Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Why are there only 20 natural amino acids?
- Can you make other non-natural amino acids? Design some new amino acids.
- Where did amino acids come from before enzymes that make them, and before life started?
- If you make an a-helix using D-amino acids, what handedness (right or left) would you expect?
- Can you discover additional helices in proteins?
- Why are most molecular helices right-handed?
- Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
ANSWERS
Question 1
As known, amino acids are the building blocks of proteins. However, meat is not composed entirely of protein; its composition varies depending on the animal and the specific cut. If we consider beef, which contains approximately 23% protein by weight, then in a 500 g portion there would be about 115 g of protein. It is stated that an average amino acid has a molecular weight of approximately 100 Daltons, and since 1 Dalton corresponds to 1 g/mol, this means an average amino acid has a molar mass of roughly 100 g/mol. For an estimate, we divide the total grams of protein by this average molar mass:
115g÷100 g/mol=1.15 mol
To convert moles into molecules, we multiply by Avogadro number:
1.15 mol x 6.022 x 10^23=6.9 x 10^23 molecules (approximately)
Therefore, an estimated 6.9 x 10²³ amino acid molecules are consumed in a 500 g portion of beef.
This estimation assumes that:
- The protein content is accurately represented by the 23% value.
- The average amino acid mass is approximately 100 g/mol.
- The digestive system fully hydrolyzes all proteins into individual amino acids. While simplified, the order of magnitude (~10²³) correctly reflects the enormous molecular scale involved in biological systems.
Question 2
Life is supported by four principal classes of biomolecules: proteins, carbohydrates, lipids, and nucleic acids. Each fulfills distinct structural and functional roles. Although beef contains DNA and proteins from the cow, consuming them does not transfer “cow identity” to the human body. First, biological identity is determined by organized genetic information and regulated gene expression, not by the mere presence of biomolecules. The muscle tissue we consume does not carry an active developmental program capable of altering human genetic regulation. Second, during digestion, macromolecules are broken down into their basic components. Proteins are hydrolyzed into amino acids, DNA into nucleotides, and lipids into fatty acids. What the intestine absorbs are these small molecular building blocks not intact cow genes, regulatory networks, or functional tissues. Therefore, when we eat beef, we obtain matter (carbon, nitrogen, amino acids, nucleotides), but not biological information in an operational sense. Species identity depends on genomic organization, developmental programming, and tightly regulated cellular systems. Digestion reduces complex biomolecules to reusable components, which our own cells incorporate according to human genetic instructions.
Question 3
From a chemical point of view, there are virtually limitless possibilities for amino acids, since many different side chains could theoretically be synthesized. Therefore, the limitation to 20 is not due to chemical constraints but rather biological selection. In the standard genetic code, there are 64 codons, of which 3 are stop codons. Although 61 codons encode amino acids, they do not correspond to 61 different amino acids. Instead, the code contains redundancy (degeneracy), meaning multiple codons specify the same amino acid. This redundancy helps protect protein synthesis against point mutations, since some nucleotide changes do not alter the amino acid sequence. If life had used fewer than 20 amino acids, proteins might lack essential chemical diversity, such as hydrophobic, charged, aromatic, and redox-active side chains, limiting structural and catalytic capabilities. On the other hand, having many more amino acids would require a more complex translational machinery (more tRNAs, synthetases, proofreading systems), increasing energetic and regulatory costs. Therefore, evolution likely settled on approximately 20 amino acids as a balance between chemical diversity and translational efficiency — enough functional variety to build complex proteins, but not so many as to make the system unnecessarily complex or energetically expensive.
