To make things tidy, I decided to answer most of the questions about the biological engineering application in sections of a separate project idea page.
PLEASE NOTE: on the project page accessible through the link I have been unable to show two images: an image of Van Gogh’s Sunflowers painting and a diagram showing how I’ve scored the actions I’ve suggested. It doesn’t appear to load the image, yet the build log indicates it’s a success. I’m not sure what the problem is but it means you won’t see those two pictures.
Assignment 2 I created a Benchling account, loaded up the Lambda DNA, and then tried different combinations of the following restriction enzymes.
EcoRI HindIII BamHI KpnI EcoRV SacI SalI I note that the Automation Art tools produces randomly created electrophoresis ladders, but I excluded Ndel, Pvull and Xhol - because they were not in the list we were supposed to use.
Assignment 3 Following on from last week’s assignment, I decided to use Vincent van Gogh’s Sunflowers painting (the one hanging in the National Gallery) for my art subject. I tried to download an image of the painting and upload it to the Opentrons automated art interface. Importing it made some artistic effects I didn’t want - it flooded the background with yellow, left out the blue streaks and didn’t do much to distinguish between orange and yellows. Importing it created something that wasn’t recognisable.
Part A: Conceptual Questions Choosing 9 of 11 questions to answer
Q1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) I’ve made some assumptions:
The meat mass does not include air or water. The meat mass does not include organic materials that are not amino acids A Dalton is a unit of mass used to express atomic and molecular weights. I used this converter to determine how many Daltons are in 500 grams of organic material. 500 g = 3.011086821E+26 daltons. If an amino acid is 100 daltons, then 500 g would contain 3.011086821E+24 molecules.
Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. I visited the Uniprot page for (P00441)[https://www.uniprot.org/uniprotkb/P00441/entry#sequences] and the normal sequence is:
