genome, the EPIP named Bernie, to investigate its potential use as a microbial community editor. Specificially, I will use Golden Gate Assembly to re-assemble Bernie with the red fluorescence protein mCherry and the strong psmyc promoter optimized for myco, so that engineered Bernie DNA could be electroporated into the M. aichiense host, the phage satellite could reboot and infect, and the bacterial host would then express the red fluorescence gene newly integrated into its genome (as assessed using plate reader techniques).
Project Aims
I. Experimental Aims
Use Golden Gate Assembly to reassemble the Bernie phage satellite (that can infect and integrate into Mycolicibacterium aichiense) with the mCherry red fluorescence gene and psmyc strong promoter optimized for mycobacteria. Reboot engineered Bernie into M. aichiense and assess for fluorescence expressed by the bacteria
II. Developmental Aims
Expand this engineering approach to swap in more complex genetic circuits, such as a CRISPR/Cas9 system that could target virulence genes in the host or a pAH (polycyclic aromatic hydrocarbons, a common organic pollutant in environments such as soil) pollutant degradation system.
III. Visionary Aims
Deploy engineered phage satellites into real-world environments such as soil to rapidly grant restorative capabilities to the microbiome.
Background
Phage satellites–mobile genetic elements which require a helper phage to propagate–have great potential to revolutionize synthetic biology applications. Biomanufacturing efforts often struggle with phage infection killing off bacterial populations (Kaminsky and Paczensky 2024), and phage satellites can defend against phage-induced lysis (Boyd and Seed, 2024). Phage satellites have also been employed to deliver genetic circuits to the bacterial chromosome, including CRISPR/Cas9 cassettes that can target virulence genes to weaken pathogens (Ram et al 2018). Additionally, one ongoing limitation of using phage therapy to treat antibiotic resistant infections is the limited host range of each phage (Cook and Hynes 2025); phage satellites have been identified to expand the host range of certain helper phages (He et al 2025).
Bacteriophage (phage) are viruses that infect bacteria, and many phage are temperate, meaning they can switch between either a lytic life cycle, replicating within and then killing bacteria to release phage particles, or a lysogenic life cycle, integrating into the bacterial chromosome and suppressing expression until a signal to induce (then excising from the bacterial genome and returning to the lytic cycle). Phage satellites are generally smaller than their helper phage and often lack the core machinery necessary for packaging and infection in their own genomes, such as capsid heads or tail fibers. They are not found in plasmids and have little homology to known phage, indicating a distinct evolutionary path (Sousa and Rocha 2021, Penades et. al 2025). Different categories of phage satellites hijack the helper phage in unique ways, but they all propagate by exploiting these phage resources.
Extracellular Prophage-Inducing Particles (EPIPs) are a newly-identified class of phage satellites that target Mycobacteriaceae and thus have particularly relevant applications for engineering native soil microbial communities to improve environmental health as well as treating common mammalian pathogens causing tuberculosis and leprosy (Qian et al., 2025). Like other phage satellites, EPIPs lack foundational proteins such as capsid and tail proteins, however they are genetically distinct from previously-characterized phage satellites, including genes such as Tape Measure Proteins that have not been found in any other phage satellites. These EPIPs are able to induce the prophage HerbertWM in Mycolicibacterium aichiense, and one EPIP, Bernie, has been identified to additionally independently integrate into the M. aichiense bacterial chromosome. My project thus seeks to use Bernie as a delivery tool for genetic circuits into Mycobacteriaceae.
Important ethical considerations for this project include the biosafety risks of deploying engineered bacteria, particularly for long-term goals with field usage, as the engineered circuit release would be difficult to reverse and phage satellite infection and excision could result in horizontal gene transfer. I might apply the ethical principles of Beneficiance, as the benefits of addressing soil pollution or deadly pathogens seems to outweigh the biosafety risks, especially given this is an improvement on the antibiotic-resistance risks existing in the status quo. Additionally, relevant precautions under the principle of non-maleficence would be utilized, such as introducing kill-switches to control circuits and assessing horizontal gene transfer before deployment.
Experimental Design
I. DNA Design of Engineered Bernie Constructs
While several engineering tools could be employed to modify EPIP phage satellites, Golden Gate Assembly has proven recently successful in assembling high GC% mycobacteriophage (Ko et al 2025) and thus would be particularly applicable for Bernie with a GC content of 63.4%. Other alternatives such as Gibson Assembly requires long overlap regions of 30-50 bp between fragments, which can be challenging to synthesize due to the high GC content in mycobacteriophage and EPIP genomes and is thus rarely attempted. The Hatfull lab, a leader in mycobacteriophage engineering efforts due to their work in the HHMI-Sponsored SEA-PHAGES program, has historically developed BRED, and by extension CRISPY-BRED, as the foundation for modifying mycobacteriophage. BRED works by prompting homologous recombination with a phage and synthetic DNA containing the mutation co-electroporated into the host (Marinelli et al 2008). CRISPY-BRED adds a selection mechanism to improve screening given BRED’s limited transformation efficiency by using CRISPR/Cas9 and an sgRNA complementary to the phage gene targeted for deletion/replacement in order to kill any phage that have not undergone recombination (Wetzel et al 2021). That said, CRISPY-BRED is limited by the constraints of PAM sites, and while screening is streamlined, still suffers from the low transformation efficiency of BRED. Likely due to these limitations, Hatfull recently transitioned efforts to focus on Golden Gate engineering of several mycobacteriophages (Ko et al 2025), and thus I selected that approach to test engineering of Bernie.