Question 4
Amino acids can indeed be generated through chemical synthesis or incorporated into proteins using expanded genetic code technologies. These approaches allow the introduction of new chemical functionalities not present in the canonical 20 amino acids. I would design a side chain containing two thiol (SH) functional groups positioned so they can act as a bidentate chelator, coordinating a metal ion through two sulfur atoms simultaneously. My target would be high affinity and selectivity for mercury (Hg²⁺), which is a soft metal ion and preferentially binds to soft donor atoms like sulfur. Compared to a single thiol group, such as in cysteine, a two-thiol side chain would increase binding strength through the chelate effect. This should enhance affinity for Hg²⁺ while reducing interaction with harder, biologically essential ions such as Mg²⁺ or Ca²⁺. This design could enable environmental applications, such as engineering proteins or microbial systems capable of capturing mercury from contaminated water or soil, thereby reducing its bioavailability and facilitating bioremediation. !image[]
Question 5
Before life existed, the early Earth had a chemically reactive atmosphere composed of simple molecules such as methane (CH₄), ammonia (NH₃), water vapor (H₂O), carbon dioxide (CO₂), nitrogen (N₂), and hydrogen (H₂). Energy sources such as lightning, ultraviolet radiation, and volcanic heat provided the conditions necessary for chemical reactions. One classic experiment demonstrating this possibility is the Miller Urey experiment, which simulated early Earth atmospheric conditions and showed that amino acids can form spontaneously from simple inorganic precursors when energy is applied. Chemically, amino acids can be formed through reactions such as Strecker synthesis, in which an aldehyde, ammonia, and hydrogen cyanide react to form an amino acid precursor. Similar chemistry may have occurred in hydrothermal vent systems, where high temperature and mineral surfaces could catalyze organic synthesis. Additionally, amino acids have been detected in meteorites, suggesting that prebiotic organic molecules may also have been delivered to early Earth from extraterrestrial sources. Therefore, amino acids likely originated through abiotic chemical processes driven by energy and simple carbon-containing molecules, before enzymes or living systems existed. Life later adopted these molecules because they were already chemically available and capable of forming polymers with diverse functional properties.
Question 6
Most a-helical proteins in nature are composed of L amino acids, and these helices are predominantly right handed. Since D-amino acids are the mirror image of L-amino acids, it follows that a helix formed entirely from D-amino acids would also be the mirror image of the natural a-helix. Therefore, a helix built from D-amino acids would be expected to be left-handed. This reasoning follows directly from molecular chirality: if the monomeric units are mirrored, the resulting secondary structure will also be mirrored.
Question 7
I interpret “discovering” additional helices as identifying rare but naturally occurring structural motifs that have not yet been fully characterized. In principle, new helices can exist because a helix is defined by a repeating pattern of backbone dihedral angles (φ and ψ) and a consistent hydrogen-bonding arrangement. However, for a new helix to be considered real and stable, it must satisfy strict structural constraints:
- Favorable backbone torsion angles (allowed Ramachandran regions)
- Consistent hydrogen-bond geometry
- Minimal steric clashes
- Reproducibility in multiple protein structures While environmental stress or unusual contexts might locally distort existing helices, classification as a new helix would require a repeatable and energetically stable pattern, not just a temporary deformation. Thus, discovering additional helices is possible, but only if they meet structural and thermodynamic criteria that allow them to exist reproducibl
Question 8
Amino acids are chiral monomers, meaning their three-dimensional orientation is not superimposable on their mirror image. Because proteins are built almost exclusively from L-amino acids, the system is inherently asymmetric. This chirality breaks symmetry between left- and right-handed helices. For L-amino acids, the right-handed a-helix provides more favorable backbone geometry, better hydrogen-bond alignment, and fewer steric clashes between side chains and the backbone. In contrast, a left-handed a-helix formed from L-amino acids would introduce steric strain and less favorable torsion angles. As a result, biology favors the right-handed helix because it represents the lower-energy, more stable configuration. Evolution naturally selects the structure that is energetically more favorable and functionally robust.
Question 9
An isolated β-strand is unstable because its backbone contains many hydrogen bond donors (NH) and acceptors (C=O) that are not satisfied. In water, these groups can interact with solvent molecules, but this is less stable than forming hydrogen bonds with another peptide backbone. When two β-strands align side by side, they form an extended network of intermolecular hydrogen bonds. This satisfies the backbone donors and acceptors, lowering the free energy of the system. The alignment also creates a repetitive structure that is energetically favorable. In addition to hydrogen bonding, the hydrophobic effect plays an important role. Many β-strands have alternating hydrophobic side chains. When strands stack together, hydrophobic residues can pack against each other and exclude water, which further stabilizes the structure. Therefore, β-sheets tend to aggregate because:
- Backbone hydrogen bonds become satisfied.
- Hydrophobic side chains pack together.
- The overall free energy of the system decreases. In simple terms: β-strands are “sticky” because leaving their hydrogen bonding groups exposed is energetically unfavorable, and pairing them reduces that instability.
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it. T4 lysozyme is an enzyme encoded by bacteriophage T4 that plays a crucial role during infection. Its primary function is to degrade the peptidoglycan layer of the bacterial cell wall, either facilitating DNA injection or enabling host cell lysis at the end of the viral replication cycle. Structurally, T4 lysozyme is a relatively small, predominantly a-helical protein, and its three-dimensional structure has been extensively characterized, making it a classical model in structural biology.
