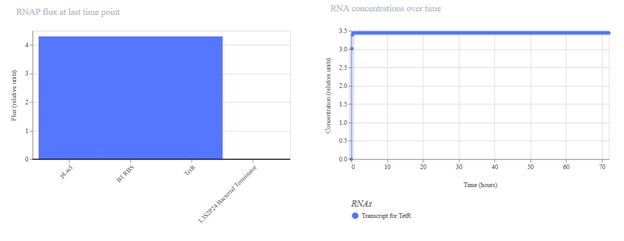
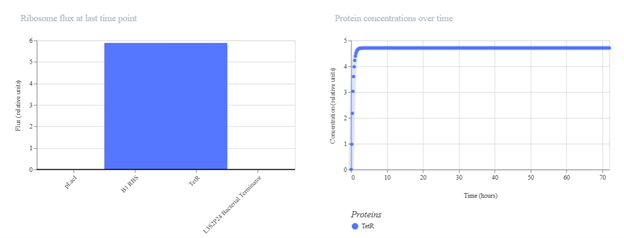
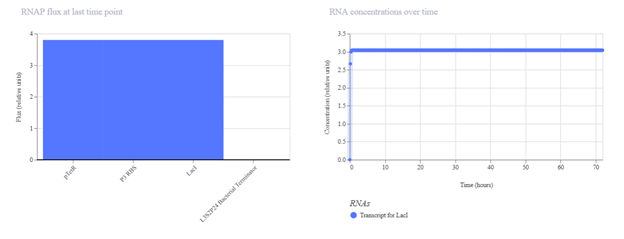
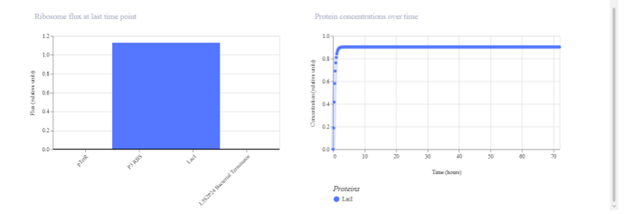
First, describe a biological engineering application or tool you want to develop and why. Paratransgenic symbiont to block dengue transmission in Aedes aegypti Mosquito-borne dengue is a global threat, yet current control measures have a vector elimination focus increasingly undermined by insecticide resistance. Vaccines have shown limited efficacy, and with no broadly effective antivirals, dengue prevention still relies heavily on mosquito and larvae control (Hu et al., 2025). Considering this, researchers are leveraging synthetic biology to develop paratransgenic strategies that render A. aegypti mosquitoes refractory to infection by delivering anti-pathogen molecules inside the mosquito, thereby blocking virus replication and transmission (Gao et al., 2025). The biological engineering tool proposed is a synthetic paratransgenic bacterial symbiont designed to live in the gut of A. aegypti mosquitoes and actively block dengue virus transmission. The purpose is to use a naturally mosquito-associated bacterium (such as Asaia spp.) genetically engineered to sense mosquito feeding conditions and secrete antiviral effector molecules directly into the midgut lumen. The gut of A. aegypti offers a strategic intervention point. Dengue virus first encounters the midgut epithelium after a blood meal, and if viral entry is blocked at this stage, systemic infection of the mosquito can be prevented by secretion of viral entry inhibitors, such as peptides. It is an ecologically targeted solution, because it doesn’t intend to eradicate the mosquito populations from their ecosystems, as every part of the trophic network needs to stay in balance.
Final Project For this project, several elements will be measured across the experimental, computational, and synthetic biology stages in order to evaluate the performance of the proposed platform. Because the project is structured as a pipeline, the measurable outputs include nucleic acid quality, sequence-derived features, predicted protein properties, and candidate prioritization metrics.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork what you contributed to the community bioart project I tried to transform a hexagon into a bacteriophage by adding some details in the exterior area. I think they were restored to the original picture before deadline what you liked about the project I was reminded of another collaborative project that was funny what about this collaborative art experiment could be made better for next year. Maybe a bigger canvas Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each components role is in the cell-free reaction. BL21 (DE3) Star Lysate (includes T7 RNA Polymerase):
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit" answer the following questions in each faculty member’s section Homework Questions from Professor Jacobson
Opentron Art Post-Lab Questions Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. Summary This study introduces Pyhamilton, an open-source Python framework that enables flexible programming of liquid-handling robots for high-throughput biological experimentation. Unlike traditional robotic automation, which merely replicates hand-pipetting protocols, Pyhamilton allows for dynamic decision-making, asynchronous execution, and real-time feedback integration.
Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Why do humans eat beef but do not become a cow, eat fish but do not become fish? Why are there only 20 natural amino acids? Can you make other non-natural amino acids? Design some new amino acids. Where did amino acids come from before enzymes that make them, and before life started? If you make an a-helix using D-amino acids, what handedness (right or left) would you expect? Can you discover additional helices in proteins? Why are most molecular helices right-handed? Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? ANSWERS Question 1 As known, amino acids are the building blocks of proteins. However, meat is not composed entirely of protein; its composition varies depending on the animal and the specific cut. If we consider beef, which contains approximately 23% protein by weight, then in a 500 g portion there would be about 115 g of protein. It is stated that an average amino acid has a molecular weight of approximately 100 Daltons, and since 1 Dalton corresponds to 1 g/mol, this means an average amino acid has a molar mass of roughly 100 g/mol. For an estimate, we divide the total grams of protein by this average molar mass:
Part A: SOD1 Binder Peptide Design Peptide Perplexity ipTM score N terminus B-barrel Dimer interface WRYPAAAAALKX 4.30808 0.3 Close No Surface bound WRYGATVAAHKX 5.811953 0.48 Far No Partially buried WLSGAAALALKX 5.716131 0.45 Close No Surface bound WLYPAAALALKX 8.30171 0.36 Far No Partially buried FLYRWLPSRRGG 0.38 Far No Surface bound The predicted protein–peptide complexes produced relatively low ipTM scores overall, indicating weak confidence in the modeled interactions. The PepMLM-generated peptides showed ipTM values ranging from 0.30 to 0.48. The highest score was observed for the peptide WRYGATVAAHKX (ipTM = 0.48), followed by WLSGAAALALKX (ipTM = 0.45), both of which exceeded the ipTM score of the known SOD1-binding peptide FLYRWLPSRRGG (ipTM = 0.38). Despite these slightly higher scores, none of the predicted peptides appeared to strongly interact with the β-barrel region of SOD1, and most were either surface-bound or only partially buried on the protein surface. Overall, while some PepMLM-generated peptides showed marginally higher ipTM scores than the known binder, the predicted interactions remain weak and uncertain.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity DNA Polymerase A proofreading polymerase with 3′→5′ exonuclease activity, which ensures very low error rates during DNA synthesis. Phusion HF or GC Buffer Provides optimal ionic conditions (Mg²⁺, salts, pH). HF buffer: for standard templates GC buffer: improves amplification of GC-rich or difficult templates dNTPs (400 µM each) Building blocks (dATP, dTTP, dCTP, dGTP) required for DNA strand synthesis. Mg²⁺ (within the buffer) Essential cofactor for polymerase activity and influences enzyme fidelity and efficiency. What are some factors that determine primer annealing temperature during PCR? The annealing temperature in PCR is determined by several factors: Primer melting temperature (Tm) Calculated based on primer sequence (GC content, length). Annealing temperature is typically ~3–5°C below Tm Primer length Longer primers = higher Tm GC content Higher GC = stronger binding = higher annealing temperature Primer sequence composition Secondary structures (hairpins, dimers) affect binding Salt concentration Higher salt stabilizes primer-template binding Polymerase type Some enzymes (like Phusion) require higher annealing temperatures due to their buffer system There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR Restriction Enzyme Digests Starting materials Template DNA and primers DNA with restriction sites Key reagents Polymerase, primers and dNTPs Restriction enzyme and buffer Mechanism DNA amplification DNA cutting Temperature profile Multiple cycles Single temperature Control of fragment Defined by primer Defined by enzyme sites Output Many copies of a single fragment Multiple fragments Critical design step Primer design Enzyme selection Time 1 to 3 hours Roughly 1 hour Flexibility High Limited by sequence PCR is generally preferable when you need to generate a specific DNA fragment with precise boundaries or added sequences, such as overlaps for Gibson Assembly, because it allows high flexibility through primer design and can amplify even very small amounts of DNA. In contrast, restriction enzyme digestion is preferable when the DNA already contains suitable restriction sites, making it a simpler and faster method for cutting plasmids or generating fragments without the need for amplification. Therefore, PCR is favored for custom design and low DNA availability, while restriction digestion is best for routine cloning tasks where appropriate sites are already present.
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs provide graded and analog computation rather than a strict ON/OFF logic. Enabling cells to integrate multiple inputs with tunable weights and produce continuous outputs that reflect the signal strength, not just presence/absence. IANNs can implement thresholding, nonlinear decision boundaries, and noise tolerance, making them more robust in heterogeneous biological environments. They also allow combinatorial regulation, which is difficult to achieve with simple Boolean gates without increasing the circuit complexity.
Homework Part A: General and Lecturer-Specific Questions General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis offers greater flexibility and control compared to in vivo systems because it allows precise manipulation of reaction conditions such as component concentrations, temperature, and reaction time. Additionally, it eliminates cellular interference, such as metabolic regulation, toxicity effects, and competing pathways, enabling more efficient and tunable protein production. One case where cell-free expression is advantageous is in the production of toxic proteins, such as toxins or antimicrobial peptides, which would otherwise damage or kill the host cell. Another case is the synthesis of proteins requiring non-natural amino acids or specialized conditions, which are difficult to achieve in living cells due to their tightly regulated environment.
Subsections of Homework
Week 1 HW: Principles and Practices
1. First, describe a biological engineering application or tool you want to develop and why.
Paratransgenic symbiont to block dengue transmission in Aedes aegypti
Mosquito-borne dengue is a global threat, yet current control measures have a vector elimination focus increasingly undermined by insecticide resistance. Vaccines have shown limited efficacy, and with no broadly effective antivirals, dengue prevention still relies heavily on mosquito and larvae control (Hu et al., 2025). Considering this, researchers are leveraging synthetic biology to develop paratransgenic strategies that render A. aegypti mosquitoes refractory to infection by delivering anti-pathogen molecules inside the mosquito, thereby blocking virus replication and transmission (Gao et al., 2025).
The biological engineering tool proposed is a synthetic paratransgenic bacterial symbiont designed to live in the gut of A. aegypti mosquitoes and actively block dengue virus transmission. The purpose is to use a naturally mosquito-associated bacterium (such as Asaia spp.) genetically engineered to sense mosquito feeding conditions and secrete antiviral effector molecules directly into the midgut lumen.
The gut of A. aegypti offers a strategic intervention point. Dengue virus first encounters the midgut epithelium after a blood meal, and if viral entry is blocked at this stage, systemic infection of the mosquito can be prevented by secretion of viral entry inhibitors, such as peptides. It is an ecologically targeted solution, because it doesn’t intend to eradicate the mosquito populations from their ecosystems, as every part of the trophic network needs to stay in balance.
Sources
Gao, H., Hu, W., Cui, C., Wang, Y., Zheng, Y., Jacobs-Lorena, M., & Wang, S. (2025). Emerging challenges for mosquito-borne disease control and the promise of symbiont-based transmission-blocking strategies. PLoS Pathogens, 21(8), e1013431. https://doi.org/10.1371/journal.ppat.1013431
Hu, W., Gao, H., Cui, C., Wang, L., Wang, Y., Li, Y., Li, F., Zheng, Y., Xia, T., & Wang, S. (2025). Harnessing engineered symbionts to combat concurrent malaria and arboviruses transmission. Nature Communications, 16(1), 2104. https://doi.org/10.1038/s41467-025-57343-2
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals
1. Minimize Ecological Disruption.
Ecuador’s laws have a strong precautionary approach. The 2008 Constitution explicitly bans any genetically modified organisms (GMOs) that may be harmful to human health, food sovereignty or ecosystems and requires precautionary measures against activities that could drive species to extinction or destroy ecosystems. At a regulatory level, the Organic Environmental Code from 2017 mandates that competent authorities issue detailed biosafety regulations and conduct case-by-case risk assessments for all modern biotechnology products to prevent impacts on biodiversity and the environment
1.1 Ensure that genetically modified symbionts do not unintentionally affect non-target mosquito species or other organisms through horizontal gene transfer or ecological spillover.
1.2 Implement long-term ecological monitoring of mosquito populations and their predators to confirm that the intervention does not disrupt local food webs or biodiversity
2. Contain and Control Engineered Microorganisms
Ecuador’s biosafety regulations require rigorous containment and risk management for any GMOs. The Environmental Code’s biosafety chapter in articles 229 to 233 states the requirement for institutions to evaluate and manage risks of GMOs to prevent or avoid any adverse effects on the environment, biodiversity or public health. Proponents of any GMO activity must submit comprehensive risk assessments and follow government‐defined risk-management parameters at each stage. Locally, the Comisión Nacional de Bioseguridad (CONABIO) has been established to coordinate interagency oversight of such activities, and Galápagos Biosecurity Agency (ABG) would similarly screen any exotic microbes for release.
2.1 Develop and enforce biosafety standards that require the engineered microbial strains to have built-in biocontainment systems to prevent uncontrolled environmental spread.
2.2 Require pre-release risk assessments and phased field trials under regulatory oversight to evaluate microbial persistence, gene stability, and potential unintended interactions.
3. Promote Transparency and Public Engagement
The Ecuadorian Constitution guarantees that “all persons have the right to freely access information generated by public entities. No information shall be withheld except as established by law.”. Environmental laws require public consultation. The secondary Environmental Code regulations mandate coordination of citizen participation mechanisms and formal public consultations for decisions on living modified organisms. In practice this means communities, including indigenous and local stakeholders, must be informed and consulted before releases.
3.1 Establish open communication channels with affected communities, including clear explanations of the goals, risks, and safeguards of the paratransgenic approach.
3.2 Include local stakeholders in ethical review and governance frameworks to ensure culturally appropriate consent and benefit sharing.
4. Align with Global Health and Equity Principles
Ecuador’s strategies for biotechnology are framed by broader commitments to public health and social equity. Internationally, the Sustainable Development Goals and WHO’s health strategies call for universal, affordable access to health innovations. WHO’s latest vector-control frameworks explicitly focus on safety, affordability and effectiveness of new tools
4.1 Ensure that the tool is accessible and affordable to dengue-endemic low- and middle-income countries (LMICs) and not monopolized by private patent holders.
4.2 Align the deployment strategy with WHO guidelines and regional vector control programs to ensure coordinated, ethically governed interventions
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Aspects
1. Tiered Registry and Information‐Sharing System
2. Mandatory Regulatory Standards and Risk Protocols
3. Stakeholder and Community Consent Processes
Purpose
Establish a public registry for all engineered-symbiont research and releases. Currently there aren´t comprehensive database for engineered vector organisms; so a tiered registry is proposed to track lab and field activities, list sites and organisms, and inform regulators and principally the public. The proposed change is to mandate registration of any released or planned paratransgenic symbionts so stakeholders can coordinate and anticipate impacts.
Enact new rules requiring thorough risk assessment, phased testing, and monitoring for any field release of engineered symbionts. Currently, most countries rely on existing GMO frameworks which may not address symbiont-specific issues (horizontal transfer, ecosystem effects). The change would be to adopt vector-control–specific guidelines (drawing on WHO and national GMO guidelines) that spell out required studies, containment levels, and post-release surveillance.