012345… MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V represents one mutation where the ‘A’ changes to a ‘V’ at position 4: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I found additional information about the AV4 mutation on the ALS Association’s page
Genetic Circuits Part I: Assembly Technologies 1 What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? A product page for Fisher Scientific indicate: “Phusion High-Fidelity PCR Master Mix is convenient 2X mix containing Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2. Two master mix formulations are available: with HF Buffer (F-531S and F-531L) and with GC Buffer (F-532S and F-532L).”1
Genetic Circuits Part II: Neuromorphic Circuits Part 1: Intracellular Artificial Neural Networks (IANNs) 1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Week 9 — Cell-Free Systems General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The greatest advantage the cell-free protein synthesis has over in vivo methods is that the viability of the cell does not have to be maintained in order to maintain the viability of the protein synthesis. By allowing proteins to be grown in an open environment, greater control can be exercised over the key factors that produce the protein. Materials can be added to help protein production which may otherwise be toxic to cell populations. Scientists can control ion concentrations, cofactors and energy sources. Enhancing and prohibiting materials can also be added. Growing proteins in a cell-free environment also means protein production does not have to accommodate cloning of cells.
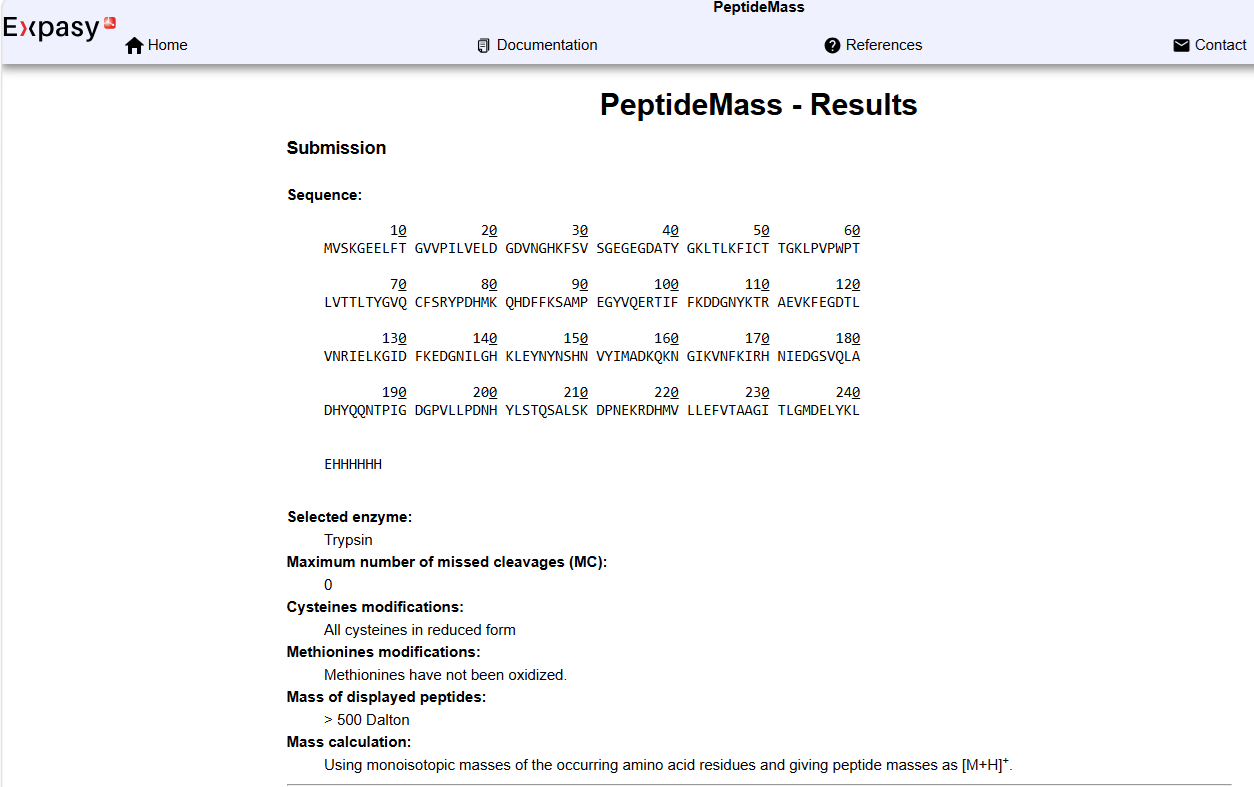
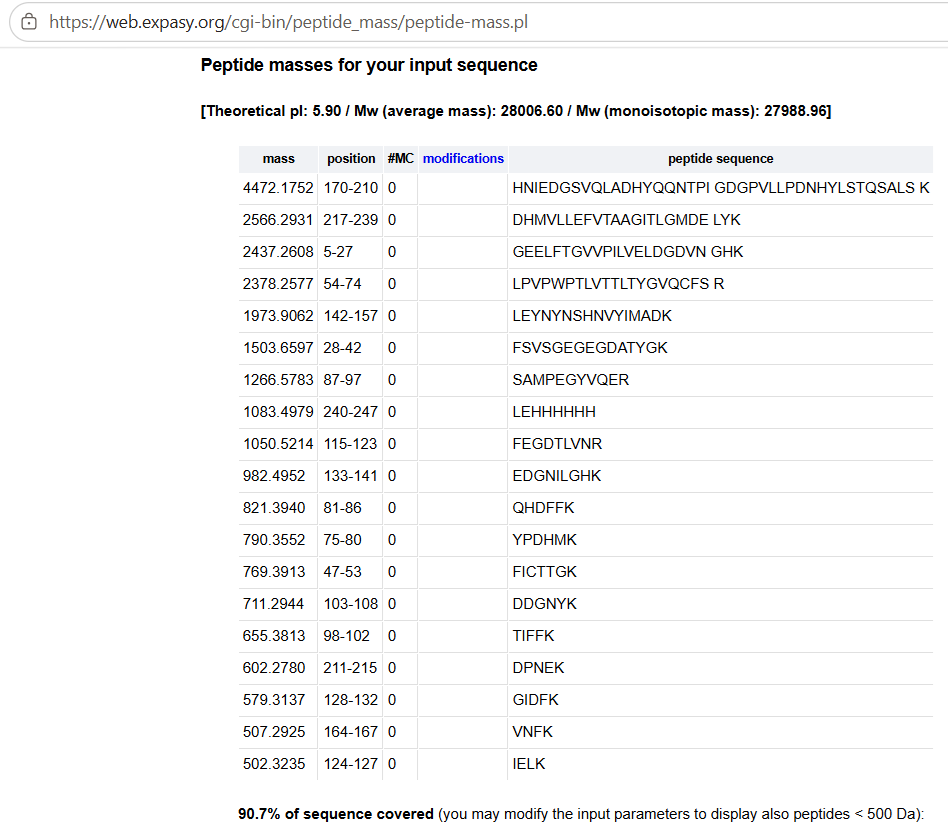
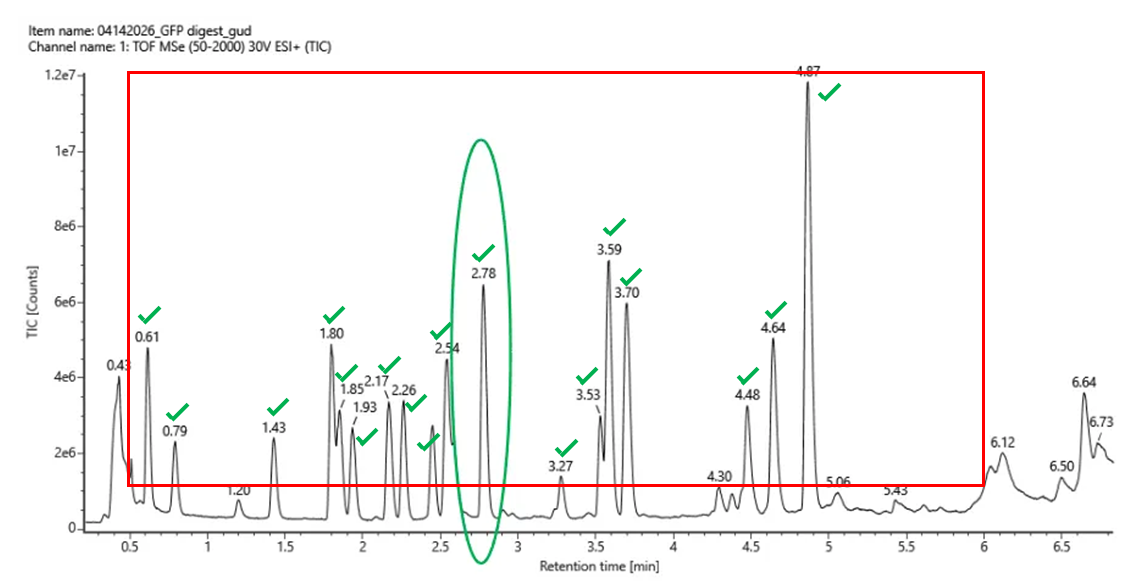
Homework: Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail
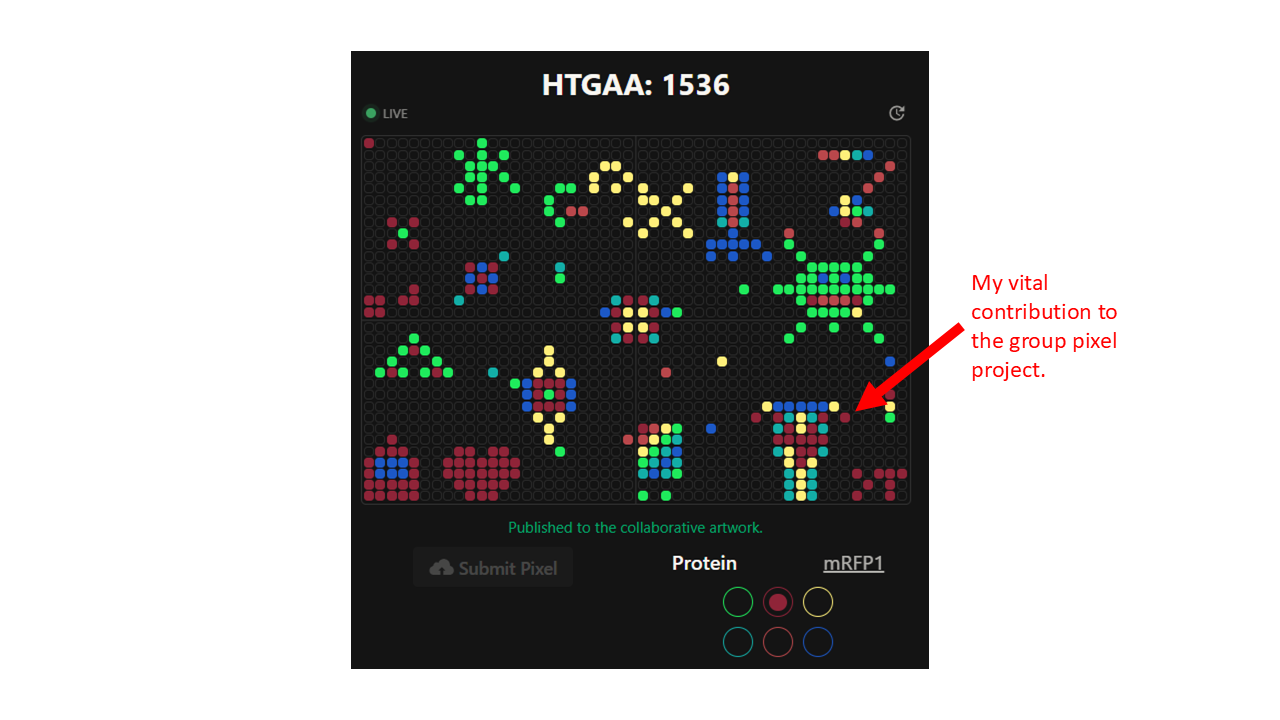
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse. If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉 Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”) what you liked about the project, and what about this collaborative art experiment could be made better for next year.