I selected T4 lysozyme because it is directly related to bacteriophages, which align with my academic interests, and because it represents a well-studied protein with a clearly defined structure-function relationship. Its simplicity and availability of high-resolution structural data make it ideal for visualization and analysis.
- Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? Sequence Length: 97 amino acids
Amino Acid Frequencies: A: 10 (10.31%) V: 9 (9.28%) I: 8 (8.25%) S: 8 (8.25%) X: 6 (6.19%) N: 6 (6.19%) K: 6 (6.19%) T: 6 (6.19%) L: 6 (6.19%) Q: 5 (5.15%) E: 5 (5.15%) P: 5 (5.15%) G: 5 (5.15%) R: 4 (4.12%) Y: 3 (3.09%) C: 2 (2.06%) M: 2 (2.06%) D: 1 (1.03%)
How many protein sequence homologs are there for your protein? 250 244 viruses 3 eukaryota 3 bacteria 1 bacteroidota
Does your protein belong to any protein family? T4 lysozyme belongs to the lysozyme superfamily, specifically the phage-type lysozymes, which share a conserved catalytic function but may differ structurally from other lysozyme families such as hen egg-white lysozyme.
- Identify the structure page of your protein in RCSB When was the structure solved? Is it a good quality structure?
Deposited: 2024-08-21
Released: 2026-02-18
Resolution: 1.45 Å Are there any other molecules in the solved structure apart from protein? The structure includes:
Copper ion (Cu²⁺)
NTA (nitrilotriacetic acid ligand)
Likely water molecules Does your protein belong to any structure classification family?
Member of the lysozyme superfamily
T4 lysozyme-like fold
Predominantly a-helical protein
Two-domain architecture
- Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
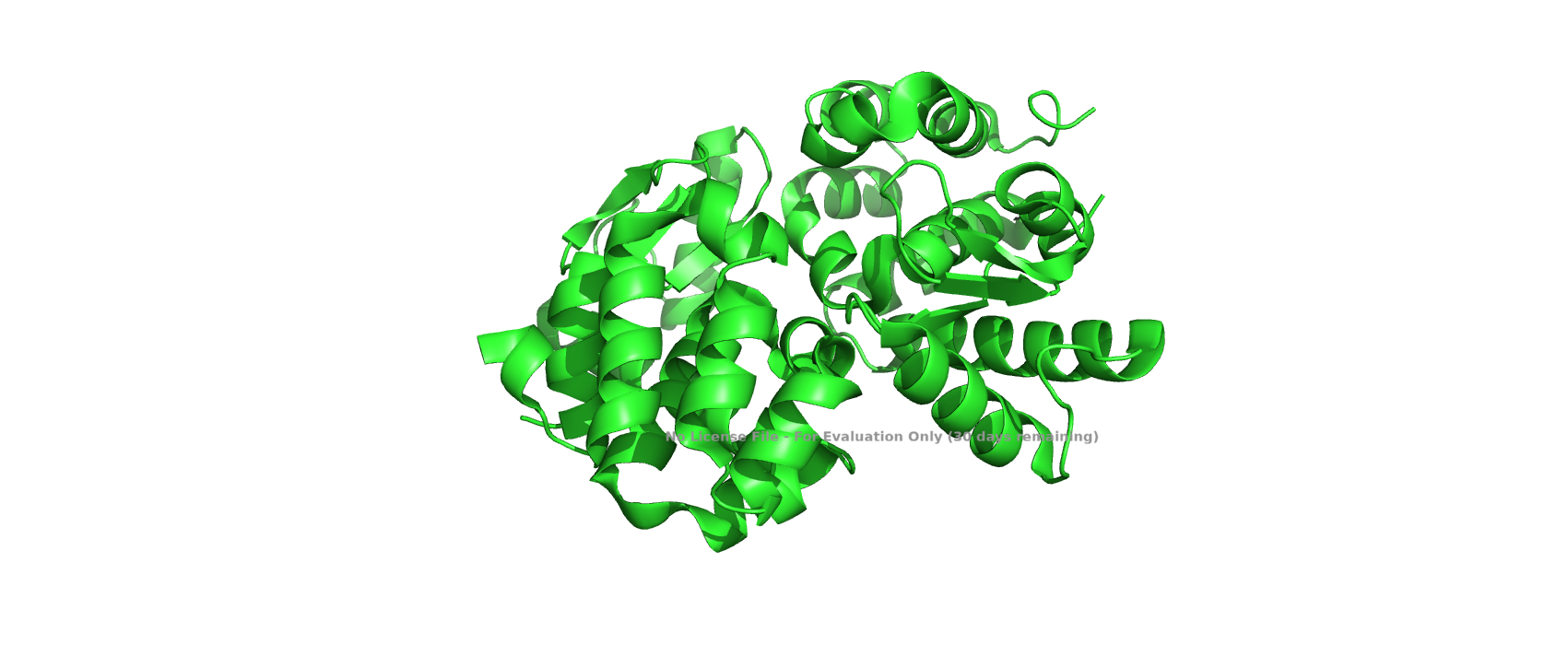

 Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by secondary structure. Does it have more helices or sheets?