Implement mandatory procedures for social engagement to earn a “social license” before any release. The idea is to involve local communities, NGOs, and the public early and continuously, rather than later in permitting. Traditionally regulators have allowed only formal comment periods, but advocates propose deeper consultation and even consent
Design
Develop the registry via government with the help of researchers and companies to submit details of strains, release locations, and monitoring plans. Like drug trials, entries would be tiered by risk or scale: small lab tests vs. large releases. Responsible actors include national regulators, funding agencies, and possibly a CBD Biosafety Clearing-House platform. The system relies on open-access infrastructure and clear legal mandates. It also demands data standards like genetic characterization and risk assessment data, so entries are meaningful.
Regulators would issue rules or guidance documents requiring stepwise trials like contained lab studies, then small confined field trials, then larger releases. Risk assessment protocols would specify endpoints. Oversight might involve multi-agency review committees and public comment periods. Technical protocols would be developed by scientists in concert with regulators. International harmonization could produce common benchmarks. Actors include government regulators, scientific advisory panels, and companies, who must perform the studies and comply.
Can take the form of legal requirements or funding conditions. For instance, governments or donors might require a community advisory board, public meetings in local languages, and independent social science studies as prerequisites for approval. Developers and regulators would be responsible for organizing dialogue supported by facilitators or anthropologists. Transparency rules would be part of the design. Civil-society actors, as NGOs, might be enlisted to monitor the process.
Assumptions
Actors will comply and report honestly, sharing information reduces conflicting releases, and regulators have capacity to use the data. It is assumed that registry data will not be misused and that publicly listing projects won’t discourage innovation. It also assumes the registry can keep up with fast-moving research.
Scientific risk assessments can anticipate key hazards and regulators can interpret novel synthetic-biology data. It’s assumed that agencies have the expertise and resources to evaluate complex ecological risks. Policy will define “safe enough” thresholds and that risk models are valid. A hidden assumption is that stricter standards won’t stifle useful innovation.
Communities want to be involved and that two-way communication is possible. It is presumed that expressed public concerns are informed and constructive, and that engagement leads to buy-in. There is an implicit belief that “consent” improves legitimacy. It also assumes that implementing agencies and companies are willing and able to conduct genuine dialogue, not just box-checking
Risks of Failure & “Success”
If few groups register or data are incomplete, the registry fails to improve oversight. Overly burdensome entry requirements could drive researchers overseas or into informal channels. On the other hand, success could create a false sense of security; regulators might defer to the database rather than actively evaluate risks. Publicizing releases might also provoke alarm or opposition even if data is purely informational.
If guidelines are too vague or under-resourced, they may be ignored in practice. Inflexible rules might prevent beneficial interventions. Even well-designed protocols could fail to catch rare effects. There is also risk of “Type I” versus “Type II” errors: being too risk-averse may block a life-saving tool, whereas too lax regulation could allow environmental ham. On the flip side, if rules become “successful” and streamlined, developers might rely on checklists without true scrutiny. Overconfidence in regulations could delay independent monitoring or adaptive management.
Engagement efforts can backfire if superficial or one-sided, leading to mistrust, misinformation, or public backlash. Demanding individual consent from all residents near a release site is often unfeasible and may hinder scientific progress. Excluding communities can trigger legal or political resistance, while even well-executed engagement may not yield consensus but can help clarify values and tradeoffs. However, relying on engagement alone—without strong safety measures—risks undermining trust if problems arise. The GMO experience shows that genuine transparency and trust-building are more critical than simply sharing information.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
2
• By helping respond
1
2
3
Foster Lab Safety
• By preventing incident
N/A
1
N/A
• By helping respond
N/A
2
N/A
Protect the environment
• By preventing incidents
2
1
2
• By helping respond
2
2
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
2
• Feasibility?
1
2
2
• Not impede research
1
2
2
• Promote constructive applications
2
1
1
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the scoring and analysis, I would prioritize a combined governance approach—anchored in Option 2 (Mandatory Risk Protocols) as the foundation, supported by Option 1 (Tiered Registry) and Option 3 (Community Engagement)—to be recommended to international biosafety regulators and global health bodies, such as the Secretariat of the Cartagena Protocol, the World Health Organization (WHO), and national biosafety authorities in dengue-endemic countries like Ecuador’s Ministry of Environment and Health.
Why prioritize Option 2 as the foundation?
Option 2 received the strongest scores for biosecurity, lab safety, and environmental protection, reflecting its robust capacity to prevent and respond to incidents. Requiring phased field trials, genetic stability checks, and ecological risk modeling ensures that synthetic paratransgenic tools like engineered Asaia strains are deployed cautiously and adaptively. It builds scientific credibility and trust while laying down consistent benchmarks for safety.
Why support it with Option 1?
A tiered registry system enables transparency and coordination without heavy regulatory delays. It enhances biosecurity by enabling early detection of overlaps, duplicate trials, or potential cross-contamination. It also supports scientific collaboration and reduces redundant risk assessments. Crucially, it helps regulatory agencies in LMICs and oversee releases with limited infrastructure.
Why include Option 3?
Though more variable in impact, community engagement is critical for legitimacy and long-term sustainability. As shown in other biocontrol trials, scientific rigor alone cannot overcome public opposition. Option 3 helps align the intervention with local values, reduces misinformation, and opens channels for adaptive governance. As trade-off, engagement may increase costs and time, and consensus is not always guaranteed. However, these are acceptable trade-offs when weighed against the potential for social backlash.
Weekly Reflections
This week’s class opened my eyes to the ethical complexities of deploying engineered biological tools like synthetic symbionts in real-world environments. While nearly everything was new to me, one concern stood out most: how weak or misaligned regulatory systems can unintentionally hinder national scientific progress.
As someone who has interned at Ecuador’s Ministry of the Environment, I’ve seen firsthand how delays in permits and biosafety evaluations, especially for research involving genetic engineering does not come from bad intentions but from a lack of technical expertise and understaffing. These issues have worsened since the Ministry was merged with the Ministry of Energy and Mines, creating additional bureaucratic burden without increasing biosafety capacity. This disconnect risks turning regulation into a barrier rather than a guide for safe innovation.
This raises an ethical concern I hadn’t considered before: when poor governance prevents life-saving science, especially in countries heavily affected by vector-borne diseases, it becomes a form of structural injustice. Innovation should not be a privilege reserved for countries with better infrastructure.
Proposed Governance Actions
Re-establish a dedicated, well-funded national biosafety office, independent from industrial portfolios like mining or energy.
Develop specialized biosafety training programs for regulatory personnel, in partnership with universities and international biosafety experts.
Streamline approval pathways for public-interest research, with fast-track options for projects aligned with national health or environmental priorities.
Create a scientific advisory board to support regulators with risk assessments, especially for synthetic biology proposals.
For this project, several elements will be measured across the experimental, computational, and synthetic biology stages in order to evaluate the performance of the proposed platform. Because the project is structured as a pipeline, the measurable outputs include nucleic acid quality, sequence-derived features, predicted protein properties, and candidate prioritization metrics.
1. Metagenomic DNA quality and quantity
The first elements to be measured are the concentration, purity, and integrity of extracted metagenomic DNA obtained from Andean environmental samples. These measurements are essential to ensure that the genetic material is suitable for sequencing and downstream bioinformatic analysis.
Quantity and purity will be measured using spectrophotometry or fluorometry, depending on instrument availability.
Integrity will be evaluated by agarose gel electrophoresis, which allows visualization of DNA fragmentation or degradation.
That way it is ensured only high-quality samples move forward into sequencing workflows.
2. Metagenomic sequence data and predicted ORFs
A central output of this project is the set of nucleotide sequences and protein-coding open reading frames (ORFs) recovered from the metagenomic datasets. At this stage, what will be measured is not a physical biomarker, but rather the presence, number, and characteristics of predicted coding sequences.
DNA sequencing will be the main technology used to generate raw sequence data, either from real samples or curated public datasets.
After sequencing, bioinformatic preprocessing will measure:
Number of reads,
Read quality,
Assembly statistics such as contig length and coverage,
Number of predicted ORFs.
3. Protein sequence features and functional predictions
Once protein sequences are predicted, the project will measure sequence derived features associated with antimicrobial potential. These include properties such as sequence length, amino acid composition, charge, hydrophobicity, and similarity or divergence relative to known proteins.
These measurements will be performed computationally using:
Protein language models such as ESM or ProtBERT for sequence embeddings,
Machine learning classification tools to estimate antimicrobial potential,
Clustering or dimensionality reduction methods such as PCA or UMAP to detect novelty in latent space.
4. Structural properties of selected protein candidates
For prioritized candidates, the project will measure predicted structural stability and the presence of functional motifs relevant to antimicrobial activity.
These measurements will be obtained through:
Computational protein structure prediction, such as AlphaFold,
And structural inspection tools for identifying motifs, folds, or possible interaction surfaces.
The measurable outputs may include:
Predicted three dimensional structure,
Confidence metrics from structural models,
And inferred features related to protein stability or function.
Technologies to be used
The main technologies used in this project will include:
DNA extraction protocols for environmental samples
Spectrophotometry or fluorometry for DNA quantification and purity assessment
Agarose gel electrophoresis for evaluating DNA integrity
DNA sequencing for generating metagenomic datasets
Bioinformatic assembly and ORF prediction tools for recovering coding sequences
Protein language models and machine learning tools for functional prediction
Dimensionality reduction and clustering methods for novelty detection
Protein structure prediction tools such as AlphaFold for evaluating candidate proteins
Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight?
28006.60 Da
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation
Selected Values
m1 = 875.4421
m2 = 903.7148
Determination of charge state z
The charge state is calculated using:
z = (m2 - H) / (m2 - m1)
where:
H = 1.0073 Da (mass of a proton)
Substituting values:
z = (903.7148 - 1.0073) / (903.7148 - 875.4421)
z = 902.7075 / 28.2727 ≈ 31.93 ≈ 32
Therefore:
m1 = 875.4421 → z = 32
m2 = 903.7148 → z = 31
Molecular weight calculation
The molecular weight is calculated using:
MW = z x (m/z - H)
Using m1 = 875.4421, z = 32:
MW = 32 x (875.4421 - 1.0073)
MW = 32 x 874.4348 ≈ 27981.9 Da
Using m2 = 903.7148, z = 31:
MW = 31 x (903.7148 - 1.0073)
MW = 31 x 902.7075 ≈ 27983.9 Da
Final experimental molecular weight: MW ≈ 27.98 kDa
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, the exact charge state cannot be confidently observed from the zoomed in peak alone. Although its high m/z suggests a low charge state, the peak is too weak and lacks a clearly resolved neighboring charge-state or isotopic pattern needed for definite assignment.
Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses?
Native proteins are compact and have fewer accessible ionizable sites, resulting in lower charge states and peaks at higher m/z values. When proteins denature, they unfold and expose more residues, allowing them to acquire more charges during ionization. This leads to a broader charge distribution with peaks at lower m/z values. Thus, the shift in charge state distribution in the mass spectrum reflects protein unfolding.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
The charge state of the peak at approximately 2800 m/z can be determined by analyzing the spacing between the isotopic peaks in the zoomed in spectrum.
In mass spectrometry, the spacing between isotopic peaks is equal to:
Δ(m/z) = 1 / z
From the zoomed in region, the distance between adjacent peaks is approximately 0.33 m/z.
Using this relationship:
z = 1 / Δ(m/z)
z ≈ 1 / 0.33 ≈ 3
Therefore, the charge state of the peak is:
z = 3
Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Based on the LC MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance
Between 0.5 and 6 minutes, approximately 18 - 19 chromatographic peaks above 10% relative abundance can be observed in the eGFP peptide map. Only prominent peaks were counted, while smaller signals near the baseline were excluded. The exact number may vary slightly depending on the threshold interpretation, but the total is approximately 18 peaks.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The predicted number of peptides was 19, which is approximately consistent with the number of chromatographic peaks observed above the selected threshold. Therefore, the chromatogram shows about the same number of peaks as the predicted peptides. Any small difference would likely be due to co elution, low-abundance peptides, or non peptide signals.
Identify the mass to charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([ M + H ]+) based on its m/z and z.
The peptide has a most abundant peak at m/z 525.76712. The isotope spacing is approximately 0.5 m/z, which indicates a charge state of z = 2 because delta(m/z) = 1/z. Using this charge state, the singly charged form is calculated as (M+H)+ = 2 x 525.76712 - 1.0073 = 1050.53 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
The experimental mass (~1050.53 Da) matches the theoretical peptide mass of 1050.5214 Da corresponding to the sequence FEGDTLVNR. The mass error is calculated as:
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
what you contributed to the community bioart project
I tried to transform a hexagon into a bacteriophage by adding some details in the exterior area. I think they were restored to the original picture before deadline
what you liked about the project
I was reminded of another collaborative project that was funny
what about this collaborative art experiment could be made better for next year.
Maybe a bigger canvas
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each components role is in the cell-free reaction.
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase):
Provides the complete transcription translation machinery (ribosomes, tRNAs, enzymes) required for protein synthesis; the incorporated T7 RNA polymerase enables high- fficiency transcription from T7 promoters.
Acts as a buffering agent to maintain a stable physiological pH optimal for enzymatic activity during transcription and translation.
Magnesium Glutamate
Supplies Mg²⁺ ions, which are essential cofactors for ribosome assembly, ATP utilization, and nucleic acid stability.
Potassium phosphate monobasic
Contributes to buffering capacity and provides phosphate ions required for nucleotide metabolism.
Potassium phosphate dibasic
Works with the monobasic form to stabilize pH and maintain phosphate balance for energy transfer reactions.
Ribose: Serves as a precursor for nucleotide biosynthesis and contributes to maintaining energy metabolism.
Glucose: Functions as a primary energy source, fueling ATP regeneration through glycolytic enzymes present in the lysate.
AMP: Acts as a nucleotide precursor and participates in energy recycling pathways within the system.
CMP: Provides cytidine nucleotides required for RNA synthesis.
GMP: Supplies guanosine nucleotides necessary for transcription and translation processes.
UMP: Contributes uridine nucleotides for RNA synthesis.
Guanine: Serves as an additional base precursor to support nucleotide pool balance and synthesis.
17 Amino Acid Mix: Provides the majority of amino acids required for protein synthesis, excluding those prone to instability or oxidation.
Tyrosine: Supplied separately due to limited solubility, ensuring sufficient availability for incorporation into proteins.
Cysteine: Added independently because of its susceptibility to oxidation, maintaining proper redox conditions for protein synthesis.
Nicotinamide: Functions as a precursor for NAD⁺/NADH, supporting redox balance and metabolic reactions necessary for sustained protein synthesis.
Nuclease Free Water: Adjusts the final reaction volume and maintains reagent concentrations without introducing nucleases that could degrade DNA or RNA.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1 hour PEPNTP system uses direct high-energy substrates (PEP) and fully supplied NTPs, enabling rapid transcription and translation but with fast energy depletion and shorter reaction lifetimes. In contrast, the 20 hour NMP ribose glucose system relies on metabolic regeneration, where nucleotides are built from NMPs and ribose and ATP is regenerated via glucose driven pathways, supporting longer, more sustainable protein synthesis.