Homework: Finish your Final Project. Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners) Done!
Subsections of Homework
Week 1 HW: Principles and Practices
To make things tidy, I decided to answer most of the questions about the biological engineering application in sections of a separate project idea page.
PLEASE NOTE: on the project page accessible through the link I have been unable to show two images: an image of Van Gogh’s Sunflowers painting and a diagram showing how I’ve scored the actions I’ve suggested. It doesn’t appear to load the image, yet the build log indicates it’s a success. I’m not sure what the problem is but it means you won’t see those two pictures.
Q1. First, describe a biological engineering application or tool you want to develop and why.
Q2 Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Q3 Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of “Failure” and “Success”).
Q5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answer 1
The slides and other sources indicate DNA polymerases have a error frequencies of about 10-6 mutations/bp and the human genome has between 3,100,000,000 and 3,200,000,000 base pairs of DNA. 12 Cells employ various repair mechanisms. Some errors are corrected during replication through a process called proofreading. After replication, mismatch repairs can reduce the rate even further. 3
Answer 2 There are very many ways that proteins could be coded, but most of the combinations do not result in stable three dimensional conformations. The 4th edition of the Molecular Biology of the Cell notes that:
“Since each of the 20 amino acids is chemically distinct and each can, in principle, occur at any position in a protein chain, there are 20 × 20 × 20 × 20 = 160,000 different possible polypeptide chains four amino acids long, or 20n different possible polypeptide chains n amino acids long. For a typical protein length of about 300 amino acids, more than 10390 (20300) different polypeptide chains could theoretically be made. This is such an enormous number that to produce just one molecule of each kind would require many more atoms than exist in the universe.
Only a very small fraction of this vast set of conceivable polypeptide chains would adopt a single, stable three-dimensional conformation—by some estimates, less than one in a billion. The vast majority of possible protein molecules could adopt many conformations of roughly equal stability, each conformation having different chemical properties. And yet virtually all proteins present in cells adopt unique and stable conformations.” 4
Questions relating to Dr. Le Proust’s slides:
What’s the most commonly used method for oligo synthesis currently?
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Why can’t you make a 2000bp gene via direct oligo synthesis?
Answer 1:
Solid-phase phosphoramidite chemistry. According to Twist Bioscience:
“Phosphoramidite chemistry is the gold standard method for DNA synthesis that has been used in the industry for almost 35 years. Since its discovery, its simplicity and high efficiency have allowed large volumes of oligonucleotide sequences to be synthesized up to 200 base pairs in length. Currently, it is the only commercially viable chemistry able to provide the volume of DNA required by the synthetic biology market.” 5
Answer 2
According to Pichon, it is difficult to synthesise sequences longer than 150 nucleotides because the theoretical yields for larger sequences is low. When solid phase phosphoramidite chemistry is used to make the sequences, the efficiency of coupling between successive oligonucleotides becomes less efficient as the sequence becomes longer. In using this process, the longer the sequence, the more likely that the resulting sequence will contain insertion or deletion errors. 6
Answer 3
If two nucleotides are used to make a single base pair, then a 2000 bp gene would require 4000 nucleotides. If synthesising sequences of more than 200 nucleotides is difficult, much more than that would be impractical. Yin describes how “…the state of the art chemical synthesis methods cannot reliably produce oligos longer than 200 nt.” The author explains that for some longer sequences, “…if a sequence contains higher order structures with unusual stability, the PCR assembly method may not function effectively.” 7
Question about Prof. Church’s slide #4
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Answer
According to the Cleveland Clinic page on amino acids, the human body needs 20 amino acids to make the all the proteins that make it function properly. However the page and a few other sources only count nine rather than ten essential amino acids. These amino acids are needed by all animals:
One article names Arginine as a conditionally essential amino acid that is essential in certain life stages or when certain physiological stresses are present. 9
If the genetically altered dinosaurs could not produce lysine, they would presumably die before they even reached maturity.
McInerney P, Adams P, Hadi MZ. Error rate comparison during polymerase chain reaction by DNA polymerase. Molecular biology international. 2014;2014(1):287430. ↩︎
Base Pair, National Human Genome Research Institute, February 8, 2026. ↩︎
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002. The Shape and Structure of Proteins. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26830/↩︎
Yin Y, Arneson R, Yuan Y, Fang S. Long oligos: direct chemical synthesis of genes with up to 1728 nucleotides. Chem Sci. 2024 Dec 18;16(4):1966-1973. doi: 10.1039/d4sc06958g. PMID: 39759933; PMCID: PMC11694485. ↩︎
The project proposal is to create a bacterial painting of Vincent van Gogh’s Sunflowers (1888), which hangs in London’s National Gallery. Bacterial pigments will be applied to an agar medium inside a petri plate. The choice of which biopigments to use will be based on those whose hues best correspond to the pigments which appear in parts of the original painting.
I would like to use the art work as an example of showing how living bacteria species can produce colours that could be analogous to those which have traditionally been provided by chemical pigments. I would take time lapse photographs at different points between finishing the work and when the bacteria had died. This is meant to reflect the natural change in colours shown in the original artwork.
By using bacteria to help make the art work, I am hoping:
it encourages people to develop an interest in micro organisms, who often goes unnoticed in our day-to-day living.
it encourages discussions about whether microorganisms could - or should - provide a viable alternative to synthetic colourants
it encourages people to appreciate the aspect of transience in Bioart by referring to an original artwork which itself showing transience in its colours
I would like to do this particular project for two reasons:
bacterial painting is a well-established activity that appears to have gathered significant recommendations relating to ethics, biosafety and biosecurity.
it is a piece of Bioart, which would seem to encounter different benefits-vs-harms issues because it balances an aesthetic rather than a utility-based outcome versus potential risks.
I’ve chosen to use van Gogh’s Sunflowers painting for the following reasons:
its subject matter is likely to remain recognisable if it becomes more abstracted in the process of drawing it on a petri dish with a low resolution of ‘bacterial pixels’.
the pigment composition of the painting is well-understood
it is an example of an art work which is undergoing colour change, which may make it more relatable to the changing nature of a work made with living organisms.