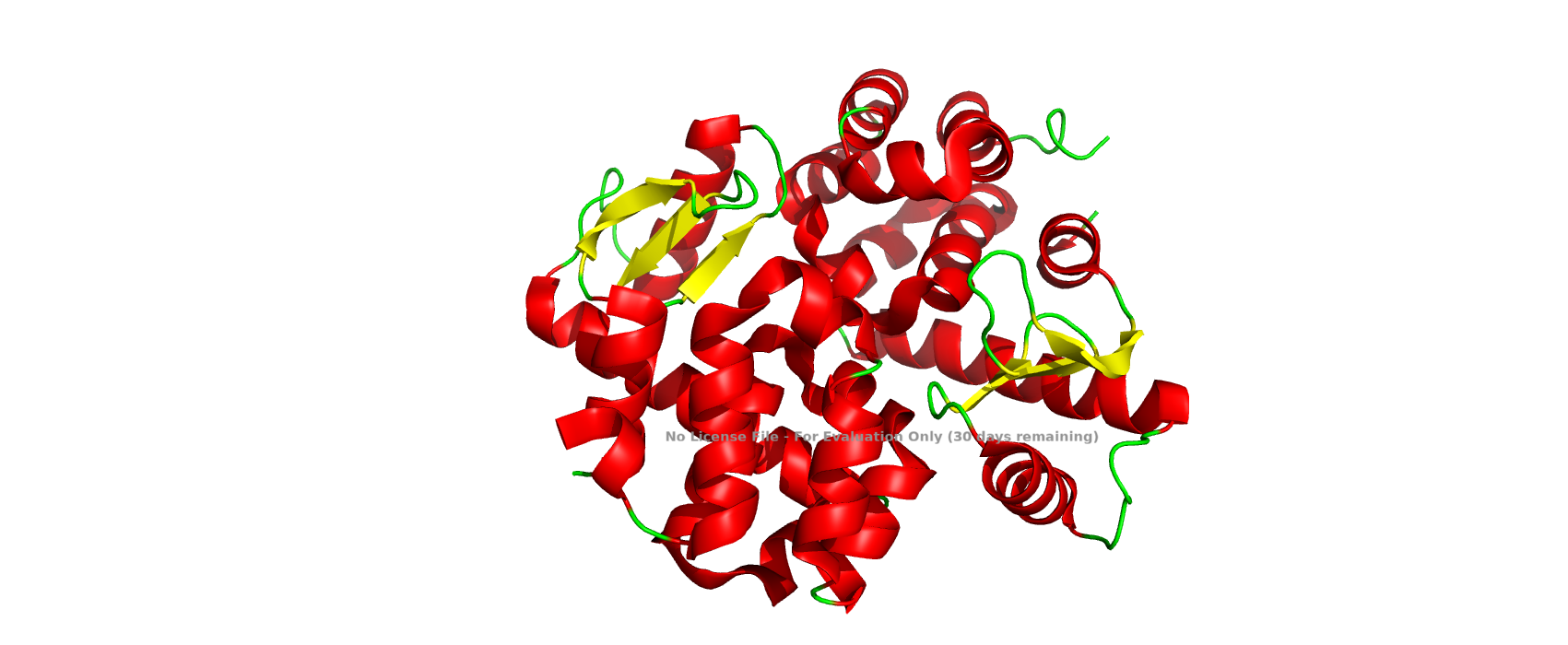 Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
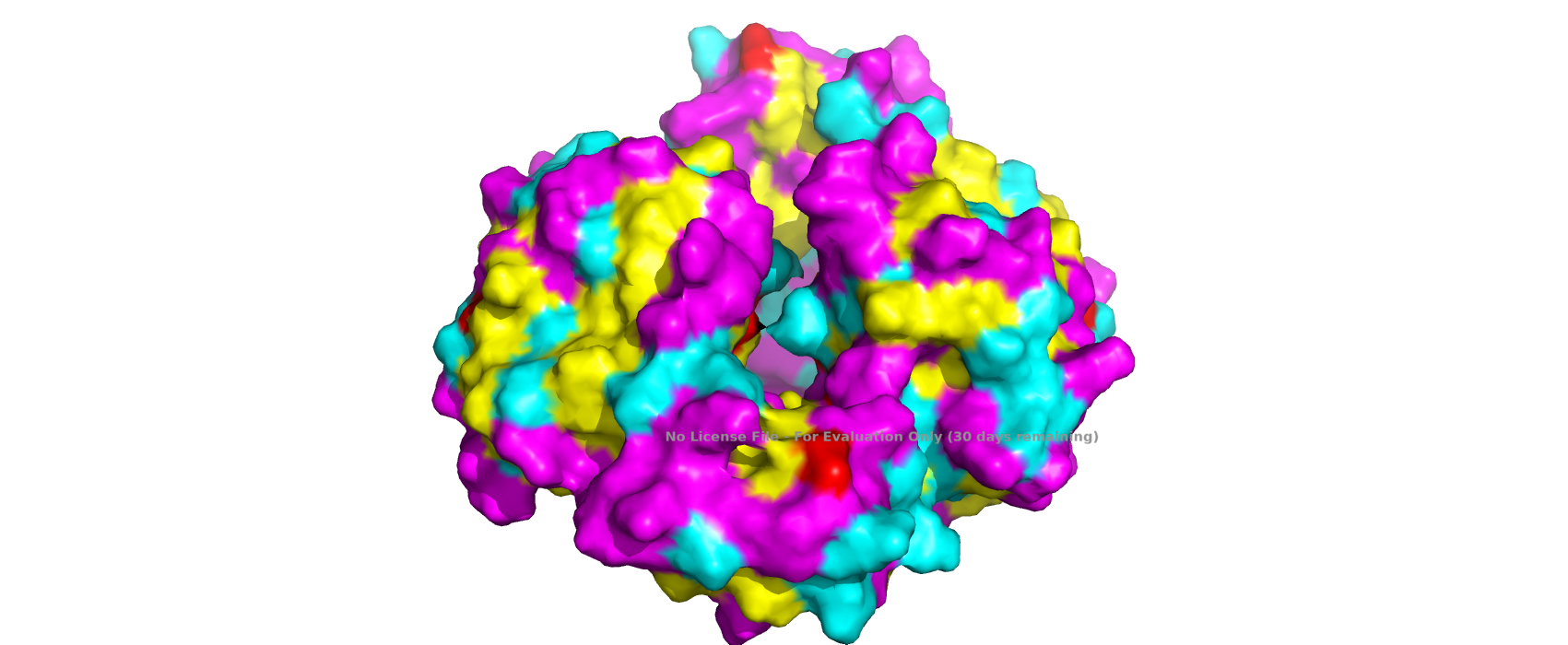
 Visualize the surface of the protein. Does it have any “holes”?