Additionally, the 20-hour system is simplified and more balanced (fewer additives, inclusion of phosphate buffering and nicotinamide), prioritizing stability and longevity over the high initial reaction speed seen in the PEP-based mix.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
sfGFP
Superfolder GFP is engineered for enhanced folding robustness, allowing efficient chromophore formation even under suboptimal conditions typical of cell-free systems. This makes it one of the most reliable reporters with strong signal output.
mRFP1
mRFP1 has a relatively slow maturation time, meaning fluorescence appears later after translation. In CFPS, this can lead to underestimation of expression at early timepoints.
mKO2
mKO2 is acid-sensitive, with fluorescence decreasing at lower pH. Since CFPS reactions can acidify over time due to metabolism, its signal may diminish during long incubations.
mTurquoise2
mTurquoise2 has a high quantum yield and efficient chromophore formation, producing bright fluorescence even at lower protein concentrations. This improves
sensitivity in CFPS readouts.
mScarlet_I
mScarlet I is optimized for fast maturation among red fluorescent proteins, enabling earlier fluorescence detection compared to older RFPs. This is advantageous for time-course measurements in CFPS.
Electra2
Electra2 is oxygen dependent for chromophore formation, like most fluorescent proteins. Limited oxygen availability in CFPS (especially in closed reactions) can reduce or delay fluorescence development.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Hypothesis: For mKO2, increasing the HEPES-KOH buffer concentration and optimizing the phosphate buffer ratio in the 36 hour mastermix will better maintain pH near 7.5, reducing acid driven loss of fluorescence during long incubation.
Expected effect: Because mKO2 fluorescence is acid sensitive, stronger buffering should preserve chromophore brightness and produce a higher final fluorescent signal over 36 hours.
Week 2 HW: DNA Read, Write and Edit
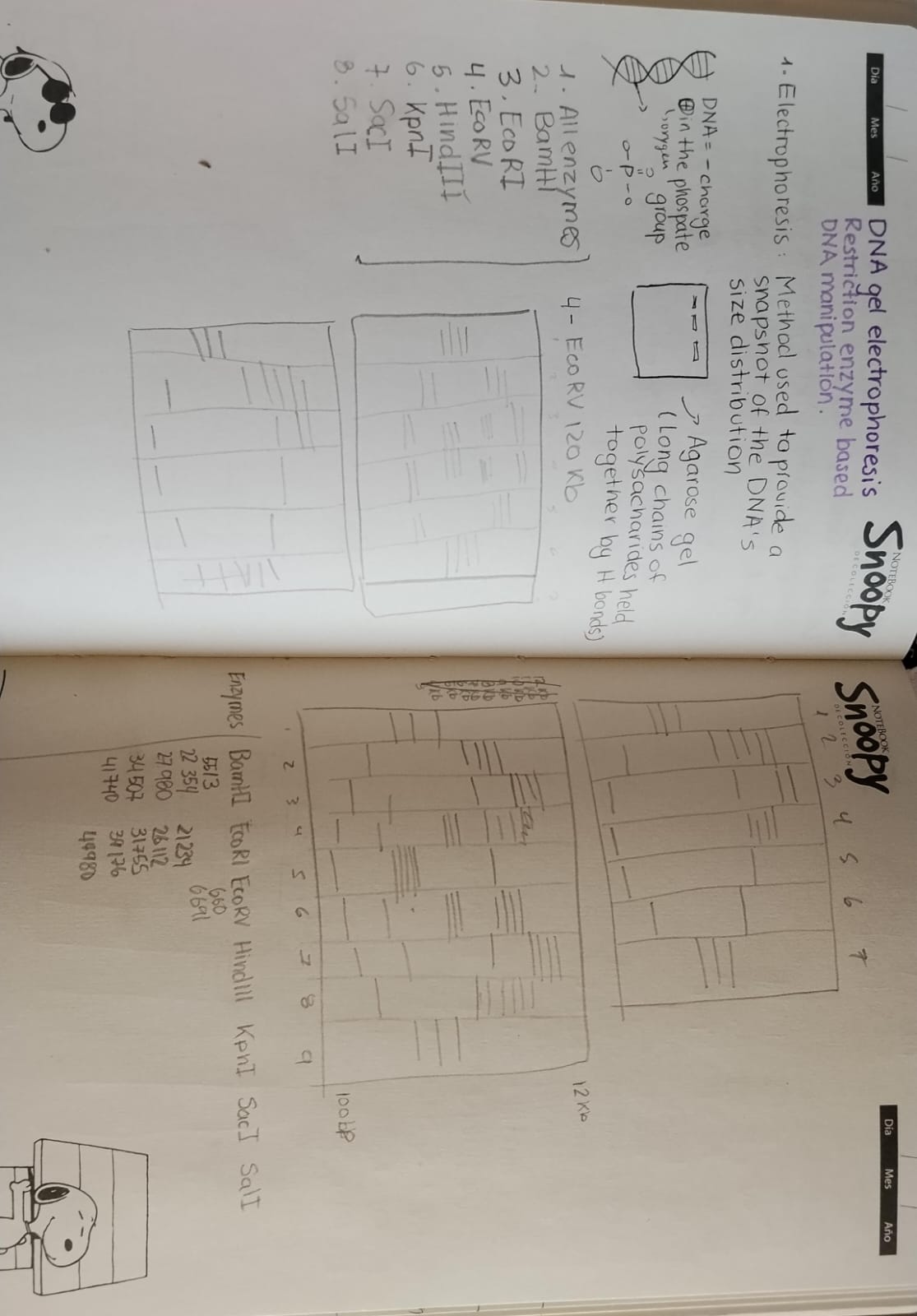
Part 1: Benchling & In-silico Gel Art
Preliminary notebook sketches illustrating the conceptual design process for the intended latent figure.
What DNA would you want to sequence (e.g., read) and why?
I would like to sequence viral metagenomic DNA (environmental virome) collected from aquatic ecosystems, such as wastewater effluent, hospital discharge sites, and agricultural runoff. Rather than focusing exclusively on bacterial genomes, this approach prioritizes complete bacteriophage genomes present in these environments.
Phages are major drivers of bacterial evolution, influencing antimicrobial resistance dissemination, virulence modulation, and horizontal gene transfer. By sequencing environmental phage DNA, it becomes possible to identify functional genetic modules such as receptor-binding proteins, depolymerases, integrases, and transducing elements that shape bacterial populations.
This intends shifts surveillance from reactive pathogen detection to a predictive aproach. Environmental virome sequencing could serve as an early-warning system, detecting emerging resistance dynamics or novel virulence-associated genetic elements before they become clinically dominant.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
To sequence environmental viral metagenomic DNA, It would be ideal to use a combination of third-generation long-read sequencing (Oxford Nanopore or PacBio) and second-generation high-throughput short-read sequencing (Illumina) in a hybrid strategy.
I think that this approach would leverage the strengths of both platforms: long reads enabling assembly of complete phage genomes and the resolution of structural variants, while short reads provide high accuracy for polishing and variant correction.
Input: purified viral DNA extracted from environmental water samples.
Essential preparation steps:
viral particle enrichment (filtration and DNase treatment to remove non-viral DNA)
viral DNA extraction
library preparation
fragmentation (for short-read platforms)
end repair and adapter ligation
quality control and quantification
Illumina (second generation):
DNA fragments bind to a flow cell.
Bridge amplification creates clusters.
Sequencing by synthesis occurs using fluorescently labeled reversible terminator nucleotides.
After each nucleotide incorporation, fluorescence is detected.
Base calling is determined by the emitted fluorescent signal.
Nanopore (third generation):
Single DNA molecules pass through a protein nanopore.
Each nucleotide alters ionic current differently.
Electrical signal changes are recorded in real time.
Machine learning algorithms convert signal patterns into base calls.
The output consists of:
FASTQ files containing sequence reads with quality scores
Assembled phage genomes
Annotated functional gene predictions
Comparative genomic datasets for surveillance
What DNA would you want to edit and why?
For this project, I would love to synthesize a phage-derived receptor-binding domain (RBD) fused to a fluorescent reporter, essentially creating a highly specific bacterial detection module inspired by bacteriophages.
Phages are incredibly precise when it comes to recognizing their bacterial hosts — their tail fibers or tailspikes bind very specific surface structures like capsules or LPS. Instead of synthesizing a whole phage genome (which would be unnecessary and unsafe), I would isolate just the receptor-binding domain of a phage tail fiber that targets a clinically relevant bacterium, such as Klebsiella pneumoniae. Then I would fuse that domain to a reporter protein like GFP.
The idea is that this synthetic gene would encode a fusion protein that binds specifically to its bacterial target and produces a fluorescent signal. So instead of using antibodies for detection, we would be using phage specificity as a biosensing tool. I think that’s incredibly powerful because phage receptor-binding proteins are often more specific than antibodies and can distinguish even subtle differences like capsule types.
To synthesize the phage receptor-binding domain–GFP fusion construct, I would use commercial gene synthesis based on phosphoramidite solid-phase DNA synthesis, followed by enzymatic DNA assembly (such as Gibson Assembly).
Phosphoramidite chemistry is the standard method used to chemically synthesize short DNA oligonucleotides. These oligos can then be assembled enzymatically into a full-length gene construct. This approach is highly accurate and allows complete sequence customization, including codon optimization and addition of regulatory elements.
I would choose this method because it enables precise design of non-replicative, modular constructs without needing a natural template, which is ideal for synthetic biology applications.
What DNA would you want to edit and why?
I would want to edit bacteriophage genomes, specifically lytic phages that infect clinically relevant bacteria such as Klebsiella pneumoniae or other multidrug-resistant pathogens.
Phages naturally evolve to recognize and infect bacteria, but their host range is often narrow and their therapeutic use can be limited by bacterial resistance mechanisms. By editing phage DNA, we could enhance desirable properties such as host specificity, lytic efficiency, or anti-virulence activity, while maintaining safety.
The types of edits I would focus on include:
Modifying tail fiber or receptor-binding protein genes to expand or retarget host range.
Inserting capsule depolymerase genes to improve penetration of protective bacterial capsules.
Deleting lysogeny-related genes (if present) to ensure strictly lytic behavior.
Optimizing regulatory elements to increase stability and predictability of infection dynamics.
The goal would not be to make phages more harmful, but rather more precise and controllable as therapeutic or ecological tools. In a One Health context, engineered phages could be used to reduce pathogenic bacteria in clinical, agricultural, or environmental settings without relying solely on antibiotics.
To edit bacteriophage genomes, I would use CRISPR-Cas–based genome editing combined with homologous recombination in bacterial host cells.
CRISPR-Cas systems are precise and programmable, making them ideal for modifying specific genes such as tail fiber or depolymerase genes. This approach allows targeted edits without randomly mutating the phage genome.
CRISPR-Cas works by using a guide RNA to direct the Cas nuclease to a specific DNA sequence. The Cas enzyme creates a cut at that location. If a repair template containing the desired modification is provided, the cell’s natural DNA repair machinery incorporates the new sequence.
For phage editing, the general process would involve:
Designing a guide RNA targeting the phage gene of interest.
Designing a donor DNA template containing the desired modification.
Introducing the CRISPR system and donor template into a bacterial host.
Infecting the bacteria with the phage.
Selecting for phages that incorporate the desired edit.
Preparation includes:
Designing guide RNAs targeting specific phage genes.
Designing a donor DNA repair template containing the edited sequence.
Cloning CRISPR components into plasmids.
Preparing competent bacterial host cells.
There are several limitations:
Editing efficiency can vary depending on the phage and target gene.
Some phages may escape editing due to rapid replication.
Off-target effects are possible if guide RNAs are not carefully designed.
Delivery of editing components must be optimized.
Week 2 LP: DNA Read, Write and Edit
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit" answer the following questions in each faculty member’s section
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Summary
This study introduces Pyhamilton, an open-source Python framework that enables flexible programming of liquid-handling robots for high-throughput biological experimentation. Unlike traditional robotic automation, which merely replicates hand-pipetting protocols, Pyhamilton allows for dynamic decision-making, asynchronous execution, and real-time feedback integration.
The authors demonstrate several novel applications:
Complex liquid transfer patterns to simulate population dynamics.
Real-time feedback-controlled turbidostats maintaining hundreds of bacterial cultures in log-phase growth.
Automated metabolic fitness landscape mapping across 100 nutrient conditions in triplicate.
Integration with plate readers to dynamically adjust media replacement based on optical density measurements.
Notably, the system enables maintenance of up to 480 parallel cultures with real-time monitoring and feedback control, transforming static protocols into adaptive experimental systems.
The paper illustrates how automation becomes transformative when paired with programmable control logic, data-driven feedback, and asynchronous task execution, enabling experiments impossible to perform manually.
Citation
Chory EJ, Gretton DW, DeBenedictis EA, Esvelt KM. Enabling high-throughput biology with flexible open-source automation. Mol Syst Biol (2021).
Write a description about what you intend to do with automation tools for your final project.
Project Title: Automated Combinatorial Optimization of Programmable Host Cell Circuits for Viral Vector Manufacturing
What I Intend to Automate
The goal is to automate the tuning and validation of a programmable host-cell control circuit designed to dynamically regulate viral vector production.
The automation workflow will focus on:
Combinatorial helper plasmid ratio optimization
Promoter and regulatory element tuning
Viral yield vs cell viability quantification
Iterative design–build–test cycles
Automated Workflow Overview
Construct Assembly & Preparation
Use Opentrons to assemble combinatorial promoter/RBS variants.
Prepare helper plasmid ratio matrices.
Generate condition libraries across 96-well format.
This automation framework transforms viral vector manufacturing optimization from static parameter tuning into a programmable, feedback-driven engineering process aligned with scalable synthetic biology platforms.
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Why are there only 20 natural amino acids?
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
If you make an a-helix using D-amino acids, what handedness (right or left) would you expect?
Can you discover additional helices in proteins?
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
ANSWERS
Question 1
As known, amino acids are the building blocks of proteins. However, meat is not composed entirely of protein; its composition varies depending on the animal and the specific cut. If we consider beef, which contains approximately 23% protein by weight, then in a 500 g portion there would be about 115 g of protein.
It is stated that an average amino acid has a molecular weight of approximately 100 Daltons, and since 1 Dalton corresponds to 1 g/mol, this means an average amino acid has a molar mass of roughly 100 g/mol. For an estimate, we divide the total grams of protein by this average molar mass:
115g÷100 g/mol=1.15 mol
To convert moles into molecules, we multiply by Avogadro number:
1.15 mol x 6.022 x 10^23=6.9 x 10^23 molecules (approximately)
Therefore, an estimated 6.9 x 10²³ amino acid molecules are consumed in a 500 g portion of beef.
This estimation assumes that:
The protein content is accurately represented by the 23% value.
The average amino acid mass is approximately 100 g/mol.
The digestive system fully hydrolyzes all proteins into individual amino acids.
While simplified, the order of magnitude (~10²³) correctly reflects the enormous molecular scale involved in biological systems.
Question 2
Life is supported by four principal classes of biomolecules: proteins, carbohydrates, lipids, and nucleic acids. Each fulfills distinct structural and functional roles. Although beef contains DNA and proteins from the cow, consuming them does not transfer “cow identity” to the human body.
First, biological identity is determined by organized genetic information and regulated gene expression, not by the mere presence of biomolecules. The muscle tissue we consume does not carry an active developmental program capable of altering human genetic regulation.
Second, during digestion, macromolecules are broken down into their basic components. Proteins are hydrolyzed into amino acids, DNA into nucleotides, and lipids into fatty acids. What the intestine absorbs are these small molecular building blocks not intact cow genes, regulatory networks, or functional tissues.
Therefore, when we eat beef, we obtain matter (carbon, nitrogen, amino acids, nucleotides), but not biological information in an operational sense. Species identity depends on genomic organization, developmental programming, and tightly regulated cellular systems. Digestion reduces complex biomolecules to reusable components, which our own cells incorporate according to human genetic instructions.