I believe that my project is ethical. Its risks can be reduced by adopting aspects of governance which considers best practice for handling biomaterials. Its benefits include promoting an interest in viewers to learn more about microorganisms. Its benefits and risks can also be better articulated by identifying the key ethical questions involved with presenting microorganisms in art.
2 Developing a policy framework to make the project contribute to an ethical future of bioengineering
I would derive a governance framework based both on the general framework provided in the assignment and a set of ethical questions that would be specifically about microbial art. Fawcett and Dumitriu, who have collaborated as scientist and artist respectively, have produced a useful commentary outlining key questions for displaying microbiological Bioart. They are:
What are the overall aims and potential benefits of the piece?
Do these aims require, or are enhanced by, the use of the ‘real thing’?
If there are risks, how can they be minimised, and how do they compare to existing public displays?
The policy goals for this project will include the following:
Beneficence of artistic expression. As a piece of Bioart, the outcome must demonstrate some benefit borne from its aesthetic.
Justification for using living microorganisms. As a piece of Bioart, the use of living microorganisms must be at least important if not necessary for creating the outcome.
Biosecurity. As a bioengineering activity, the project must prevent harm presented by biosecurity concerns.
Biosafety. As a bioengineering activity, the project must prevent harm presented by biosafety concerns.
Feasibility. As a bioengineering activity, the project must be practical enough to do and not present too great an expense of resources.
3 Ideation of actions to support policy framework
3.1 Policy goal: benificence of artistic expression actions
3.1.1 Action: acquire naturally occurring bacteria which happen to produce colours that match those in the original painting.
Purpose: to use naturally occurring species of bacteria that are not genetically altered and which produce accurate analogous hues to the hues shown in Sunflowers. These bacteria contribute both to the aesthetic of the art work and provide a more interesting variety of stories of the ecosystems in which they live.
Design: Identify ethical vendors who stock naturally occurring colour-producing bacteria. Ensure that they have quantifiable hue ranges that can be matched with those of analogous pigments used in Sunflowers.
Assumptions:
There is a great enough variety of naturally occurring bacteria that happen to produce accurate matches for Sunflower pigment colours.
Risks of Failures and Success:
This action could fail if the variety of accurate colour matches with Sunflowers pigments is low.
Effectiveness: High
3.1.2 Action: share credit for the artwork with the microorganisms
Purpose: Supports raising awareness of microorganisms with the viewer.
Design: Name and describe the species and modifications to bacteria used to produce colour. In the display, describe the species of bacteria used for the artwork. Describe the genes responsible for creating the colour. Indicate if those genes were the result of genetic modification, either to a normal gene or by inserting a gene. If relevant, describe the source organisms that provide transferred genes which are used to produce the colours.
Assumptions:
Detailed provenance for mail-ordered microbial colourants is available.
Risks of Failures and Success:
Failure to provide adequate information undermines the interest of highlighting the role and importance of the organisms which contributed to the artwork.
Effectiveness: High
3.1.3 Action: acquire genetically altered bacteria which produce colours that match the original painting.
Purpose: to use genetically altered species of bacteria that are programmed to express proteins whose colours match the hues shown in Sunflowers. These bacteria contribute would mainly contribute to the aesthetic of the artwork, but would do less to promote the biodiversity of naturally occurring bacteria.
Design: Identify ethical vendors who stock bacteria which have been genetically altered to fluoresce with specific colours. Ensure that they have quantifiable hue ranges that can be matched with those of analogous pigments used in Sunflowers.
Assumptions:
It is easy to obtain hue information about microbial pigment sources.
Risks of Failures and Success:
Without sufficient information about the hue ranges produced by the vendor’s bacterial sources, they may not relate well to the hue ranges associated with the chemical pigments used by van Gogh.
Effectiveness: Medium
3.1.4 Action: acquire naturally occurring bacteria which happen to produce colours that match the original painting.
Purpose: to use naturally occurring species of bacteria that are not genetically altered and which produce accurate analogous hues to the hues shown in Sunflowers. These bacteria contribute both to the aesthetic of the art work and provide a more interesting variety of stories of the ecosystems in which they live.
Design: Identify ethical vendors who stock naturally occurring colour-producing bacteria. Ensure that they have quantifiable hue ranges that can be matched with those of analogous pigments used in Sunflowers.
Assumptions:
There is a great enough variety of naturally occurring bacteria that happen to produce accurate matches for Sunflower pigment colours.
Risks of Failures and Success:
This action could fail if the variety of accurate colour matches with Sunflowers pigments is low.
Effectiveness: Medium
3.2 Policy goal: minimising need for using natural resources
3.2.1 Action: use only the protein colourants produced by the bacteria rather than the bacteria themselves in the art work.
Purpose: Provides a way of producing colourant that better supports biosecurity and biosafety concerns.
Design: Order colourants which contain only the coloured byproducts of bacteria rather than the bacteria themselves. Use a normal paintbrush, bacteria-derived watercolourants and paper to reproduce van Gogh’s Sunflowers painting.
Assumptions:
It is possible to obtain colourants which only contain the colour-producing proteins but not microorganisms themselves.
Risks of Failures and Success:
This option greatly reduces the biosafety and biosecurity risks. However, the end product is unlikely to qualify as Bioart, because many artists define Bioart as working collaboratively with a living organism, not its byproducts. It might encourage people to think of bacteria-derived colourants as an alternative to chemical paints, but the project would fail to emphasise the importance or contribution of microorganisms.
Effectiveness: High
3.2.2 Action: acquire genetically altered variants of common bacteria which produce colours that match those in the original painting.
This is essentially the same as ‘Acquire genetically altered bacteria which produce colours that match the original painting’. In this context, the main reason to use it is to reduce the amount of naturally occurring bacteria that are collected, which perhaps may come from fragile overharvested environments.
Effectiveness: Medium
Action. Acquire naturally occurring bacteria which happen to produce colours that match those in the original painting.
This action has already been defined to support the Beneficence of artistic expression policy goal. However, in this context, it is the least preferable option for reducing the reliance on natural sources of bacteria.
Effectiveness: Low
3.3 Policy goal: non-malfeasance
3.3.1. Action: obtain training about biosecurity and biosafety regulations and best practices
Purpose: to ensure that I’m able to adequately implement the practices that reduce the likelihood and impact of events relating to biosecurity and biosafety.
Design: Take the required laboratory practice training class, take notes and ask to have access to a recording.
Assumptions:
None
Risks of Failures and Success:
If this step failed I would not be allowed or want to embark on the project.
