Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
| Peptide | Perplexity | ipTM score | N terminus | B-barrel | Dimer interface |
|---|---|---|---|---|---|
| WRYPAAAAALKX | 4.30808 | 0.3 | Close | No | Surface bound |
| WRYGATVAAHKX | 5.811953 | 0.48 | Far | No | Partially buried |
| WLSGAAALALKX | 5.716131 | 0.45 | Close | No | Surface bound |
| WLYPAAALALKX | 8.30171 | 0.36 | Far | No | Partially buried |
| FLYRWLPSRRGG | 0.38 | Far | No | Surface bound |
The predicted protein–peptide complexes produced relatively low ipTM scores overall, indicating weak confidence in the modeled interactions. The PepMLM-generated peptides showed ipTM values ranging from 0.30 to 0.48. The highest score was observed for the peptide WRYGATVAAHKX (ipTM = 0.48), followed by WLSGAAALALKX (ipTM = 0.45), both of which exceeded the ipTM score of the known SOD1-binding peptide FLYRWLPSRRGG (ipTM = 0.38). Despite these slightly higher scores, none of the predicted peptides appeared to strongly interact with the β-barrel region of SOD1, and most were either surface-bound or only partially buried on the protein surface. Overall, while some PepMLM-generated peptides showed marginally higher ipTM scores than the known binder, the predicted interactions remain weak and uncertain.
| Peptide | Predicted binding affinity | Solubility | Hemolysis probability | Net charge | Molecular weight (Da) |
|---|---|---|---|---|---|
| WRYPAAAAALKX | 5.437 | Soluble | Non - hemolitic | 1.76 | 1199.6 |
| WRYGATVAAHKX | 5.440 | Soluble | Non - hemolitic | 1.85 | 1241.6 |
| WLSGAAALALKX | 6.550 | Soluble | Non - hemolitic | 0.76 | 1082.6 |
| WLYPAAALALKX | 6.693 | Soluble | Non - hemolitic | 0.76 | 1198.7 |
| FLYRWLPSRRGG | 5.96 | Soluble | Non - hemolitic | 2.76 | 1507.7 |
The peptide property predictions were broadly favorable, since all candidates were predicted to be soluble and non-hemolytic. However, the AlphaFold3 results showed only modest ipTM values, suggesting weak to moderate confidence in the predicted protein-peptide interactions. The peptide with the highest ipTM score was WRYGATVAAHKX (0.48), while the best predicted binding affinity value was observed for WRYPAAAAALKX (5.437), indicating that higher ipTM did not perfectly correlate with stronger predicted affinity. Overall, WRYGATVAAHKX appears to offer the best balance between structural binding potential and therapeutic properties, so it would be the strongest candidate to advance.
I would choose WRYGATVAAHKX, because:
- it has the highest ipTM
- it is soluble
- it is non-hemolytic
- its charge is moderate
- it outperformed the known binder in ipTM
Part C: Final Project: L-Protein Mutants
| Variant | Mutation | Region | Experimental evidence | Conservation analysis | Expected effect | Rationale |
|---|---|---|---|---|---|---|
| V1 | P13L | Soluble region | Lysis = 1; Protein level = 1 | Highly conserved; keep with caution | May alter soluble-domain behavior while preserving lysis | Selected because it retained lysis activity and detectable protein expression in the experimental mutant dataset. Although the site is conserved, it is kept as a cautious candidate because experimental data supports functionality. |
| V2 | S15A | Soluble region | Lysis = 1; Protein level = 1 | Moderately conserved / partially variable | May preserve or improve folding while maintaining lysis | Selected because it is a small amino acid change, retained lysis activity, and occurs in a less constrained region than fully conserved sites. |
| V3 | R30Q | Soluble region | Lysis = 1; Protein level = 1 | Highly conserved; keep with caution | May affect DnaJ-associated interaction or soluble-domain properties | Selected because the soluble domain is associated with DnaJ interaction, and this mutant retained lysis and protein expression experimentally. |
| V4 | L44P | Transmembrane region | Lysis = 1; Protein level = 1 | Highly conserved; keep with caution | May alter membrane-associated lysis activity | Selected because the transmembrane region affects lysis activity, and this mutation remained functional in the experimental data. |
| V5 | A45P | Transmembrane region | Lysis = 1; Protein level = 1 | Moderately conserved / partially variable | May modify transmembrane behavior while preserving lysis | Selected because it retained both lysis activity and detectable protein expression and is less strictly conserved than nearby transmembrane residues. |
To select MS2 L-protein mutant candidates, I first divided the protein into two functional regions: the soluble N-terminal region, which is associated with DnaJ interaction, and the C-terminal transmembrane region, which affects lysis activity. I prioritized mutations that retained lysis activity and detectable protein levels in the experimental mutant dataset. I then used homologous L-protein sequences from pBLAST and aligned them with Clustal Omega to evaluate whether each candidate position was conserved or variable. Highly conserved sites were interpreted with caution, while partially variable sites were considered more permissive for mutation. The final five variants include mutations in both the soluble and transmembrane regions, allowing the design to test effects on DnaJ-related behavior and membrane-associated lysis.