Question 3
From a chemical point of view, there are virtually limitless possibilities for amino acids, since many different side chains could theoretically be synthesized. Therefore, the limitation to 20 is not due to chemical constraints but rather biological selection.
In the standard genetic code, there are 64 codons, of which 3 are stop codons. Although 61 codons encode amino acids, they do not correspond to 61 different amino acids. Instead, the code contains redundancy (degeneracy), meaning multiple codons specify the same amino acid. This redundancy helps protect protein synthesis against point mutations, since some nucleotide changes do not alter the amino acid sequence.
If life had used fewer than 20 amino acids, proteins might lack essential chemical diversity, such as hydrophobic, charged, aromatic, and redox-active side chains, limiting structural and catalytic capabilities. On the other hand, having many more amino acids would require a more complex translational machinery (more tRNAs, synthetases, proofreading systems), increasing energetic and regulatory costs.
Therefore, evolution likely settled on approximately 20 amino acids as a balance between chemical diversity and translational efficiency — enough functional variety to build complex proteins, but not so many as to make the system unnecessarily complex or energetically expensive.
Question 4
Amino acids can indeed be generated through chemical synthesis or incorporated into proteins using expanded genetic code technologies. These approaches allow the introduction of new chemical functionalities not present in the canonical 20 amino acids.
I would design a side chain containing two thiol (SH) functional groups positioned so they can act as a bidentate chelator, coordinating a metal ion through two sulfur atoms simultaneously. My target would be high affinity and selectivity for mercury (Hg²⁺), which is a soft metal ion and preferentially binds to soft donor atoms like sulfur.
Compared to a single thiol group, such as in cysteine, a two-thiol side chain would increase binding strength through the chelate effect. This should enhance affinity for Hg²⁺ while reducing interaction with harder, biologically essential ions such as Mg²⁺ or Ca²⁺.
This design could enable environmental applications, such as engineering proteins or microbial systems capable of capturing mercury from contaminated water or soil, thereby reducing its bioavailability and facilitating bioremediation.
!image[]
Question 5
Before life existed, the early Earth had a chemically reactive atmosphere composed of simple molecules such as methane (CH₄), ammonia (NH₃), water vapor (H₂O), carbon dioxide (CO₂), nitrogen (N₂), and hydrogen (H₂). Energy sources such as lightning, ultraviolet radiation, and volcanic heat provided the conditions necessary for chemical reactions.
One classic experiment demonstrating this possibility is the Miller Urey experiment, which simulated early Earth atmospheric conditions and showed that amino acids can form spontaneously from simple inorganic precursors when energy is applied.
Chemically, amino acids can be formed through reactions such as Strecker synthesis, in which an aldehyde, ammonia, and hydrogen cyanide react to form an amino acid precursor. Similar chemistry may have occurred in hydrothermal vent systems, where high temperature and mineral surfaces could catalyze organic synthesis.
Additionally, amino acids have been detected in meteorites, suggesting that prebiotic organic molecules may also have been delivered to early Earth from extraterrestrial sources.
Therefore, amino acids likely originated through abiotic chemical processes driven by energy and simple carbon-containing molecules, before enzymes or living systems existed. Life later adopted these molecules because they were already chemically available and capable of forming polymers with diverse functional properties.
Question 6
Most a-helical proteins in nature are composed of L amino acids, and these helices are predominantly right handed. Since D-amino acids are the mirror image of L-amino acids, it follows that a helix formed entirely from D-amino acids would also be the mirror image of the natural a-helix. Therefore, a helix built from D-amino acids would be expected to be left-handed.
This reasoning follows directly from molecular chirality: if the monomeric units are mirrored, the resulting secondary structure will also be mirrored.
Question 7
I interpret “discovering” additional helices as identifying rare but naturally occurring structural motifs that have not yet been fully characterized. In principle, new helices can exist because a helix is defined by a repeating pattern of backbone dihedral angles (φ and ψ) and a consistent hydrogen-bonding arrangement.
However, for a new helix to be considered real and stable, it must satisfy strict structural constraints:
Reproducibility in multiple protein structures
While environmental stress or unusual contexts might locally distort existing helices, classification as a new helix would require a repeatable and energetically stable pattern, not just a temporary deformation.
Thus, discovering additional helices is possible, but only if they meet structural and thermodynamic criteria that allow them to exist reproducibl
Question 8
Amino acids are chiral monomers, meaning their three-dimensional orientation is not superimposable on their mirror image. Because proteins are built almost exclusively from L-amino acids, the system is inherently asymmetric.
This chirality breaks symmetry between left- and right-handed helices. For L-amino acids, the right-handed a-helix provides more favorable backbone geometry, better hydrogen-bond alignment, and fewer steric clashes between side chains and the backbone. In contrast, a left-handed a-helix formed from L-amino acids would introduce steric strain and less favorable torsion angles.
As a result, biology favors the right-handed helix because it represents the lower-energy, more stable configuration. Evolution naturally selects the structure that is energetically more favorable and functionally robust.
Question 9
An isolated β-strand is unstable because its backbone contains many hydrogen bond donors (NH) and acceptors (C=O) that are not satisfied. In water, these groups can interact with solvent molecules, but this is less stable than forming hydrogen bonds with another peptide backbone.
When two β-strands align side by side, they form an extended network of intermolecular hydrogen bonds. This satisfies the backbone donors and acceptors, lowering the free energy of the system. The alignment also creates a repetitive structure that is energetically favorable.
In addition to hydrogen bonding, the hydrophobic effect plays an important role. Many β-strands have alternating hydrophobic side chains. When strands stack together, hydrophobic residues can pack against each other and exclude water, which further stabilizes the structure.
Therefore, β-sheets tend to aggregate because:
Backbone hydrogen bonds become satisfied.
Hydrophobic side chains pack together.
The overall free energy of the system decreases.
In simple terms: β-strands are “sticky” because leaving their hydrogen bonding groups exposed is energetically unfavorable, and pairing them reduces that instability.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
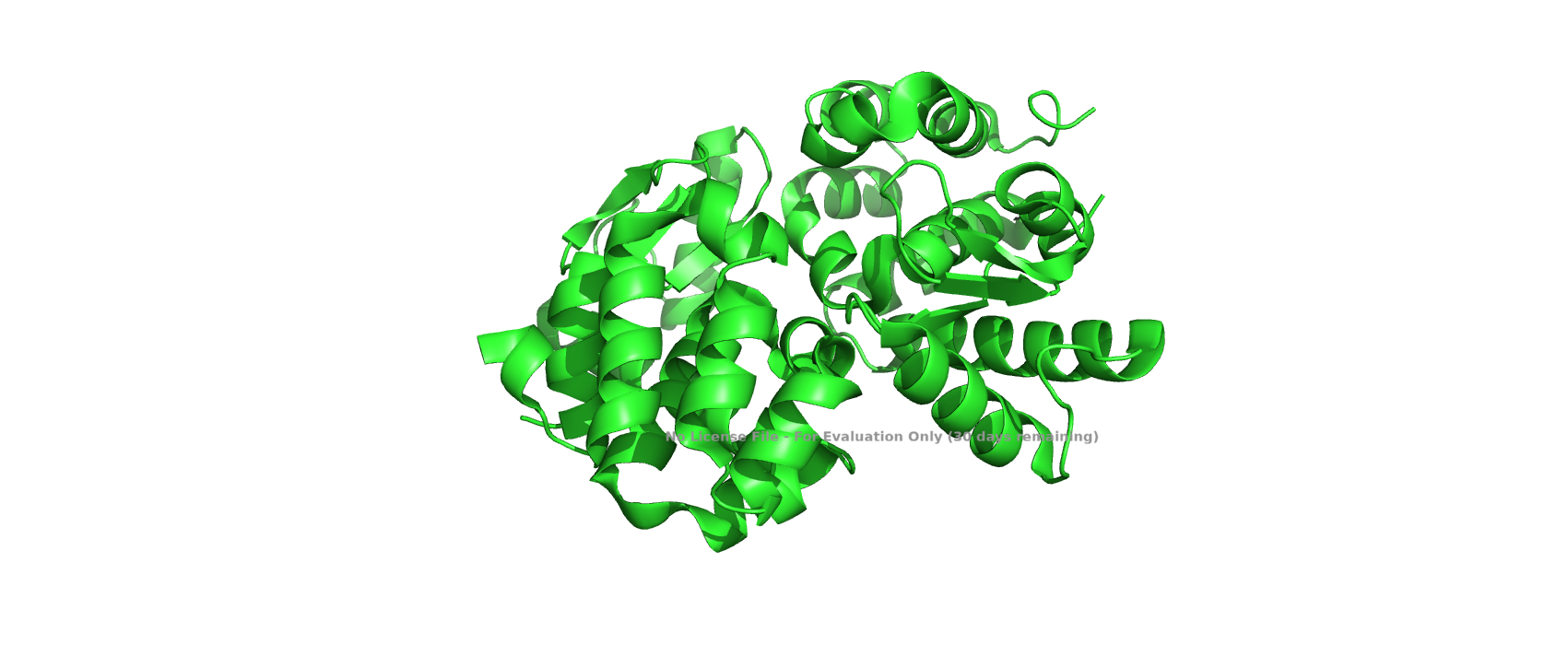
T4 lysozyme is an enzyme encoded by bacteriophage T4 that plays a crucial role during infection. Its primary function is to degrade the peptidoglycan layer of the bacterial cell wall, either facilitating DNA injection or enabling host cell lysis at the end of the viral replication cycle. Structurally, T4 lysozyme is a relatively small, predominantly a-helical protein, and its three-dimensional structure has been extensively characterized, making it a classical model in structural biology.
I selected T4 lysozyme because it is directly related to bacteriophages, which align with my academic interests, and because it represents a well-studied protein with a clearly defined structure-function relationship. Its simplicity and availability of high-resolution structural data make it ideal for visualization and analysis.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid?
Sequence Length: 97 amino acids
How many protein sequence homologs are there for your protein?
250
244 viruses
3 eukaryota
3 bacteria
1 bacteroidota
Does your protein belong to any protein family?
T4 lysozyme belongs to the lysozyme superfamily, specifically the phage-type lysozymes, which share a conserved catalytic function but may differ structurally from other lysozyme families such as hen egg-white lysozyme.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure?
Deposited: 2024-08-21
Released: 2026-02-18
Resolution: 1.45 Å
Are there any other molecules in the solved structure apart from protein?
The structure includes:
Copper ion (Cu²⁺)
NTA (nitrilotriacetic acid ligand)
Likely water molecules
Does your protein belong to any structure classification family?
Member of the lysozyme superfamily
T4 lysozyme-like fold
Predominantly a-helical protein
Two-domain architecture
Open the structure of your protein in any 3D molecule visualization software:
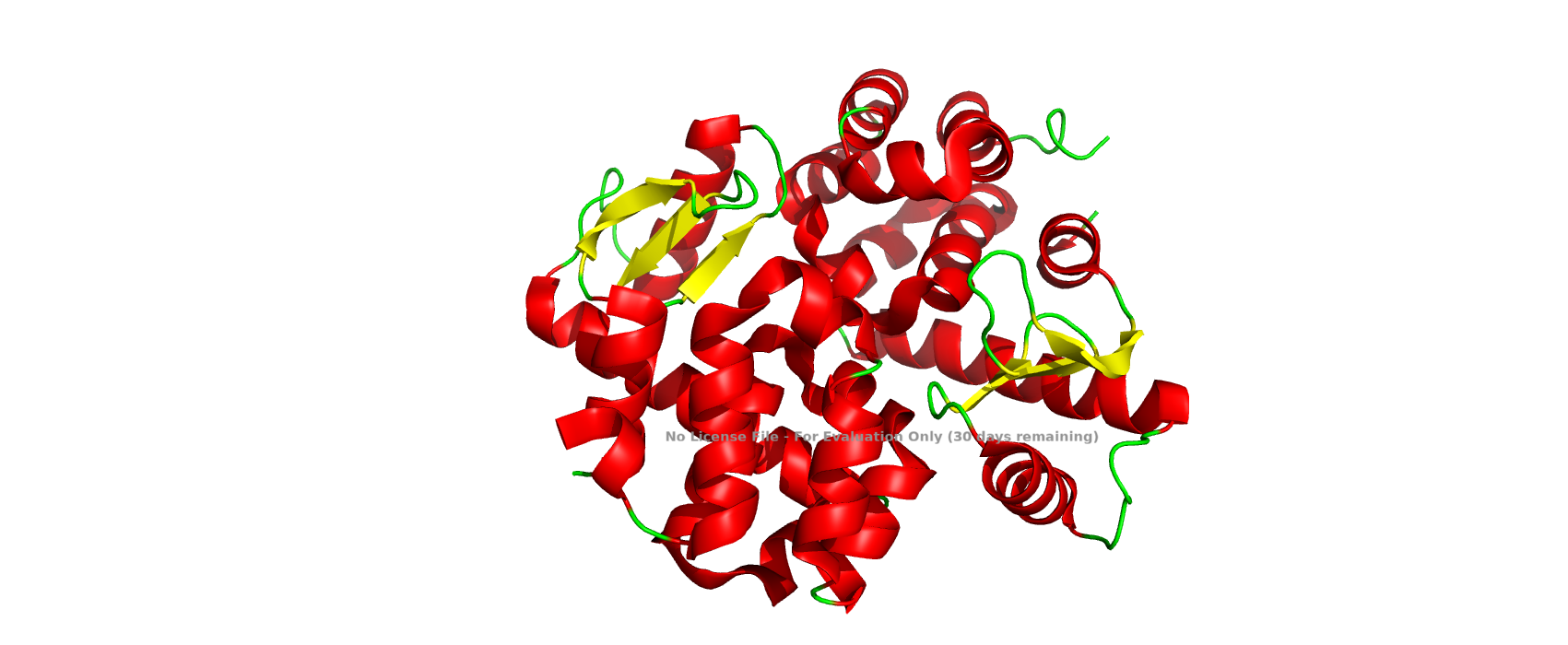
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.Color the protein by secondary structure. Does it have more helices or sheets?Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?Visualize the surface of the protein. Does it have any “holes”?
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
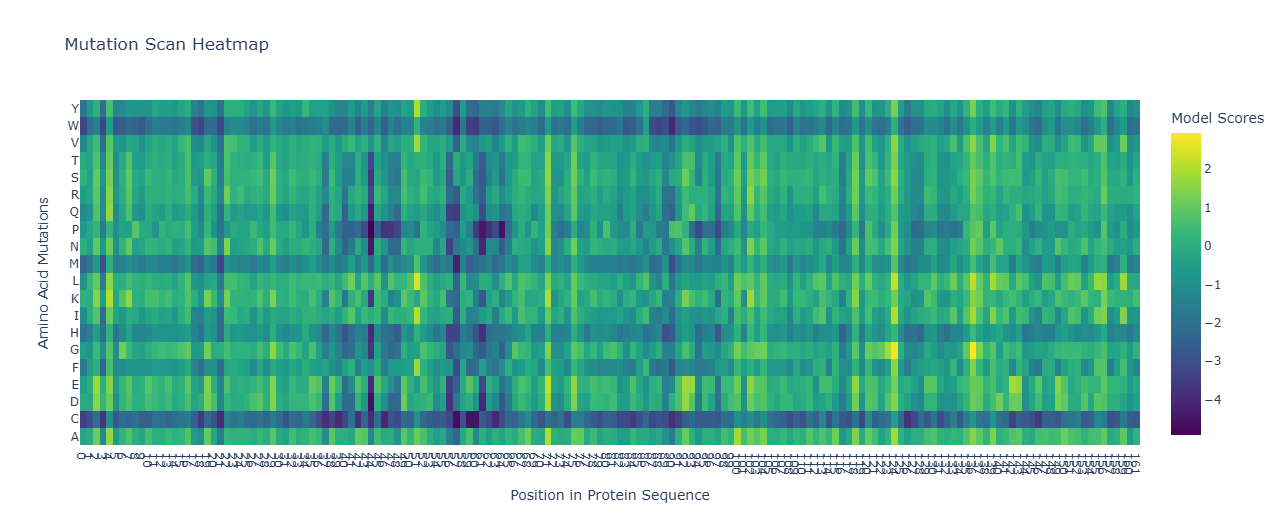
Selected Position (0-indexed): 122
Wild-type Amino Acid at Position 122:protein_sequence[122] is ‘A’.