Effectiveness: High
3.3.2 Action: consult laboratory supervisor to verify ongoing compliance with regulations and best practices about biosecurity and biosafety
Purpose: to have my own practices spot-checked by laboratory staff who have great expertise in safely handling microbiological materials in the lab.
Design: During labs, actively seek the advice from laboratory staff about how I can improve my techniques.
Assumptions:
None
Risks of Failures and Success:
If this step failed I would not be allowed or want to embark on the project.
Effectiveness: High
3.3.3 Action: record the ‘performance’ of the microorganisms evolving the painting rather than the painting itself
Purpose: Captures an important aspect of Bioart, which is co-development of an art work between artist and the bacteria.
Design: When the art work begins to show colour, take a sequence of pictures or ideally a time-lapsed video that lasts until all the bacteria have died. Perhaps consider slowly heating the finished product to accelerate decay so that it is more practical to film. When the bacteria have died, autoclave the petri dish and everything in it. Safely dispose of the remains of the Bioart and allow the work to live on only through a video or a sequence of timed snapshots.
Assumptions:
A video camera can track the decay of the bacterial painting.
Risks of Failures and Success:
If the video camera footage or still shots don’t turn out well, it may compromise the Bioart goals.
Effectiveness: Medium
3.4 Policy goal: feasibility
3.4.1 Action: prefer colour-producing bacteria that may already be in stock in the lab.
Purpose: to reduce the time needed to order new materials when I could buy materials that are already in stock.
Design: Check with the London Lab whether it already has some colour-producing bacteria in stock.
Assumptions:
The lab retain excess stock.
The colours will match.
Risks of Failures and Success:
This action could fail if there are no bacterial sources kept in stock in the London Lab, or if the ones that are retained do not match the pigment colours shown in Sunflowers.
Effectiveness: High
3.4.2 Action: prefer colour-producing bacteria that are made in the UK.
Purpose: to reduce the time and cost associated with obtaining reagents from other countries.
Design: Try to obtain colour-producing bacteria sources from the UK.
Assumptions:
It is cheaper to obtain desired bacterial sources from the UK than ordering it from abroad.
Risks of Failures and Success:
Failure will mean the bacteria have to be obtained from sources abroad.
Effectiveness: Medium
3.4.3 Action: consider heating the bacterial painting to make it decay quickly.
Purpose: to make photographic recording of how the painting decays cheaper to do.
Design: Once the painting has been finished and begins to show colour, perhaps slowly heat it to help speed up the evolution and then decay of bacterial growth. Denaturing coloured proteins may cause them to change hue and thereby make the work relatable to the changes found in the original Sunflowers.
Assumptions:
Heating will adequately simulate what would happen to the painting if it were left to evolve until all the bacteria died.
Risks of Failures and Success:
If heating just rapidly denatures the painting, it may not have a meaningful decay and therefore would not be worth doing.
Effectiveness: Low
4 Evaluating effectiveness of actions that support policy goals
Based on these actions, I created the chart below and assigend High = 3, Medium = 2 and Low = 1.
5 Discussions of priorities and assumptions
Let’s assume that the list of prioritised actions will be viewed by a project review board that would allow me to do this project. Initially the most important part of the project is to promote the non-malfeasance policy goal. Without demonstrating those actions for minimising risks associated with biosecurity and biosafety the project should not proceed. In fact, for this exercise I’m not sure whether it makes sense to view either of these with a priority. Priority tends to suggest to me the idea of optionality and I can’t imagine a project being able to proceed without adequate training in laboratory techniques to minimise biosecurity and biosafety concerns.
Feasibility actions would be the next priority area of policy actions to consider because the project must be practical to implement in the lab. For example, I may find out that the variety of hues available through colour-producing bacteria is very limited or would be prohibitively expensive to diversify.
Beneficence of artistic expression actions would be the third most important area to prioritise. Once I become familiar with catalogues for ordering colour-producing bacterial sources, I should be able to acquire extra information about each species and at least some understanding of the genes which are responsible for making coloured proteins. I would not be suprised if matching colours with the original painting pigments may be a very rough guess! But, I do expect there would be enough basic bacterial hues to work with to make the work recognisable as a version of Van Gogh’s Sunflowers painting.
The lowest priority area is the actions for minimising the use of natural resources. I wouldn’t be going out to obtain colour-producing bacteria myself. These would be ordered from a catalogue. Let’s assume that the provider will do ethical sourcing of bacteria in a way which will not undermine fragile ecosystems. Once they obtain a sample of bacteria, presumably it is easy to replicate them as much as they want. This use of natural resources then would seem very different than for example, trying to make Bioart using loose fallen feathers gathered from endangered birds living at the edge of existence in a dwindling patch of rainforest.
In the process of identifying areas of policy that would govern the project, I encountered two issues. The first is that when I initially considered the need to minimise natural resources, I realised the ethics of using animals to produce art seems different for microbes than large animals. When Eduardo Kac produced a genetically engineered fluorescent rabbit as a piece of Bioart, it caused great controversy. However, if someone wants to use genetically engineered bacteria to produce Bioart, this seems to have already become acceptable.
From my own previous research into Bioart, I would conclude that human beings will be more empathetic about perceived harm to animals if they live at our scale of living (e.g. rabbits) and would appear to have the ability to experience pain. I also think that whereas humans have had thousands of years to work out their own sense of morality towards animals that live at their scale, they are still trying to figure out what is ethical to allow in relation to creatures that only became visible a few hundred years ago, and the world’s major religions had already long developed.
Another issue I encountered was observing a tension in goals between providing the best aesthetic outcome for bacterial pigments versus providing the most impactful message about biodiversity in the microbial world. I suspect that it is probably easier to get accurate colour matches between bacterial and oil paint pigments through genetically engineered bacteria versus using naturally occurring bacteria that produce different colours. I suspect that it is more cost effective to cultivate versions of common bacteria that have been altered in a specific gene which produces a coloured protein of a specific range of hues.
Mapping original pigments to biopigments
In the table below, the first two columns come from:
Roy A. National Gallery Technical Bulletin, Volume 37. Yale University Press; 2016, p. 68.