Visualize the surface of the protein. Does it have any “holes”?
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
- Deep Mutational Scans
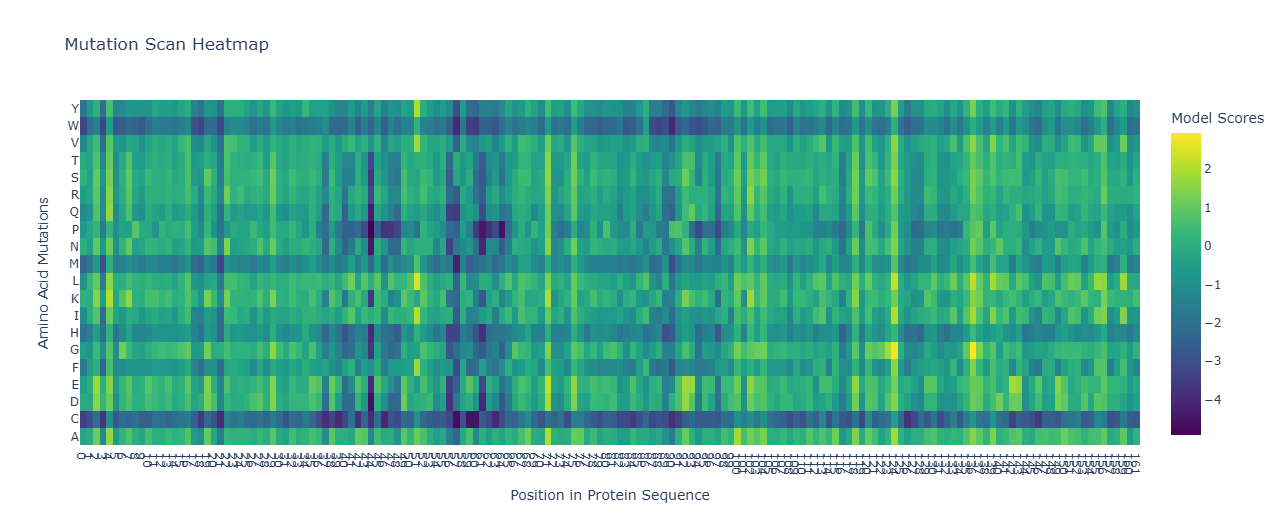 Selected Position (0-indexed): 122
Selected Position (0-indexed): 122