Mutated Amino Acid: ‘W’ (Tryptophan)
Log-Likelihood Ratio (LLR): Approximately -19.07
Latent Space Analysis
C2. Protein Folding
C3. Protein Generation
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Peptide
Perplexity
ipTM score
N terminus
B-barrel
Dimer interface
WRYPAAAAALKX
4.30808
0.3
Close
No
Surface bound
WRYGATVAAHKX
5.811953
0.48
Far
No
Partially buried
WLSGAAALALKX
5.716131
0.45
Close
No
Surface bound
WLYPAAALALKX
8.30171
0.36
Far
No
Partially buried
FLYRWLPSRRGG
0.38
Far
No
Surface bound
The predicted protein–peptide complexes produced relatively low ipTM scores overall, indicating weak confidence in the modeled interactions. The PepMLM-generated peptides showed ipTM values ranging from 0.30 to 0.48. The highest score was observed for the peptide WRYGATVAAHKX (ipTM = 0.48), followed by WLSGAAALALKX (ipTM = 0.45), both of which exceeded the ipTM score of the known SOD1-binding peptide FLYRWLPSRRGG (ipTM = 0.38). Despite these slightly higher scores, none of the predicted peptides appeared to strongly interact with the β-barrel region of SOD1, and most were either surface-bound or only partially buried on the protein surface. Overall, while some PepMLM-generated peptides showed marginally higher ipTM scores than the known binder, the predicted interactions remain weak and uncertain.
Peptide
Predicted binding affinity
Solubility
Hemolysis probability
Net charge
Molecular weight (Da)
WRYPAAAAALKX
5.437
Soluble
Non - hemolitic
1.76
1199.6
WRYGATVAAHKX
5.440
Soluble
Non - hemolitic
1.85
1241.6
WLSGAAALALKX
6.550
Soluble
Non - hemolitic
0.76
1082.6
WLYPAAALALKX
6.693
Soluble
Non - hemolitic
0.76
1198.7
FLYRWLPSRRGG
5.96
Soluble
Non - hemolitic
2.76
1507.7
The peptide property predictions were broadly favorable, since all candidates were predicted to be soluble and non-hemolytic. However, the AlphaFold3 results showed only modest ipTM values, suggesting weak to moderate confidence in the predicted protein-peptide interactions. The peptide with the highest ipTM score was WRYGATVAAHKX (0.48), while the best predicted binding affinity value was observed for WRYPAAAAALKX (5.437), indicating that higher ipTM did not perfectly correlate with stronger predicted affinity. Overall, WRYGATVAAHKX appears to offer the best balance between structural binding potential and therapeutic properties, so it would be the strongest candidate to advance.
I would choose WRYGATVAAHKX, because:
it has the highest ipTM
it is soluble
it is non-hemolytic
its charge is moderate
it outperformed the known binder in ipTM
Part C: Final Project: L-Protein Mutants
Variant
Mutation
Region
Experimental evidence
Conservation analysis
Expected effect
Rationale
V1
P13L
Soluble region
Lysis = 1; Protein level = 1
Highly conserved; keep with caution
May alter soluble-domain behavior while preserving lysis
Selected because it retained lysis activity and detectable protein expression in the experimental mutant dataset. Although the site is conserved, it is kept as a cautious candidate because experimental data supports functionality.
V2
S15A
Soluble region
Lysis = 1; Protein level = 1
Moderately conserved / partially variable
May preserve or improve folding while maintaining lysis
Selected because it is a small amino acid change, retained lysis activity, and occurs in a less constrained region than fully conserved sites.
V3
R30Q
Soluble region
Lysis = 1; Protein level = 1
Highly conserved; keep with caution
May affect DnaJ-associated interaction or soluble-domain properties
Selected because the soluble domain is associated with DnaJ interaction, and this mutant retained lysis and protein expression experimentally.
V4
L44P
Transmembrane region
Lysis = 1; Protein level = 1
Highly conserved; keep with caution
May alter membrane-associated lysis activity
Selected because the transmembrane region affects lysis activity, and this mutation remained functional in the experimental data.
V5
A45P
Transmembrane region
Lysis = 1; Protein level = 1
Moderately conserved / partially variable
May modify transmembrane behavior while preserving lysis
Selected because it retained both lysis activity and detectable protein expression and is less strictly conserved than nearby transmembrane residues.
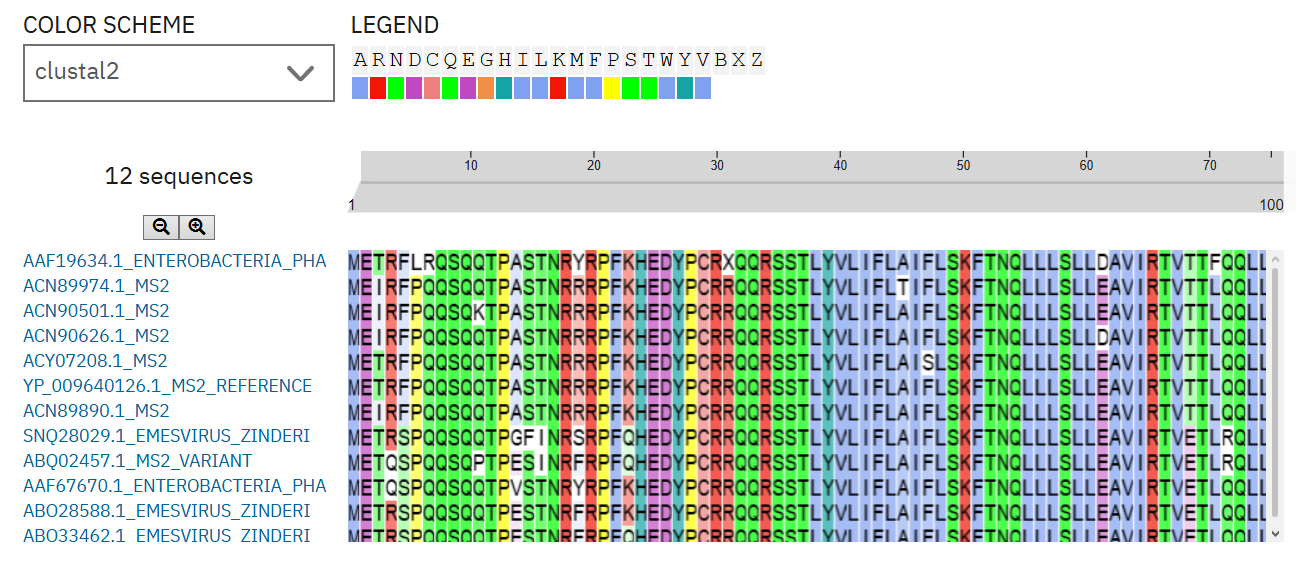
To select MS2 L-protein mutant candidates, I first divided the protein into two functional regions: the soluble N-terminal region, which is associated with DnaJ interaction, and the C-terminal transmembrane region, which affects lysis activity. I prioritized mutations that retained lysis activity and detectable protein levels in the experimental mutant dataset. I then used homologous L-protein sequences from pBLAST and aligned them with Clustal Omega to evaluate whether each candidate position was conserved or variable. Highly conserved sites were interpreted with caution, while partially variable sites were considered more permissive for mutation. The final five variants include mutations in both the soluble and transmembrane regions, allowing the design to test effects on DnaJ-related behavior and membrane-associated lysis.
The expected outcome is that at least one of these L-protein variants will maintain lysis activity while changing properties related to folding, DnaJ interaction, or membrane-associated lysis. Soluble-domain mutants may help test whether the protein can become less dependent on DnaJ-mediated processing, while transmembrane mutants may affect the speed or efficiency of bacterial lysis. Because some selected residues are conserved, these mutations should be interpreted as candidates for testing rather than guaranteed improvements. The next experimental step would be to synthesize the mutant genes, clone them into the appropriate construct, and compare their lysis activity against the wild-type L-protein.
Figure 1. Clustal Omega alignment of selected MS2 L-protein homologs. The alignment was used to evaluate whether candidate mutation sites were highly conserved or partially variable before selecting final L-protein mutant variants.
Note. Candidate mutations were interpreted using conservation patterns across homologous L-protein sequences. Highly conserved residues were kept only with caution when supported by experimental lysis and protein-expression data.
Week 6 — Genetic Circuits Part I: Assembly Technologies
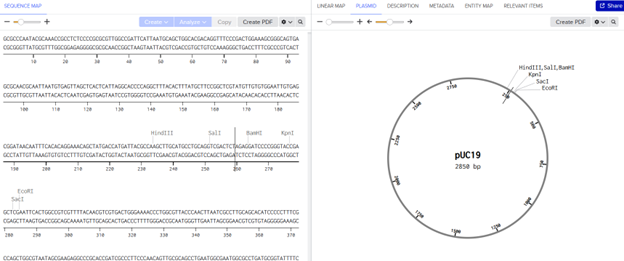
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity DNA Polymerase
A proofreading polymerase with 3′→5′ exonuclease activity, which ensures very low error rates during DNA synthesis.
Primer sequence composition
Secondary structures (hairpins, dimers) affect binding
Salt concentration
Higher salt stabilizes primer-template binding
Polymerase type
Some enzymes (like Phusion) require higher annealing temperatures due to their buffer system
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR
Restriction Enzyme Digests
Starting materials
Template DNA and primers
DNA with restriction sites
Key reagents
Polymerase, primers and dNTPs
Restriction enzyme and buffer
Mechanism
DNA amplification
DNA cutting
Temperature profile
Multiple cycles
Single temperature
Control of fragment
Defined by primer
Defined by enzyme sites
Output
Many copies of a single fragment
Multiple fragments
Critical design step
Primer design
Enzyme selection
Time
1 to 3 hours
Roughly 1 hour
Flexibility
High
Limited by sequence
PCR is generally preferable when you need to generate a specific DNA fragment with precise boundaries or added sequences, such as overlaps for Gibson Assembly, because it allows high flexibility through primer design and can amplify even very small amounts of DNA. In contrast, restriction enzyme digestion is preferable when the DNA already contains suitable restriction sites, making it a simpler and faster method for cutting plasmids or generating fragments without the need for amplification. Therefore, PCR is favored for custom design and low DNA availability, while restriction digestion is best for routine cloning tasks where appropriate sites are already present.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that DNA fragments are appropriate for Gibson Assembly, several critical criteria must be met:
Presence of overlapping regions (20–40 bp)
Fragments must share homologous sequences at their ends to allow correct assembly.
Correct sequence design
Overlaps must be: Specific, In the correct orientation, Free of mismatches
Proper fragment size and integrity
Verified by gel electrophoresis
High purity of DNA
Removal of primers, enzymes, and contaminants (e.g., via PCR cleanup)
Correct concentration (stoichiometry)
Balanced molar ratios improve assembly efficiency (this is where your answer fits)
Absence of unwanted sequences
No internal overlaps or conflicting regions
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli cells through artificially induced membrane permeability:
Heat shock transformation
Cells are treated with CaCl₂ to make membranes more permeable
A sudden temperature increase (e.g., 42°C) creates a thermal imbalance
This allows DNA to enter the cell
Electroporation
A short electrical pulse creates temporary pores in the membrane
DNA enters through these pores
After entry the membrane reseals, the cell recovers and begins expressing the plasmid
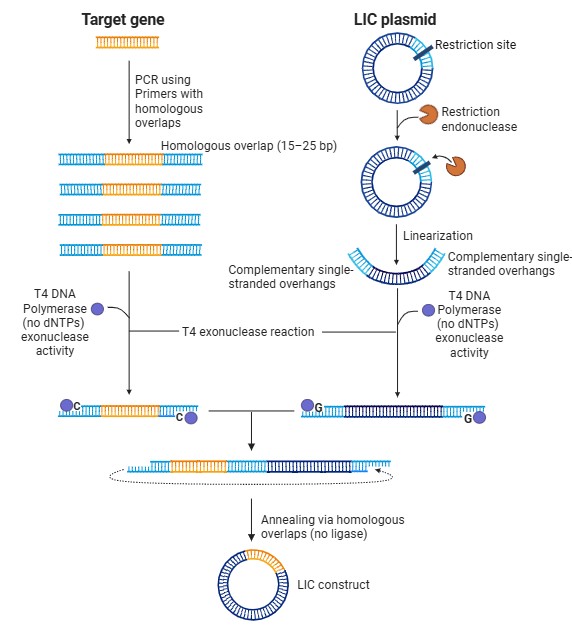
Describe another assembly method in detail
SLIC is a DNA assembly method that joins fragments based on short homologous overlaps, similar to Gibson Assembly but with less enzyme requirements. In this method, DNA fragments are first designed with overlapping regions (15–25 bp). A 3′→5′ exonuclease activity (often from T4 DNA Polymerase) is used to chew back the ends of the DNA, generating complementary single-stranded overhangs. These overhangs allow fragments to anneal to each other without ligase initially, forming a stable intermediate. The assembled DNA is then transformed into bacteria, where the host’s repair machinery seals the nicks in the backbone. Unlike Gibson Assembly, SLIC does not require a ligase or multiple enzymes in the reaction mix, making it more cost-effective. However, it can be slightly less efficient and more sensitive to experimental conditions. Therefore, SLIC is a useful low-cost, overlap-based assembly method.
Model this assembly method with Benchling or Asimov Kernel!
SLIC was modeled in Benchling by designing 20 bp homologous overlaps via PCR primers and assembling fragments using an overlap-based assembly tool (Gibson simulation), since exonuclease processing and in vivo repair cannot be directly simulated.
ASIMOV kernel
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Construct 1
pTac → B1 RBS → TetR → Terminator
I expected pTac to constitutively drive TetR production. In simulation, TetR should increase over time and possibly stabilize at a plateau. If expression is lower than expected, it may be due to weak promoter activity, translation efficiency, or high degradation settings.
Construct 2
pTetR → P3 RBS → LacI → Terminator
I expected this construct to produce LacI continuously. The simulation output should show rising LacI concentration, although with dynamics that may differ from Construct 1 due to promoter and RBS differences. If the observed signal is weak, it may reflect lower promoter strength or insufficient simulation time.
Construct 3
pTac → B1 RBS → LacI → Terminator + pLacI → P1 RBS → TetR → Terminator
I expected the first cassette to express LacI, which would repress the pLacI promoter in the second cassette and keep TetR expression low. In simulation, LacI should increase while TetR should remain low compared with an unrepressed expression construct. If TetR remains high, possible causes include promoter leakiness, delayed LacI accumulation, or insufficient repression strength.