Painting Feature
Pigment Analysis
Potential Biopigment
Biopigment Context
The light brownish-grey ground, left-hand edge
Pb Lead white
Mid light blue of edge of table, left
French ultramarine
Intense dark blue streak on sunflower
Pb,Si,Al,Cr,Cu,Zn. Chrome yellow: French ultramarine
Intense dark blue, centre of sunflower
French ultramarine
Intense cold green of sunflower
Cr,Zn (Pb). Viridian, some chrome yellow
Mid yellow-reen petal
Cu,As,Pb,Cr,Zn Emerald green: chrome yellow
Mid yellow-green leaf
Cu, As, Pb, Cr, Zn. Emerald green:chrome yellow
Light dull greenish-yellow petal
Pb, Cr Chrome yellow
Pale yellow slightly greenish background,right-hand side
Zn Contains zinc white
Dark yellow tabletop
Pb, Cr (Zn) Chrome yellow
Dark yellow of sunflower (brighter orange-yellow below surface)
Yellow-green thickest impasto of uppermost sunflower
Pb, Cr, Zn, Fe(Mn, Al, Si). Chrome yellow: ochre
Very intense deep red glaze from sunflower, left-hand side
Red lake; red ochre; French ultramarine
Pale yellow of vase over pale pink
Traces of vermilion beneat the surface
Notes:
French ultramarine is also known as synthetic ultramarine.
Viridian is also known as Hydrated chromium (III) oxide
Emerald green is also known as copper acetoarsenite
Week 2 DNA Read, Write and Edit
Assignment 2
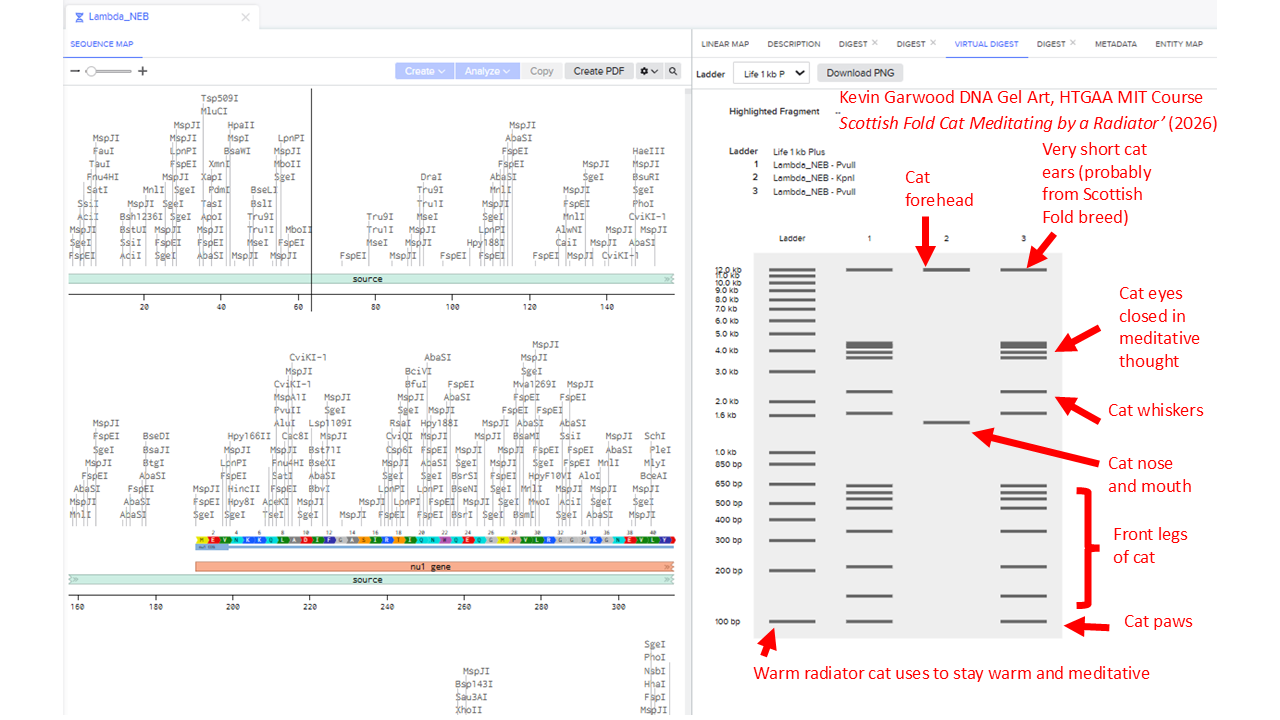
I created a Benchling account, loaded up the Lambda DNA, and then tried different combinations of the following restriction enzymes.
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
I note that the Automation Art tools produces randomly created electrophoresis ladders, but I excluded Ndel, Pvull and Xhol - because they were not in the list we were supposed to use.
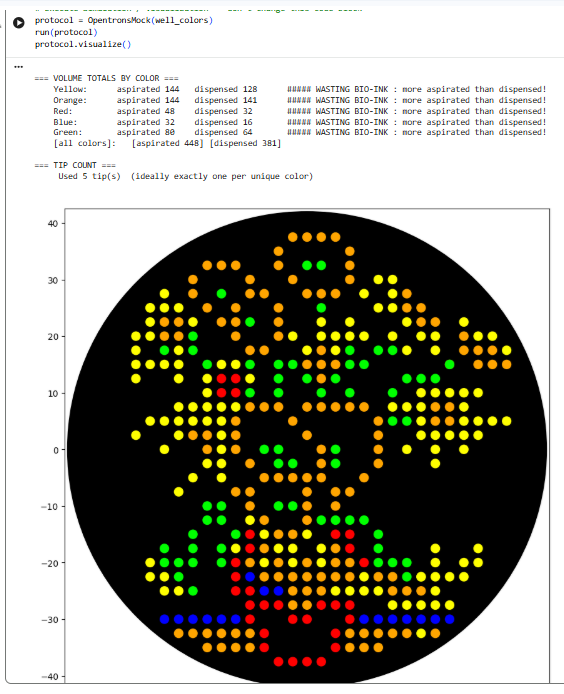
I found it difficult to produce virtual digest ladders that could be combined into recognisable artistic shapes. To get an idea of the range of possible patterns, I systematically looked at choosing combinations of 1, 2, 3, 4 and 5 enzymes.
I’ll include some examples of this brute force way of assessing potential patterns. But that didn’t work well. So I widened my enzyme list to include the enzyme Pvull as well. I ended up with this pattern:
Part 3: DNA Design Challenge
3.1
I’ve chosen the sequence for staphyloxanthin, the protein found in the bacteria Staphylococcus aureus that creates a deep yellow colour that might be similar to the chrome yellow pigment van Gogh used to paint the centres of his sunflowers in the ‘Sunflowers’ painting hanging in the National Gallery.