The expected outcome is that at least one of these L-protein variants will maintain lysis activity while changing properties related to folding, DnaJ interaction, or membrane-associated lysis. Soluble-domain mutants may help test whether the protein can become less dependent on DnaJ-mediated processing, while transmembrane mutants may affect the speed or efficiency of bacterial lysis. Because some selected residues are conserved, these mutations should be interpreted as candidates for testing rather than guaranteed improvements. The next experimental step would be to synthesize the mutant genes, clone them into the appropriate construct, and compare their lysis activity against the wild-type L-protein.
Figure 1. Clustal Omega alignment of selected MS2 L-protein homologs.
The alignment was used to evaluate whether candidate mutation sites were highly conserved or partially variable before selecting final L-protein mutant variants.
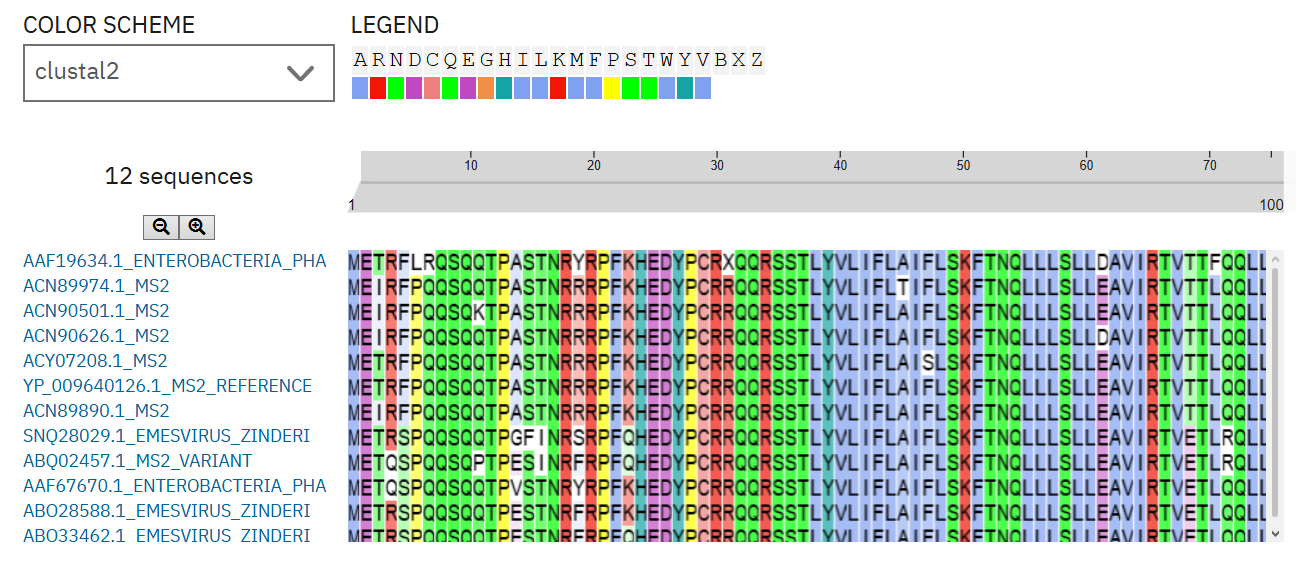
Note. Candidate mutations were interpreted using conservation patterns across homologous L-protein sequences. Highly conserved residues were kept only with caution when supported by experimental lysis and protein-expression data.