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs provide graded and analog computation rather than a strict ON/OFF logic. Enabling cells to integrate multiple inputs with tunable weights and produce continuous outputs that reflect the signal strength, not just presence/absence. IANNs can implement thresholding, nonlinear decision boundaries, and noise tolerance, making them more robust in heterogeneous biological environments. They also allow combinatorial regulation, which is difficult to achieve with simple Boolean gates without increasing the circuit complexity.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Goal: Detect a pathogenic state only when a specific combination of biomarkers is present at sufficient levels.
Inputs (continuous, not binary)
X1: Inflammatory cytokine level (Proxy via promoter responsive to NF-κB)
X2: Bacterial quorum signal (AHL-responsive promoter)
X3: Hypoxia signal (HIF-responsive promoter)
Network behavior (IANN)
Each input drives expression of regulatory RNAs/proteins that act as weights (RNA regulators).
The network computes a weighted sum:
High X1 and X2, moderate X3 → output ON (pathogenic infection)
High X1 alone → output low (inflammation but not infection)
A thresholding layer converts the summed signal into a measurable output.
Output
Fluorescent protein (GFP) or therapeutic gene (antimicrobial peptide). Output intensity reflects strength of classification
Why IANN is useful here
Avoids false positives from single signals
Integrates multiple noisy biomarkers
Produces graded output for better diagnostics
Limitations of IANNs
Biological noise: stochastic gene expression affects weights and outputs
Tuning difficulty: precise control of weights (promoter/RBS strength) is nontrivial
Crosstalk: regulatory parts may interfere with each other
Metabolic burden: multi-layer circuits can slow growth
Latency: transcription/translation delays limit response time
Scalability: adding layers increases complexity and failure modes
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium-based composites
Basidiomycetes dominate for composites and “pure mycelium” sheets.
A concrete example are insulation boards made with Pleurotus ostreatus and Ganoderma lucidum grown on wheat straw to generate mycelium-based boards. A widely cited commercial packaging example is Mushroom® Packaging (MycoComposite™), marketed as a regenerative alternative to plastic foams; the company describes it as “mycelium + hemp hurd,” home-compostable in 45 days under appropriate conditions.
Compared with petroleum foams (e.g., EPS), MBCs offer biodegradable end-of-life and the possibility of using waste feedstocks, but typically require moisture-protective coatings or application environments that limit water exposure. Risk-focused analysis also emphasizes that durability under humid weathering is a key performance gap; coatings help but may not fully seal porous composites.
Fungal leather and leather-like mycelium sheets
A premium commercial entrant is MycoWorks, which markets “Reishi™” made via its patented “Fine Mycelium™” process (engineered interlocking cellular structures) and states it uses chrome-free tanning/dyeing technologies in finishing.
In contrast, Bolt Threads states it has discontinued development and manufacturing of its mycelium leather product Mylo™, illustrating the real-world challenge of financing and scaling novel biomaterials through commodity-like production economics.
Mycelium leather aims to reduce reliance on livestock and fossil based polymers while enabling biodegradability; however, most formulations still require finishing chemistry (tanning/crosslinking, coatings) to match abrasion/water resistance of incumbent leathers, and consistent quality at scale remains an open challenge in the literature.
Fungal-derived chitin and chitosan
KitoZyme positions itself as a major manufacturer of fungal-origin chitosan and chitin-glucan, targeting cosmetics, agriculture, healthcare, and winemaking.
Regulatory signals also exist: the U.S. Food and Drug Administration GRAS notices database lists “Chitosan from Aspergillus niger” and “Chitin-glucan from Aspergillus niger” among reviewed notices, indicating pathway precedent for food-contact/ingredient contexts depending on use case and dossier.
Fungal chitosan often compares favorably on allergen/mineral contamination risk and supply-chain stability (fermentation vs seafood seasonality), but can still face higher production costs for bulk commodity uses (e.g., large-scale water treatment) and must meet rigorous purity/toxin specifications for biomedical and food uses.
Fungal pigments
Carotenoids deliver color and antioxidant functionality and are used in food/feed/cosmetics; the review emphasizes broad carotenoid diversity in fungi and notes fungi can be strong hosts for carotenoid production relative to some alternative platforms.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would engineer fungi to produce enhanced mycelium-based biomaterials with increased mechanical strength and environmental responsiveness, by modifying cell wall composition and introducing synthetic protein scaffolds. This leverages fungi’s natural ability to form structured materials while enabling sustainable alternatives to plastics and leather.
A core reason is that fungi combine eukaryotic cell biology with an industrially proven capacity for secretion and materials formation. Filamentous fungi are eukaryotes with ER/Golgi pathways and PTMs that bacteria generally lack; reviews emphasize strategies such as engineering glycosylation sites, unfolded protein response (UPR) management, and secretion pathway optimization to improve heterologous protein output.
A cell-wall biotechnology review states that filamentous fungi can outperform bacteria and yeasts in secretion efficiency, reaching reported secreted protein levels up to ~100 g/L in some contexts. Unlike bacteria, fungi naturally build macroscopic fibrous networks (mycelia) that act as binders and scaffolds. MBCs explicitly exploit this to bind particles into solids; biomineralized ELM work likewise uses mycelium as a scaffold for functional composites. Filamentous fungi are specialized decomposers; reviews emphasize their ability to depolymerize complex substrates externally by secreting enzymes, allowing direct use of lignocellulosic wastes and diverse sugars, feedstocks that many bacteria cannot access without extensive pre-processing.
Fungal systems are generally slower-growing and introduce additional process complexity due to morphology (pellets vs dispersed hyphae) affecting viscosity, mixing, and oxygen transfer. Tool maturity is improving but still uneven: a domestication-focused review highlights challenges such as locus biases, transformation efficiencies, and limited cross-species part libraries, issues that are often less severe in mature bacterial chassis ecosystems.
Week 9 — Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis offers greater flexibility and control compared to in vivo systems because it allows precise manipulation of reaction conditions such as component concentrations, temperature, and reaction time. Additionally, it eliminates cellular interference, such as metabolic regulation, toxicity effects, and competing pathways, enabling more efficient and tunable protein production.
One case where cell-free expression is advantageous is in the production of toxic proteins, such as toxins or antimicrobial peptides, which would otherwise damage or kill the host cell. Another case is the synthesis of proteins requiring non-natural amino acids or specialized conditions, which are difficult to achieve in living cells due to their tightly regulated environment.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system is composed of several essential components that enable protein synthesis outside of living cells. First, it includes a genetic template (DNA or mRNA) that contains the gene of interest. If DNA is used, transcription machinery such as RNA polymerase is required to synthesize mRNA.
The system also contains the translation machinery, including ribosomes, tRNAs, amino acids, and translation factors, which work together to synthesize the protein. Additionally, an energy regeneration system is required, supplying molecules such as ATP, GTP, and other energy substrates to sustain the reaction.
Finally, buffers and salts are included to maintain optimal physicochemical conditions, such as pH and ionic strength, which are necessary for proper enzyme activity and protein synthesis.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision and regeneration are critical in cell-free systems because protein synthesis is a highly energy-intensive process that consumes ATP and GTP during transcription and translation. Since cell-free systems lack metabolic pathways to recycle energy, ATP is rapidly depleted, leading to an early توقف of protein production. Therefore, continuous energy regeneration is necessary to sustain the reaction and achieve higher protein yields.
One common method to ensure continuous ATP supply is the use of an energy regeneration system based on phosphoenolpyruvate (PEP), which acts as a high-energy phosphate donor to regenerate ATP from ADP, maintaining the reaction over time.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic and eukaryotic cell-free expression systems differ mainly in their complexity and ability to perform post-translational modifications. Prokaryotic systems, such as those derived from E. coli, are simpler, faster, and more cost-effective, but they lack the machinery for most post-translational modifications. In contrast, eukaryotic systems are more complex and can perform modifications such as glycosylation and proper protein folding.
A suitable protein for production in a prokaryotic system is Green Fluorescent Protein (GFP), as it is relatively simple and does not require post-translational modifications. On the other hand, monoclonal antibodies are better produced in eukaryotic systems because they require correct folding, disulfide bond formation, and glycosylation to be functional.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize the expression of a membrane protein in a cell-free system, I would design an experiment that combines a suitable cell-free extract with a membrane-mimicking environment, because membrane proteins tend to aggregate or misfold in aqueous solution if their hydrophobic regions are not stabilized during synthesis. It would begin with an E. coli extract for simple bacterial membrane proteins, or a eukaryotic/microsome-containing system for more complex proteins requiring eukaryotic folding or post-translational processing. Then I would test additives such as mild detergents, liposomes, or nanodiscs to promote co-translational insertion and stabilize the protein in a native-like environment.
The main challenges here are low solubility, aggregation, incorrect folding, and loss of activity. To address these, It would be good to run a small optimization screen varying temperature, Mg²⁺/K⁺ concentrations, DNA template amount, incubation time, and the type and concentration of membrane mimic. Lowering the temperatures can reduce aggregation, while liposomes or nanodiscs often improve folding and functionality better than detergents alone. If yield is low, I would use a continuous-exchange cell-free format to extend reaction time and improve protein production.
To evaluate success, I would not only measure total protein yield, but also test whether the membrane protein is soluble, properly inserted, and functional. This could be done using SDS-PAGE for expression, centrifugation to compare soluble versus insoluble fractions, and activity or ligand-binding assays depending on the protein. In this way, the experiment would optimize both expression and functional quality, not just the amount of protein produced.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield in a cell-free system can result from several factors. First, the DNA template may be poor or suboptimal, for example if it is degraded, contains inhibitors, or has weak regulatory elements, which reduces transcription and translation efficiency. A good troubleshooting strategy would be to verify template quality, adjust DNA concentration, and include a positive control to confirm that the system itself is working properly.
Second, the reaction conditions may not be optimal, such as incorrect Mg²⁺ or K⁺ concentrations, unsuitable temperature, or an incubation time that is too short. These variables strongly affect ribosome activity and protein synthesis. To troubleshoot this, I would run a small optimization screen varying salt concentrations, temperature, and reaction time to identify the best expression conditions.
Third, the target protein itself may be unstable, misfolded, or prone to aggregation, especially if it is a difficult protein such as a membrane or disulfide-bonded protein. In that case, even if it is synthesized, it may not accumulate properly. A good strategy would be to add folding-supportive components such as detergents, liposomes, nanodiscs, redox helpers, or chaperone-like additives depending on the protein type.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
c. Could this function be realized by genetically modified natural cell?
d. Describe the desired outcome of your synthetic cell operation.
2. Design all components that would need to be part of your synthetic cell.
e. What would be the membrane made of?
f. What would you encapsulate inside? Enzymes, small molecules.
g. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
h. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
3. Experimental details
i. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
j. How will you measure the function of your system?
A useful synthetic minimal cell could be designed to detect Salmonella enterica contamination on food and release bacteriophages only when the pathogen is present. The function of this system is targeted antimicrobial delivery. The input would be a Salmonella-associated quorum-sensing molecule (AI-2), and the output would be the release of lytic anti-Salmonella enterica phages.
This function cannot be fully realized by cell-free Tx/Tl alone without encapsulation, because the system requires physical containment and controlled release of phage particles. However, a genetically modified natural cell could potentially perform a similar sensing-response function, although it would be less controllable and may raise biosafety and regulatory concerns, especially in food applications.
The desired outcome of this synthetic cell is to improve food safety by reducing Salmonella contamination, while avoiding unnecessary phage release in clean food, making the system more efficient and cost-effective.
The synthetic cell would be based on a liposome membrane composed of phospholipids and cholesterol, providing structural stability. Inside the liposome, lytic anti-Salmonella enterica phages and an inactive phospholipase A2-type enzyme would be encapsulated. The membrane would display the LsrB protein, which binds the quorum-sensing molecule AI-2. When AI-2 is detected, binding to LsrB would trigger activation of the phospholipase, which then destabilizes the membrane and causes the release of phages.
This design does not require an internal Tx/Tl system, since the phages are pre-formed and only need to be released. If needed, a bacterial system would be sufficient, as no complex post-translational modifications are required.
The synthetic cell communicates with the environment through surface-level detection, meaning the signal (AI-2) does not need to enter the cell but only bind to the membrane protein. The main components include phospholipids, cholesterol, the lsrB gene (for the binding protein), and a gene encoding a phospholipase enzyme.
To evaluate the system, its function can be measured by quantifying the reduction of Salmonella in treated food samples using qPCR, comparing results with untreated controls. A successful system would show a significant decrease in Salmonella only when the trigger molecule is present.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
- Write a one-sentence summary pitch sentence describing your concept.
A smart childrens beanie incorporating freeze-dried cell-free systems that detect high UV exposure, trigger a cooling hydrogel response, and send alerts to caregivers to prevent heat stress and sun overexposure.
- How will the idea work, in more detail? Write 3-4 sentences or more.
The beanie contains freeze-dried cell-free sensor modules embedded within the fabric that are activated by moisture from sweat or environmental humidity. These systems are designed to detect high levels of UV radiation using UV-responsive genetic elements. When a threshold level of exposure is reached, the system triggers two responses: activation of a compartment containing a cooling hydrogel that expands or releases stored moisture to reduce temperature, and generation of a signal that can be detected by a small integrated electronic module, which sends a notification to a caregiver phone. This allows real-time monitoring and immediate response to potentially harmful exposure conditions.
- What societal challenge or market need will this address?
This system addresses the risk of heat stress and excessive sun exposure in children, who are more vulnerable to dehydration and sun damage and may not recognize early warning signs. It provides a preventive, wearable solution for parents and caregivers, especially in outdoor settings such as parks, schools, and sports activities. The product also aligns with increasing demand for smart textiles and health-monitoring wearables.
- How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
One limitation of cell-free systems is that they require activation, which in this case is solved by using sweat and moisture as natural triggers during wear. Stability is addressed through freeze-drying (lyophilization), allowing long-term storage within the fabric until activation. Since cell-free reactions are typically single-use, the beanie could be designed with replaceable or modular sensing patches that can be swapped after activation. Additionally, integrating the system into protected compartments within the textile helps maintain functionality and prevents environmental degradation.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Biofilms are a major concern in spacecraft because they can help bacteria persist on cabin surfaces, resist cleaning, and potentially threaten astronaut health and equipment. In closed environments such as space habitats, microbes may respond differently to stressors like radiation and microgravity. Understanding whether space-like conditions increase the biofilm potential of common surface bacteria is important for long-term missions. This topic is significant for humanity because safer microbial control will be essential as people spend longer periods in space, and it is scientifically interesting because bacterial adaptation in space is still not fully understood.
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The molecular target is the icaA gene in Staphylococcus epidermidis, a biofilm-associated gene involved in polysaccharide matrix production and surface colonization.