- Wild-type Amino Acid at Position 122:
protein_sequence[122]is ‘A’. - Mutated Amino Acid: ‘W’ (Tryptophan)
- Log-Likelihood Ratio (LLR): Approximately -19.07
- Latent Space Analysis
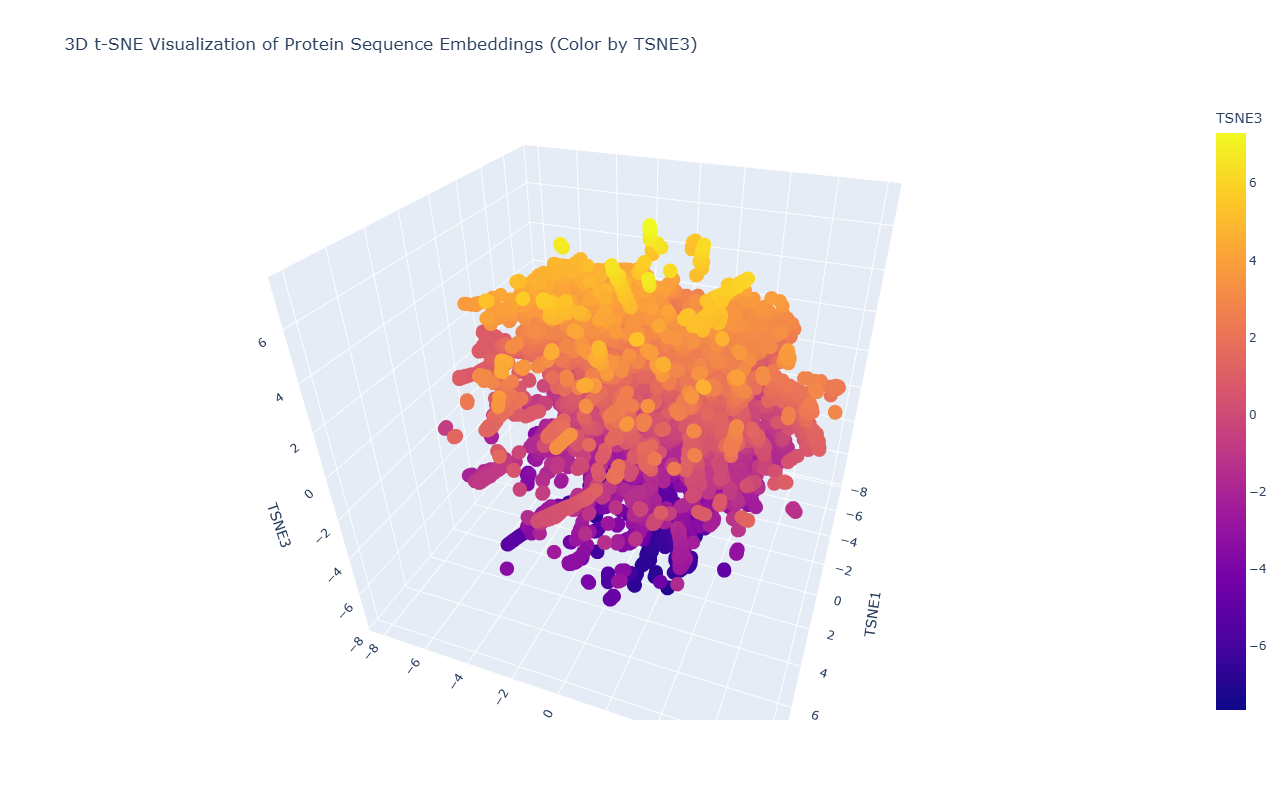
C2. Protein Folding