To achieve a high-fidelity assembly of Bernie with the strong constitutive psmyc promoter and mcherry gene, the Type IIS Restriction Enzyme PaqCI was selected to minimize internal cut sites. The Bernie genome was divided into 5 approximately-equal-sized fragments for scarless assembly using New England Biolabs’ Ligase Fidelity tool NEBridge SplitSet, where the tool predicted the best junction sites to split without overhang scarring when given ranges between gene gaps and Bernie’s two internal PaqCI cut sites. Primers were then designed using the IDT Oligo Analyzer tool to include a region complementary to these recommended junction sites as well as an overhang with the PaqC1 cut site and according filler bases.
II. Amplification of Bernie Parts for GGA
Bernie DNA was isolated using 1 ml of previously-isolated lysate and Phenol:Chloroform:Isoamyl Alcohol extraction with ethanol precipitation techniques (Phagehunting Program PCI/SDS DNA Extraction). 2 ng of said isolated DNA was utilized for each PCR fragment amplification according to standard NEB protocol alongside 0.5 μl of each forward and reverse primer, 5 μl of Q5 Hot Start 2X Master Mix, and NFW to a total of 10 μl (NEB). The annealing temperature for each reaction was determined using the corresponding primers and the NEB Tm calculator. PCR was performed on a Thermocycler with initial denaturation for 30 seconds at 98℃; 30 cycles of 98℃ for 7 seconds, annealing for 20 seconds, and extending at 72℃ for 40 seconds per amplicon kb length; and a final extension of 72℃ for 2 minutes. Products were run on a 1% agarose gel to confirm successful amplification of the expected length. PCR Products were cleaned up using NEB’s Monarch® PCR & DNA Cleanup Kit (5 μg) (NEB #T1030) following manufacturer’s instructions with 15 μl of elution buffer used.
To clone amplified fragments into plasmid backbones for preservation and use in Golden Gate Assembly, NEB’s PCR Cloning Kit (NEB #E1202S) was used following manufacturer instructions with the exception of scaling down the volume of NEB 10-beta Competent E. coli from 50 μl per reaction to 14 μl per reaction and corresponding NEB 10-beta/Stable Outgrowth Medium from 950 μl to 266 μl per reaction. Transformation steps occurred immediately after ligation. Transformed cells were plated at both 100 and 10-1 dilutions on LB agar plates containing 100 µg/mL ampicillin. Colonies were picked with sterile streaking needles, resuspended in 9 ul of NFW and then immediately dipped into 5 ml of LB media containing 100 µg/mL ampicillin in a glass tube which were placed on a shaking incubator at 37℃ and 250 rpm overnight. 1 ul of the NFW resuspension was used for insert screening PCR according to NEB PCR Cloning Kit protocols, using Q5 as the DNA polymerase and the given manufacturer Cloning Analysis primers designed around the pMiniT 2.0 Toxic Minigene Cloning Site to assess whether amplicons matched expected fragment lengths. The overnight cultures of successfully screened colonies were then miniprepped using the NEB Monarch Plasmid Miniprep Kit following manufacturer instructions. 50 μl of Elution Buffer was used given the pMiniT 2.0 backbone is a high copy plasmid.
III. Golden Gate Assembly Testing
For Golden Gate Assembly with NEB PaqC1 (#R0745S), 75 ng of each miniprepped plasmid, 7.5 U of PaqC1, 5 pmol of PaqC1 Activator, 300 U of T4 DNA Ligase, 2 μl of 10X T4 DNA Ligase Buffer, and NFW to a 20 μl total volume. Reaction was then run for 45 cycles of 37℃ for one minute and 16℃ for one minute, followed by a 5 minute incubation at 60℃. 2 μl of product was run with 2 μl of 6X loading dye and 8 μl NFW on a 1% agarose gel for 40 minutes at 150V and approximately 185 mA to assess assembly success.
IV. List of HTGAA Techniques Relevant To Project
The following HTGAA Techniques were employed:
Pipetting
Lab Safety
Bioethical Considerations
DNA Editing
DNA Construct Design
Gel Electrophoresis
Databases (NCBI)
Use of Benchling
Chassis Selection
Plasmid Preparation
Bacterial Culturing
Primer Design
PCR Reactions
Other Cloning Methods
Primer design for Golden Gate Assembly was fundamental to my project in order to design around each fragment from the phage satellite template DNA as well as the pCherry3 template DNA to include the PaqC1 recognition site and the according overhang. I also had the opportunity to employ real wet lab techniques, particularly gel electrophoresis which was employed at many steps (verifying initial PCR amplification of fragments, screening transformed colonies, running Golden Gate Assembled DNA).