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The icaA gene is directly related to biofilm formation, which allows bacteria to attach to surfaces and persist under harsh conditions. In a spacecraft cabin, this is especially relevant because bacteria growing as biofilms may be harder to remove and more resistant to cleaning procedures. By focusing on icaA, this project investigates whether space-like stress conditions are associated with stronger biofilm-related adaptation in Staphylococcus epidermidis. Detecting this gene under simulated space conditions would help us understand whether bacteria in spacecraft environments may become more persistent and create greater risks for both crew health and spacecraft materials.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis is that Staphylococcus epidermidis exposed to space-like stress conditions, specifically microgravity-like conditions and radiation stress, will show a stronger biofilm-associated genetic signature related to icaA than bacteria grown under normal Earth conditions. The reasoning is that bacteria in stressful environments often adapt to improve survival, and biofilm formation is one of the main strategies used to resist environmental stress. If space-like conditions favor this adaptation, then bacteria commonly found on spacecraft surfaces could become more persistent and harder to eliminate. The goal of this project is to test whether simulated space stress changes the detectability of icaA using a compact molecular workflow that combines miniPCR, the BioBits cell-free system, and fluorescence readout. This could help identify biofilm risk early during future space missions.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would culture Staphylococcus epidermidis under two conditions: normal Earth conditions and simulated space-like stress conditions including microgravity-like growth and radiation exposure. DNA would be extracted from both groups, and icaA would be amplified using miniPCR. The amplified products would then be linked to a BioBits cell-free fluorescent reporter reaction and visualized with the P51 Molecular Fluorescence Viewer. Controls would include a no-template control and a known icaA-positive control. Data collected would include PCR amplification success, fluorescence intensity, and comparison of signal strength between stressed and unstressed samples.
HTGAA 2026: Individual Final Project Documentation AI-Driven Mining of Healthcare Wastewater Metagenomes for Novel Phage-Derived Antimicrobial Peptides Against Salmonella enterica SECTION 1: ABSTRACT The rapid rise of antimicrobial resistance has intensified the need for new antibacterial strategies that move beyond conventional antibiotic discovery pipelines. Healthcare-associated wastewater is a particularly relevant One Health environment because it can contain clinically important bacteria, antimicrobial residues, resistance-associated genes, and diverse bacteriophages. Within this context, bacteriophages represent a valuable but underexplored source of antimicrobial molecules, including endolysin-derived peptides, membrane-active regions, and proteins associated with bacterial envelope disruption. This project aims to develop an AI-guided synthetic biology workflow to identify bacteriophage-derived antimicrobial peptide candidates from healthcare wastewater metagenomes in Andean regions and prioritize them for predicted activity against Salmonella enterica.
HTGAA 2026: Individual Final Project Documentation
AI-Driven Mining of Healthcare Wastewater Metagenomes for Novel Phage-Derived Antimicrobial Peptides Against Salmonella enterica
SECTION 1: ABSTRACT
The rapid rise of antimicrobial resistance has intensified the need for new antibacterial strategies that move beyond conventional antibiotic discovery pipelines. Healthcare-associated wastewater is a particularly relevant One Health environment because it can contain clinically important bacteria, antimicrobial residues, resistance-associated genes, and diverse bacteriophages. Within this context, bacteriophages represent a valuable but underexplored source of antimicrobial molecules, including endolysin-derived peptides, membrane-active regions, and proteins associated with bacterial envelope disruption. This project aims to develop an AI-guided synthetic biology workflow to identify bacteriophage-derived antimicrobial peptide candidates from healthcare wastewater metagenomes in Andean regions and prioritize them for predicted activity against Salmonella enterica.
The central hypothesis is that healthcare-associated wastewater metagenomes harbor previously uncharacterized phage-derived peptide sequences with potential antibacterial activity against S. enterica, and that these candidates can be detected using protein language models, antimicrobial peptide classifiers, structural prediction, and synthetic DNA design. To test this hypothesis, metagenomic sequences will be processed to identify viral and phage-associated open reading frames, extract peptide candidates from relevant phage proteins, and encode these sequences using AI-based protein representation tools. Candidate peptides will then be ranked according to predicted antimicrobial activity, structural stability, novelty, and biosafety-related filters. Finally, selected candidates will be incorporated into synthetic expression construct designs suitable for future cell-free or chassis-based production. The expected outcome is a focused computational-to-design platform that transforms healthcare wastewater metagenomic data into prioritized phage-derived antimicrobial peptide candidates, contributing to future synthetic biology strategies against Salmonella enterica within a One Health framework.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of this project is to identify and prioritize bacteriophage-derived antimicrobial peptide candidates from healthcare-associated wastewater metagenomic data from Andean regions, using metagenomic analysis, AI-based sequence screening, and in silico peptide–target interaction prediction against Salmonella enterica.
To achieve this aim, healthcare wastewater metagenomic sequences will be obtained from publicly available datasets, simulated inputs, or environmental sequencing data, depending on data availability. The metagenomic data will be quality-controlled, assembled into contigs, and analyzed to identify viral or bacteriophage-associated open reading frames.
Candidate phage-derived proteins or peptide regions will then be screened using antimicrobial peptide prediction tools, protein language model embeddings, and novelty-based prioritization. Selected peptide candidates will be evaluated through structural prediction and in silico interaction modeling against relevant S. enterica targets using tools such as AlphaFold-Multimer or related protein–peptide docking approaches.
Finally, at least one prioritized peptide candidate will be converted into a DNA sequence, codon-optimized, and designed as a synthetic expression construct in Benchling to connect computational discovery with synthetic biology implementation.
Relevant methods and tools may include metagenomic quality control, ORF prediction, phage sequence identification, antimicrobial peptide prediction, protein language models, latent space analysis, AlphaFold-Multimer, Benchling, codon optimization, and synthetic DNA construct design.
Aim 2: Development Aim
The second aim of this project is to experimentally produce and validate selected bacteriophage-derived antimicrobial peptide candidates for activity against Salmonella enterica using in vitro expression and infection-associated assays.
Following the computational prioritization completed in Aim 1, selected peptide sequences would be synthesized as codon-optimized DNA constructs and cloned into an appropriate expression system, such as Escherichia coli or a cell-free protein expression platform. Peptide production would be confirmed through protein-level validation methods such as SDS-PAGE, affinity-tag detection, Western blotting, or another suitable analytical approach depending on the final construct design.
The antibacterial activity of the produced peptide would then be tested against S. enterica through in vitro assays measuring bacterial viability, growth inhibition, membrane disruption, or changes in infection-related behavior. In a later stage, the peptide could be evaluated in a human cell infection model, such as HEK293 cells, comparing untreated S. enterica infection with peptide-treated conditions to determine whether the candidate reduces bacterial pathogenicity, host-cell damage, or infection-associated cytotoxicity.
This aim extends the project beyond computational prediction by testing whether the designed peptide can be produced and whether it demonstrates measurable biological activity against S. enterica.
Aim 3: Visionary Aim
The third aim of this project is to pioneer a decentralized “metagenome-to-medicine” platform in which healthcare wastewater from Andean regions is transformed into a source of AI-designed, phage-derived antimicrobial peptide therapeutics for rapid response against antimicrobial-resistant pathogens.
The long-term vision is to build an autonomous discovery pipeline capable of moving from environmental sample to therapeutic prototype through an integrated workflow of metagenomic sequencing, viral sequence mining, protein language model prediction, structural simulation, synthetic DNA design, and automated cell-free peptide production. In this model, healthcare wastewater would serve as a real-time biological sensor of local infectious disease threats and as a discovery reservoir for the phage-derived molecules needed to counter them. For Salmonella enterica, this could enable the identification of peptides that reduce pathogenicity, disrupt bacterial envelope integrity, interfere with infection-associated mechanisms, or complement existing antimicrobial strategies.
At full scale, this platform could redefine antimicrobial discovery by making it local, adaptive, programmable, and predictive. Instead of waiting years for new antibiotics to emerge through traditional pipelines, hospitals and research centers could use regional metagenomic data to design candidate antimicrobials within days or weeks. This vision would connect One Health surveillance with synthetic biology manufacturing, creating a future where antimicrobial resistance is addressed through distributed biological design infrastructure. The ultimate goal is not only to discover one peptide against S. enterica, but to establish a generalizable framework for rapidly generating pathogen-specific antimicrobial solutions from the microbial dark matter present in local environments.
SECTION 3: BACKGROUND
Briefly summarizetwo peer-reviewed research citationsrelevant to your research
Antimicrobial resistance is one of the most urgent challenges in global health because it reduces the effectiveness of conventional antibiotics and increases the difficulty of treating bacterial infections. This problem is especially relevant in One Health environments, where human activity, clinical waste, microorganisms, antimicrobial residues, and resistance genes can interact. Healthcare-associated wastewater is a particularly important environment because it can contain antibiotic-resistant bacteria, clinically relevant antimicrobial resistance genes, pharmaceuticals, disinfectants, and viral communities. Petrovich et al. used shotgun metagenomics to study a hospital wastewater treatment system and found that antibiotic resistance genes and viruses persisted across the treatment process, supporting the idea that hospital wastewater is not only a waste stream but also a biologically informative reservoir for antimicrobial resistance surveillance (Petrovich et al., 2020)
At the next level, wastewater is also relevant because it contains bacteriophages, which are viruses that infect bacteria and are deeply connected to bacterial ecology. Strange et al. (2021) analyzed global sewage metagenomic data from 79 cities in 60 countries and showed that sewage can be used to study the distribution of bacteriophages and their relationship with bacterial communities and antimicrobial resistance genes. This supports the use of wastewater metagenomics as a practical approach to explore viral diversity at large scale.
For this project, healthcare-associated wastewater from Andean regions is therefore proposed as a focused discovery environment where bacteriophage-derived sequences may reveal new antimicrobial candidates relevant to local and global AMR challenges.
At the micro level, bacteriophages are not only important as whole viral particles; they also encode proteins and peptide regions that may have antimicrobial potential. These include endolysin-associated regions, membrane-active domains, tail-associated proteins, and other phage-derived molecular components that may interact with bacterial envelopes or infection-related structures. This is innovative because it shifts the project away from traditional whole-phage therapy and toward the discovery of modular phage-derived antimicrobial peptides that could be computationally predicted, optimized, and redesigned using synthetic biology. In this context, Salmonella enterica is used as the target pathogen because it is a clinically and food-safety-relevant bacterium, allowing the project to connect environmental discovery with a defined pathogenic outcome.
Metagenomics is essential for this project because many microbial and viral sequences in environmental samples cannot be easily recovered through culture-based methods. Instead of isolating one organism at a time, metagenomic sequencing allows the project to access the collective genetic diversity of an entire microbial community, including uncultured bacteria, bacteriophages, and unknown protein-coding sequences. Santos-Júnior et al. demonstrated that machine learning can be applied to large metagenomic and genomic datasets to predict antimicrobial peptides, generating a catalog of hundreds of thousands of non-redundant peptide candidates and experimentally validating a subset with antibacterial activity. This study directly supports the logic of this project: environmental sequence data can be transformed into prioritized antimicrobial candidates through computational prediction, AI-guided screening, and downstream synthetic biology design (Santos-Júnior et al., 2024).
Therefore, the gap addressed by this project is the lack of a focused workflow for discovering bacteriophage-derived antimicrobial peptide candidates from healthcare-associated wastewater metagenomes in Andean regions and prioritizing them against Salmonella enterica. Existing studies show that wastewater can be used for AMR and phage surveillance, and that metagenomic data can be mined for antimicrobial peptides. However, fewer approaches integrate these ideas into a single synthetic biology pipeline that moves from healthcare wastewater metagenomes to phage-derived peptide prediction, structural prioritization, and DNA construct design. This project addresses that gap by proposing an AI-guided metagenome-to-design workflow for identifying candidate phage-derived antimicrobial peptides with predicted relevance against S. enterica pathogenicity.
Explain how your project is novel or innovative.
This project is innovative because it treats healthcare-associated wastewater from Andean regions not only as a site of microbial risk, but as a source of therapeutic design information. Rather than searching broadly for known antimicrobial genes, the project focuses on bacteriophage-derived peptide candidates that may be hidden within complex metagenomic datasets and difficult to detect through culture-dependent methods alone. The workflow is novel because it links phage sequence discovery, AI-guided peptide prioritization, structural prediction, and synthetic DNA construct design into a single pipeline. This approach expands synthetic biology beyond building with well-characterized biological parts by using environmental viral diversity as raw material for engineering new antimicrobial strategies against S. enterica.
Explain why your project matters and what impact it could have.
This project matters because antimicrobial resistance is reducing the effectiveness of conventional treatments and creating an urgent need for alternative antimicrobial strategies. S. enterica is a relevant target because it is associated with foodborne disease, clinical infections, and increasing concerns about resistant strains, making it important for both public health and One Health surveillance. Bacteriophages are promising in this context because they naturally recognize bacteria with high specificity, which could reduce damage to beneficial microbiota compared with broad-spectrum antibiotics. However, the direct clinical use of whole phages can be limited by regulatory complexity, host-range variability, immune responses, and challenges in standardizing phage preparations. For this reason, phage-derived proteins and peptides represent an attractive alternative, because they may retain antibacterial activity while being easier to design, synthesize, optimize, and incorporate into controlled synthetic biology systems.
The impact of this project lies in creating a workflow that connects healthcare wastewater surveillance with the discovery of phage-derived antimicrobial candidates against S. enterica. By using metagenomics and AI-guided prediction, the project could help identify antimicrobial peptide candidates that would be difficult to detect through culture-based screening alone. This is especially relevant for underexplored Andean healthcare environments, where local microbial and viral diversity may contain unique biological information that has not been fully studied. If successful, the project could contribute to the development of more targeted antimicrobial strategies and support future peptide-based interventions against resistant bacterial pathogens. At a broader level, this work could help shift antimicrobial discovery from slow, generalized screening toward a more precise, predictive, and regionally informed design process.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility).
This project can raise several ethical considerations related to artificial intelligence, environmental metagenomics, synthetic biology, and the use of bacteriophage-derived sequences as potential antimicrobial agents. One major ethical principle involved is non-maleficence, because the discovery and engineering of antimicrobial peptides must avoid unintended biological or ecological harm. Although this project focuses on phage-derived peptides rather than whole-phage therapeutic deployment, bacteriophages themselves evolve rapidly and can interact dynamically with bacterial populations. For this reason, any laboratory work involving phage-associated genetic material must be carefully controlled to prevent unintended environmental release or uncontrolled biological propagation. Additional concerns involve responsibility and transparency in the use of AI-based predictions, since computational models may generate false positives, overestimate biological activity, or produce misleading functional interpretations if not critically validated. The principle of justice is also relevant because the project uses biodiversity-associated genetic information from underexplored Andean healthcare environments, making it important to consider equitable scientific benefit, responsible data use, and fair access to future applications derived from these biological resources.
To ensure ethical conduct, several biosafety and research responsibility measures should be implemented throughout the project. All candidate peptides identified through AI-guided prediction should undergo rigorous computational filtering and controlled experimental validation before considering any downstream application. Any laboratory manipulation involving bacteriophage-associated material should follow strict containment and decontamination procedures, including appropriate biological waste treatment, chemical inactivation of phage-containing materials, and systematic disposal protocols to reduce the risk of environmental release. Because phages can evolve rapidly, the project must ensure that all work involving phage-derived sequences is performed under controlled and traceable conditions with clearly defined containment practices. Potential unintended consequences include incorrect antimicrobial predictions, toxicity toward non-target organisms, disruption of microbial communities, or overreliance on computational models without sufficient experimental confirmation. To address these uncertainties, AI-generated predictions should be treated as probabilistic rather than definitive and complemented with structural analysis, experimental validation, and transparent reporting of limitations. Ethical alternatives include restricting early applications to laboratory-scale discovery, environmental monitoring, or controlled cell-free systems before any therapeutic consideration. Overall, the project should prioritize biosafety, transparency, scientific responsibility, and equitable public health impact while developing new antimicrobial discovery strategies.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Define the biological question and target pathogen
The project will focus on identifying bacteriophage-derived antimicrobial peptide candidates from healthcare-associated wastewater metagenomes and prioritizing them for predicted activity against Salmonella enterica. The expected result is a clear target framework connecting the environmental source, phage-derived peptides, and S. enterica pathogenicity.