3.2 Reverse translate
I found the Uniprot entry for 4,4’-diapophytoene synthase, which is used in the biosynthesis of the yellow-orange carotenoid staphyloxanthin. In Benchling, I imported an AA sequence and specified ‘A9JQL9’ and ‘Uniprot’ to import from a database. I selected the entire protein sequence, right-clicked and clicked ‘Backtranslate’. From there, I obtained this DNA Sequence:
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
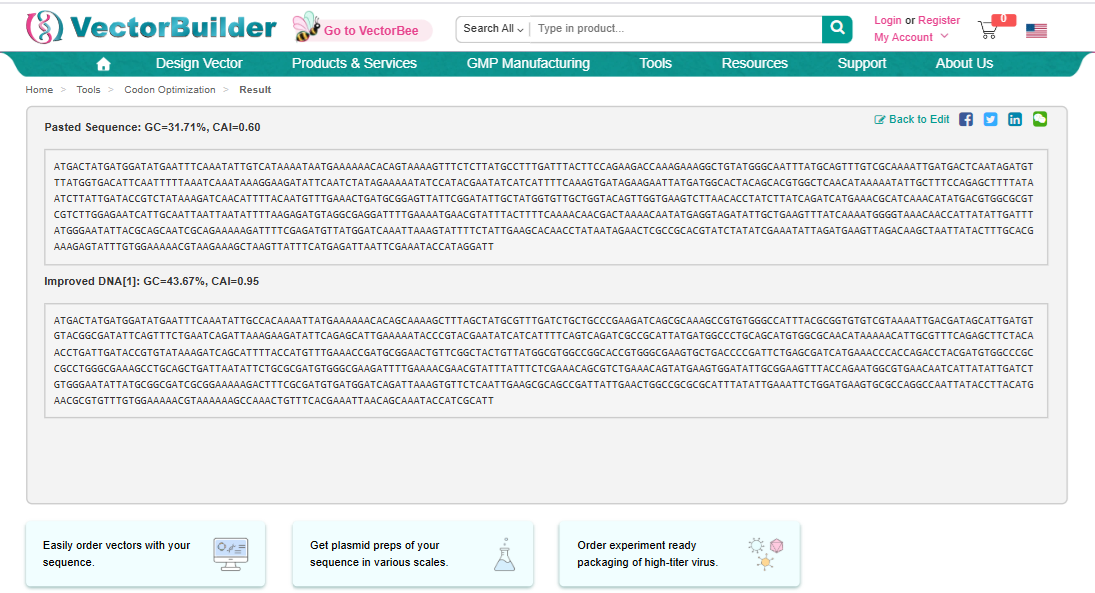
I used Vector Builder’s Codon Optimisation Tool
and specified the sequence from 3.2 to be inserted into E coli strain K-12 substr: MG1655.
I’d follow the instructions in Homework Week 2 ‘Prepare a Twist DNA Synthesis Order’ to create an expression cassette, which would be designed to drop into a plasmid vector. This would involve specifying a sequence comprising: a promoter, a Ribosome Binding Site (RBS), a start codon, the optimised codon sequence that will be designed to make the staphyloxanthin work best with E. coli, the 7x His Tag, a stop codon and a terminator. I’d carefully annotate each of these components and export a Fasta file that I would then upload to Twist. The order from twist would presumably arrive in London as the freeze-dried sequence for the cassette.
Next, I’d use PCR to amplify the insert sequence (comprising all the components I specified in Benchling) and then add it to a tube that contains the three enzymes used in Gibson Assembly cloning. First, the T5 exonuclease would cause the 5’ ends of the insert sequence and linearised vector (plasmid backbone) to be chewed back to create overlaps. Polymerase would begin to fill in at the overhangs to prevent excessive enzyme chewing. When the overhangs become stable, taq ligase would complete the fusion of the inserted staphyloxanthin sequence into the backbone sequence of the plasmid. The recommended temperature for this would be about 50’C.
In this process, the enzymes would cut at specific places on the plasmid, which would create a gap for the inserted sequence. I’d need to select appropriate primers: a forward primer that would create an overlap between the sequence leading to the beginning of the gap and the beginning of the inserted sequence. Then I’d need to pick a reverse primer that would create an overlap between the end of the inserted sequence and the beginning of the sequence that starts at the other end of the gap. The end product would be a plasmid where the sequence had been inserted into the gap.
The plasmids would be then be inserted into E. coli by applying temporary heat shock to the cells. The shock would create temporary pores in the membrane of the bacteria which would allow the plasmid to pass inside. Once inside, the E. coli’s own machinery would express the genes encoded into the plasmids as if it were its own. The promoter encoded into the insert sequence would signal the optimised staphyloxanthin sequence to be first transcribed into RNA and then later translated into a protein that would then produce the yellow colour associated with the staphyloxanthin gene.
Part 4: Prepare a Twist DNA Synthesis Order
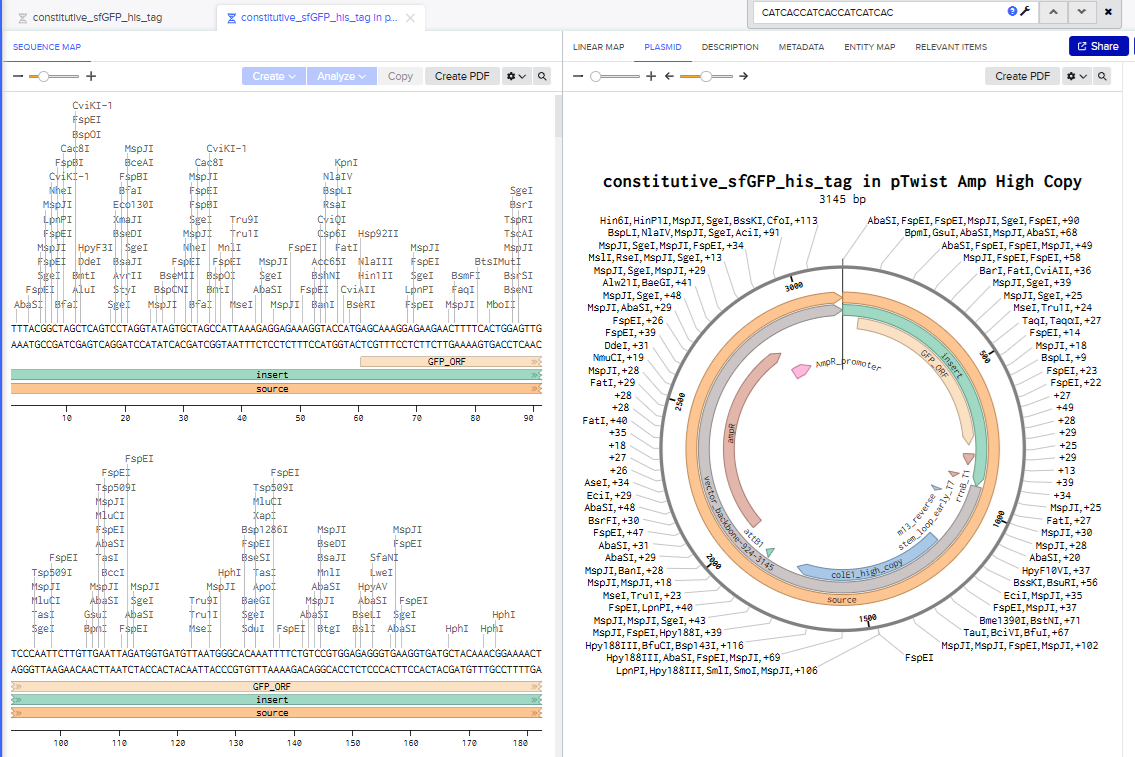
I went through all the instructions for this part using Benchling and Twist, and the final plasmid looked like this in Benchling:
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would want to sequence the protein Proteorhodopsin. When I was working on my Masters dissertation in Art History, I came across this paper: “Frangipane, Giacomo, et al. “Dynamic density shaping of photokinetic E. coli.” Elife 7 (2018): e36608.”