Results and Validation
Project Validation
I chose to validate my project using DNA Design, PCR reactions with primers, and an initial Golden Gate Assembly (protocols explained in above experimental section). Techniques such as Primer Design, PCR Reactions, Golden Gate Assembly (Other cloning methods), and Gel Electrophoresis were used in this Validation step. ‘Validated’ data refers to resulting DNA fragments of according length for each part and the expected assembled lengths.
Data Analysis
The data I generated includes DNA fragment lengths after Golden Gate Assembly as visualized on a gel (after gel electrophoresis and EtBr staining). I found partial assembly from the full reaction, hypothesized to be fragments 2-5 as the band length was approximately 8000 bp and pairwise reactions (i.e. just 2 + 3, 3 + 4, and 4 + 5) revealed bands of the expected sum of the length of the two fragments.
Overcoming Challenges
I did not see full assembly from my Golden Gate reaction, which I suspect based on my data analysis is due to complications from the 1st Fragment, which is 4000 bp and nearly twice the length of the other fragments (due to how I had to split the genome based on internal PaqC1 cut sites and avoiding splitting genes). I plan to continue to test my validation by increasing the incubation time for Golden Gate as well as testing PCRs on the Golden Gate reaction to see whether there was some assembly with fragment 1 but just not at a high enough efficiency to visualize on the gel.
Additional Material
References
Boyd, C. M., & Seed, K. D. (2024). A phage satellite manipulates the viral DNA packaging motor to inhibit phage and promote satellite spread. Nucleic Acids Research, 52(17), 10431–10446. doi:10.1093/nar/gkae675
Cook, B. W. M., & Hynes, A. P. (2025). Re-evaluating what makes a phage unsuitable for therapy. Npj Antimicrobials and Resistance, 3(1), 45. doi:10.1038/s44259-025-00117-z
de Lorenzo, V., & Martínez‐García, E. (2025). On the choice of the right plasmid vector(s) in the times of synthetic biology. Microbial Biotechnology, 18(12), e70273. doi:10.1111/1751-7915.70273
Dao TO, Park HE, Lee JH, Kim KM, Trinh MP, Kang HL, Yoo HS, Shin MK. Advances and Challenges in Mycobacterial Genetic Engineering: Techniques for Knockout, Knockdown and Overexpression. J Microbiol Biotechnol. 2025 Nov 27;35:e2507051. doi: 10.4014/jmb.2507.07051. PMID: 41309382; PMCID: PMC12685580.
He, L., Patkowski, J. B., Wang, J., Miguel-Romero, L., Aylett, C. H. S., Fillol-Salom, A., . . . Penadés, J. R. (2025). Chimeric infective particles expand species boundaries in phage-inducible chromosomal island mobilization. Cell, 188(23), 6636–6653.e17. doi:10.1016/j.cell.2025.08.019
Jain, P., Hsu, T., Arai, M., Biermann, K., Thaler, D. S., Nguyen, A., . . . Jacobs, W. R. (2014). Specialized transduction designed for precise high-throughput unmarked deletions in mycobacterium tuberculosis. mBio, 5(3), 1245. doi:10.1128/mBio.01245-14
Kamiński, B., & Paczesny, J. (2024). Bacteriophage challenges in industrial processes: A historical unveiling and future outlook. Pathogens, 13(2), 152. doi:10.3390/pathogens13020152
Ko, C., Sikkema, A. P., Lauer, M. J., Amarh, E. D., Garlena, R. A., Russell, D. A., . . . Lohman, G. J. S. (2025). Genome synthesis, assembly, and rebooting of therapeutically useful high G+C% mycobacteriophages. Proceedings of the National Academy of Sciences of the United States of America, 122(46), e2523871122. doi:10.1073/pnas.2523871122
Penadés, J. R., Seed, K. D., Chen, J., Bikard, D., & Rocha, E. P. C. (2025). Genetics, ecology and evolution of phage satellites. Nature Reviews Microbiology, 23(7), 410–422. doi:10.1038/s41579-025-01156-z
Qian, H. L., Roman, A. N., Paudel, S., Hussey, G. E., Carline, K. B. R., & Saha, M. S. (2025). A novel phage satellite class induces prophage excision in mycolicibacterium aichiense openRxiv. doi:10.64898/2025.12.02.691453
Ram, G., Ross, H. F., Novick, R. P., Rodriguez-Pagan, I., & Jiang, D. (2018). Conversion of staphylococcal pathogenicity islands to CRISPR-carrying antibacterial agents that cure infections in mice. Nature Biotechnology, 36(10), 971–976. doi:10.1038/nbt.4203
Sousa, J. A., & Rocha, E. P. C. (2021). To catch a hijacker: Abundance, evolution and genetic diversity of P4-like bacteriophage satellites. Philosophical Transactions of the Royal Society B: Biological Sciences, 377(1842), 20200475. doi:10.1098/rstb.2020.0475
Yang, F., Njire, M. M., Liu, J., Wu, T., Wang, B., Liu, T., . . . Zhang, T. (2015). Engineering more stable, selectable marker-free autoluminescent mycobacteria by one step. Plos One, 10(3), e0119341. doi:10.1371/journal.pone.0119341