Timeline
Step
Methods/tools
Estimated time
Expected result
Select or obtain healthcare wastewater metagenomic data.
Metagenomic sequencing datasets will be obtained from publicly available repositories such as NCBI SRA, NMDC, MG-RAST, or ENA. If real Andean healthcare wastewater data are not available during the course, simulated or representative hospital wastewater metagenomes will be used as proof-of-concept input.
1–2 days
A selected dataset suitable for downstream viral and peptide mining.
Perform quality control of raw sequencing reads.
Raw metagenomic reads will be evaluated using tools such as FastQC or MultiQC to assess read quality, GC content, adapter contamination, and sequence length distribution. Low-quality bases and adapters will be trimmed using tools such as Trimmomatic, fastp, or Cutadapt.
1 day
A cleaned metagenomic dataset with improved read quality.
Assemble metagenomic reads into contigs.
Quality-controlled reads will be assembled using a metagenomic assembler such as MEGAHIT or metaSPAdes. Assembly statistics, including total assembly size, N50, contig length distribution, and number of contigs, will be evaluated using QUAST.
1–2 days
A set of assembled contigs representing microbial and viral genetic material from the wastewater metagenome.
Identify viral and bacteriophage-associated contigs.
Assembled contigs will be screened for viral signatures using tools such as VirSorter2, CheckV, VIBRANT, or geNomad. Contigs predicted to be viral or bacteriophage-associated will be retained for further analysis.
1–2 days
A curated subset of putative bacteriophage-derived contigs.
Predict open reading frames from phage-associated contigs.
Open reading frames will be predicted using Prodigal in metagenomic mode or another ORF prediction tool appropriate for environmental sequences. Predicted ORFs will be translated into amino acid sequences for protein-level analysis.
1 day
A protein sequence dataset derived from putative bacteriophage contigs.
Annotate bacteriophage-derived proteins.
Predicted proteins will be compared against databases such as NCBI nr, PHROGs, UniProt, Pfam, or HMMER-based viral protein databases. This step will identify proteins related to endolysins, tail fibers, depolymerases, holins, virion-associated peptidoglycan hydrolases, or hypothetical phage proteins.
2–3 days
A functional annotation table classifying candidate phage proteins.
Extract potential antimicrobial peptide regions.
Candidate peptide regions will be obtained either from short phage proteins or from predicted antimicrobial domains within larger phage-derived proteins. Peptide candidates will be filtered by length, charge, hydrophobicity, predicted solubility, and sequence complexity.
1–2 days
A list of candidate bacteriophage-derived peptide sequences for AI-based screening.
Screen peptide candidates using antimicrobial peptide prediction tools.
Peptide sequences will be analyzed using antimicrobial peptide prediction tools such as AMPScanner, AMPlify, iAMPpred, DBAASP-associated tools, or similar models. Candidates will be ranked based on predicted antimicrobial probability.
1–2 days
A ranked list of peptides with predicted antimicrobial activity.
Apply protein language model embeddings for AI-guided prioritization.
Peptide or protein sequences will be encoded using protein language models such as ESM, ProtBERT, or related embedding tools. These embeddings will be used to compare candidate sequences with known antimicrobial peptides and to detect potentially novel candidates.
2–3 days
A numerical representation of candidates that supports clustering, novelty detection, and prioritization.
Perform latent space analysis.
Dimensionality reduction methods such as PCA, UMAP, or t-SNE will be used to visualize candidate peptides in relation to known antimicrobial and non-antimicrobial peptide datasets. Clustering will help identify candidates that are both antimicrobial-like and distinct from well-characterized sequences.
1–2 days
A visual map showing candidate distribution and novelty.
Filter candidates for safety and feasibility.
Candidate peptides will be screened for predicted toxicity, hemolytic potential, allergenicity, and poor physicochemical properties using computational tools where available. Peptides with high predicted toxicity or poor synthesis feasibility will be deprioritized.
1 day
A safer and more feasible shortlist of candidate peptides.
Select Salmonella enterica molecular targets for in silico interaction analysis.
Potential S. enterica targets may include outer membrane proteins, virulence-associated surface proteins, membrane-associated receptors, or conserved envelope-related proteins. The target selection will be based on relevance to pathogenicity, accessibility, and available structural information.
1–2 days
A defined set of S. enterica target proteins for docking or interaction modeling.
Predict peptide structure.
Selected peptide candidates will be modeled using tools such as AlphaFold, ColabFold, PEP-FOLD, or related peptide structure prediction platforms. Structural outputs will be evaluated for confidence, stability, and suitability for interaction modeling.
1–2 days
Predicted 3D structures for top peptide candidates.
Model peptide–target interactions.
Top peptide candidates will be tested against selected S. enterica targets using AlphaFold-Multimer, ColabFold-Multimer, or protein–peptide docking tools. Interaction outputs will be evaluated using confidence metrics, predicted interface contacts, binding orientation, and biological plausibility.
2–4 days
Identification of peptide candidates with plausible predicted interaction with S. enterica targets.
Prioritize final peptide candidates.
A scoring matrix will be created to integrate antimicrobial prediction score, novelty, structural confidence, toxicity filtering, synthesis feasibility, and predicted interaction with S. enterica. The highest-ranked candidate will be selected for synthetic biology design.
1 day
One to three final peptide candidates selected for construct design.
Back-translate the selected peptide into a DNA sequence.
The selected amino acid sequence will be converted into a DNA sequence and codon-optimized for the intended expression system, such as E. coli or a cell-free expression system.
1 day
A codon-optimized DNA sequence encoding the selected antimicrobial peptide.
Design a synthetic expression construct in Benchling.
The construct will include a promoter, ribosome binding site, peptide coding sequence, optional affinity tag, linker if needed, terminator, and cloning or synthesis-compatible flanking regions. Benchling will be used to document the construct architecture.
1–2 days
A complete synthetic DNA construct ready for future ordering or cloning.
Evaluate construct feasibility.
The designed sequence will be checked for GC content, restriction sites, repetitive regions, synthesis complexity, reading frame accuracy, and compatibility with the selected expression system.
1 day
A validated expression-ready construct design.
Generate final data outputs and visualizations.
The final results will include a workflow diagram, candidate ranking table, peptide property table, latent space visualization, structural prediction images, peptide–target interaction model, and final construct map.
2–3 days
A complete computational and design-based validation package for the final project.
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Foundational Lab Practices
Pipetting
Lab Safety
Bioethical Considerations (must check this box)
DNA Skills & Analysis
DNA Sequencing
DNA Editing
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases (e.g., GenBank, NCBI, Ensembl, and UCSC Genome Browser)
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots (e.g., Opentrons)
Designing a Twist Order
Creating a plan to use the Autonomous lab at Ginkgo Bioworks
Protein Design
Protein Design
Use of Boltz or PepMLM
Use of Asimov Kernel
Use of Benchling
Models and Notebooks
Databases
Bioproduction
Bioproduction
Chassis Selection (e.g., DH5alpha)
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Bacterial Processing (e.g., Centrifugation, Lysis, DNA Purification)
Cell-Free Systems
Cell Free Reactions
Freeze-Dried Cell Free Systems
miniPCR Tools
Protein Purification
Gibson Assembly
Primer Design or Selection
PCR Reactions
Gibson Assembly
Other Cloning Methods (e.g., Restriction Enzyme Digestion or Gateway Cloning)
CRISPR
CRISPR/Cas9
Designing Prime Editing gRNA
I would expand on DNA Construct Design and Protein Design as both are central to my final project.
For DNA construct design, I would take the prioritized phage-derived candidate, such as the CwlA-like endolysin identified from the wastewater metagenomic data, and convert its amino acid sequence into a codon-optimized DNA sequence suitable for future expression. This DNA sequence would then be incorporated into a synthetic expression cassette containing a promoter, ribosome binding site, purification tag, coding sequence, and terminator, such as pTac → RBS → 6xHis–Candidate → L3S2P24 terminator. This step connects the computational discovery phase with synthetic biology implementation by turning a predicted antimicrobial protein into a buildable genetic design.
For protein design, I would evaluate the selected candidate using bioinformatic and structural prediction tools before deciding whether it is suitable for expression and downstream testing. This would include analyzing conserved domains, physicochemical properties, predicted structure, and possible antimicrobial activity. In my project, this technique helps prioritize candidates that are not only biologically interesting but also more likely to fold properly, remain stable, and function as phage-derived antimicrobial proteins. Together, these techniques allow the project to move from metagenomic sequence discovery toward a realistic synthetic biology workflow for future anti-Salmonella enterica applications.
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
You are required to validate at least one aspect of your final project aims.
I chose to validate the computational-to-design portion of my final project by identifying and prioritizing a phage-derived antimicrobial protein candidate from healthcare wastewater metagenomic data. Specifically, I validated whether quality-filtered viral contigs could be annotated to recover candidate phage proteins, confirm a biologically relevant lytic domain, and translate one selected candidate into a synthetic biology expression construct.
1.1 I began with healthcare wastewater metagenomic viral contigs obtained from the available dataset.
1.2 I used CheckV to evaluate viral contig quality and filtered the dataset to prioritize high-quality and medium-quality viral contigs.
1.3 From this quality-control step, I selected the best viral contigs for downstream annotation, reducing the analysis of low-confidence viral fragments.
1.4 I annotated the selected contigs using Pharokka, which generated GenBank and GFF outputs containing predicted viral coding sequences.
1.5 I screened the Pharokka annotation manually for candidate proteins related to bacterial lysis, especially endolysin-like proteins, and also included one hypothetical protein as an exploratory novelty candidate.
1.6 I extracted candidate information including CDS ID, contig, product annotation, predicted function, PHROG annotation, amino acid sequence, and approximate protein length.
1.7 I selected one promising candidate and analyzed its sequence using NCBI Conserved Domain Search.
1.8 The conserved domain analysis identified a CwlA superfamily domain, described as an N-acetylmuramoyl-L-alanine amidase, with an E-value of 1.07e-22.
1.9 This result supported the interpretation that the candidate may function as a phage-derived cell wall hydrolase or endolysin-like protein.
1.10 I then used AlphaFold/ColabFold to generate a predicted 3D structure of the selected candidate and evaluate whether the protein contained structured regions suitable for further analysis.
1.11 Finally, I designed a synthetic expression construct for future validation of the candidate.
1.13 This construct connects the computational discovery workflow with a synthetic biology implementation strategy for future expression and testing.
I utilized DNA construct design as the main synthetic biology technique to validate the implementation potential of my project. After selecting a CwlA-like phage endolysin candidate, I designed a modular expression cassette containing an inducible promoter, ribosome binding site, purification tag, coding sequence, and bacterial terminator. This design allows the candidate protein to be expressed in a controlled way in a future bacterial or cell-free expression system. I also used protein design and computational modeling by evaluating the candidate’s conserved domain, predicted structure, and suitability for downstream expression. Together, these techniques show how metagenomic discovery can be connected to a buildable synthetic biology design.
The main data generated in this validation include the CheckV quality-control output, Pharokka annotation results, conserved domain analysis, and AlphaFold structural prediction. CheckV allowed me to prioritize high- and medium-quality viral contigs before annotation, while Pharokka identified viral coding sequences from those contigs. The strongest candidate showed a CwlA superfamily domain with an E-value of 1.07e-22, which strongly supports its predicted role as a cell wall amidase or endolysin-like protein. This result is relevant because CwlA-like amidases are associated with peptidoglycan degradation, making the candidate biologically plausible for future anti-Salmonella enterica applications. The AlphaFold model further supported that the candidate contains structured regions, although experimental validation would still be required to confirm activity.
Figure 1. Quality control of viral contigs using CheckV.
Note. Viral contigs were classified by quality category, and high- and medium-quality contigs were prioritized for downstream Pharokka annotation.
Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project. (min. 4 sentences).
During the validation, one unexpected challenge was that the viral metagenomic data contained many low-quality or fragmented contigs, which could have led to unreliable annotation and false candidate selection. To overcome this, I filtered the viral contigs using CheckV and focused only on high- and medium-quality contigs before running Pharokka. Another challenge was that Pharokka annotated many CDSs as “hypothetical protein” or “unknown function,” so I could not rely only on the initial annotation to select candidates. To address this, I used conserved domain analysis with NCBI CDD, which allowed me to identify a CwlA-like N-acetylmuramoyl-L-alanine amidase domain in one candidate and support its possible role as an endolysin-like protein.
A second limitation was that structural modeling could not fully validate antimicrobial activity. AlphaFold/ColabFold can predict whether the candidate has structured regions, but it does not experimentally prove that the protein will bind or degrade the Salmonella enterica cell wall. In addition, because the expected biological target of a CwlA-like amidase is peptidoglycan rather than a single protein receptor, protein–protein docking may not fully represent the real mechanism of action. In future work, this limitation could be addressed by combining structural prediction with molecular docking against peptidoglycan-like fragments, recombinant expression, protein purification, and antimicrobial activity assays against S. enterica.
References
Petrovich, M. L., Zilberman, A., Kaplan, A., Eliraz, G. R., Wang, Y., Langenfeld, K., Duhaime, M., Wigginton, K., Poretsky, R., Avisar, D., & Wells, G. F. (2020). Microbial and Viral Communities and Their Antibiotic Resistance Genes Throughout a Hospital Wastewater Treatment System. Frontiers In Microbiology, 11, 153. https://doi.org/10.3389/fmicb.2020.00153
Santos-Júnior, C. D., Torres, M. D., Duan, Y., Del Río, Á. R., Schmidt, T. S., Chong, H., Fullam, A., Kuhn, M., Zhu, C., Houseman, A., Somborski, J., Vines, A., Zhao, X., Bork, P., Huerta-Cepas, J., De la Fuente-Nunez, C., & Coelho, L. P. (2024). Discovery of antimicrobial peptides in the global microbiome with machine learning. Cell, 187(14), 3761-3778.e16. https://doi.org/10.1016/j.cell.2024.05.013
Strange, J. E. S., Leekitcharoenphon, P., Møller, F. D., & Aarestrup, F. M. (2021). Metagenomics analysis of bacteriophages and antimicrobial resistance from global urban sewage. Scientific Reports, 11(1), 1600. https://doi.org/10.1038/s41598-021-80990-6
Supply List and Budget
Minimum version: computational validation completed in this project
Computer or laptop (already available)
Needed for sequence handling, annotation review, and figure preparation
Estimated cost: $0 if already available
Internet access
Required to access Galaxy, NCBI, Benchling, AlphaFold/ColabFold, and databases
Estimated cost: $0–$20
Public healthcare wastewater metagenomic dataset or simulated data
Used as the input for viral contig identification and downstream annotation
Estimated cost: $0
Galaxy platform
Used for CheckV filtering, sequence processing, and Pharokka annotation
Estimated cost: $0
CheckV
Used to evaluate viral contig quality and select high- and medium-quality contigs
Estimated cost: $0
Pharokka
Used to annotate viral/phage-associated open reading frames
Estimated cost: $0
NCBI Conserved Domain Search / BLASTp
Used to identify conserved domains and compare candidates with known proteins
Estimated cost: $0
ExPASy ProtParam
Used to calculate physicochemical properties such as molecular weight, pI, and stability index
Estimated cost: $0
AlphaFold / ColabFold
Used for predicted 3D structural modeling of the selected candidate
Estimated cost: $0 if using free access
Benchling
Used to design the synthetic DNA expression construct
Estimated cost: $0 for academic/free use
Estimated minimum project cost
$0–$20, assuming public datasets and free academic bioinformatics platforms are used.