The authors had created E. coli that used proteorhodopsin to make them respond to light. When light was shone on them, it provided energy that propelled the bacteria. They were able to manipulate combinations of light and darkness to help them clump together in a way that looked like a photographic negative.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
I would use Illumina Sequencing (Short-Read), because they have often been used for metagenomic and transcriptomic sequencing of the genes for Photorhodopsin.
This paper about microbial rhodopsin dynamics by Laura Gómez-Consarnau makes use of the
Illumina PE150 platform: Gómez-Consarnau, Laura, et al. “Unexpected microbial rhodopsin dynamics in sync with phytoplankton blooms.” Nature Communications (2025).
Is your method first-, second- or third-generation or other? How so?
The method uses short reads and is an example of Next Generation (Second generation) sequencing.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
I read this paper by Dahui Qin to learn more about how NGS worked. Its main steps include:
DNA Fragmentation. Involves breaking up DNA into many short segements using various methods such as enzymatic digestion. The relevant sequences of interest are isolated using complementary probes.
Library preparation. This involves preparing the segments in a uniform way that makes it easier for sequencing primers to bind to fragments.
Sequencing. Allows massive parallel sequencing of all the fragments at the same time.
Analysis. Bioinformatics analysis tools are used to support base calling, read alignment, variant identification and variant annotation. The fragments are compared with a reference sequence to identify mutations and then all the fragment sequences are stitched together to create a complete sequence.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
As part of my final project, I’m looking at doing something similar to Part 4, but using a plasmid where sfGFP can be expressed in the presence of high levels of carbon dioxide. From what I’ve been gathering, the easiest way of doing this is to make a less specific function that would make it fluoresce green in the presence of (carbonic) acid. The main drawback is that it would then react to any acid, not just acid resulting from carbon dioxide dissolved in water. Another approach involves having the bacteria detect HC03 but I’m not yet sure how to do that.
I would be looking at a whole-cell biosensing bacteria that would likely use a cadBA promoter, which is supposed to be active as an acid stress response in E. coli. The promoter would act as a kind of switch to turn the GFP on.
See some famous examples of DNA design
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
I would be using Twist to synethesise the plasmid-containing bacteria that would fluoresce green in the presence of CO2, probably indirectly triggered by a response to an acid. So perhaps I’d be building a biosensor that was more about detecting acid than CO2.
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I’m not sure if this is relevant to what I’d like to do. I’m not wanting to synthesize DNA based on something that hasn’t been sequenced before. I’m actually more interested in using a documented promoter to turn the GFP on. I’d be doing something really similar to Part 4.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I’d want to edit the E. coli sequence so I can stuff the circuit I want into it.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
I’d be using Twist to synthesise a genetically altered E. coli that would glow green in the presence of CO2 or perhaps a strong acid stress instead.
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
I would be editing a plasmid in the way Part 4 outlines and getting Twist to do it.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Week 3 Lab Automation
Assignment 3
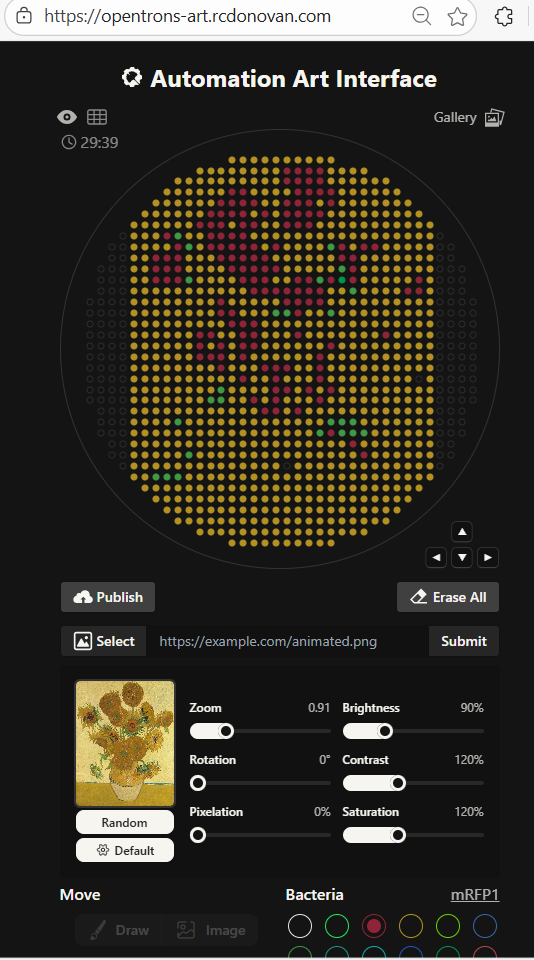
Following on from last week’s assignment, I decided to use Vincent van Gogh’s Sunflowers painting (the one hanging in the National Gallery) for my art subject. I tried to download an image of the painting and upload it to the Opentrons automated art interface. Importing it made some artistic effects I didn’t want - it flooded the background with yellow, left out the blue streaks and didn’t do much to distinguish between orange and yellows. Importing it created something that wasn’t recognisable.
I was really impressed by the tool but opted to create my own from scratch. I made some decisions like hollowing out some of the flower petals because if I filled them all with orange and yellow they would look unrecognisable. I could have used the automation art interface to hand craft the image, but I wanted to learn more about the mechanics of how the opentrons commands would work rather than immediately rely on automatically generated code.
Like the autogenerated code, I concluded it would be more efficient to do successive passes of placing droplets by colour: first one colour, then another and the next. I spaced my droplets at 2.5 mm. I tried using a larger distance between drops, but the resolution dropped and I couldn’t make a recognisable painting of Sunflowers with the space I had available. If they bleed together, that might actually work - because the painting is itself slightly abstract and it wasn’t meant to appear to be too realistic.
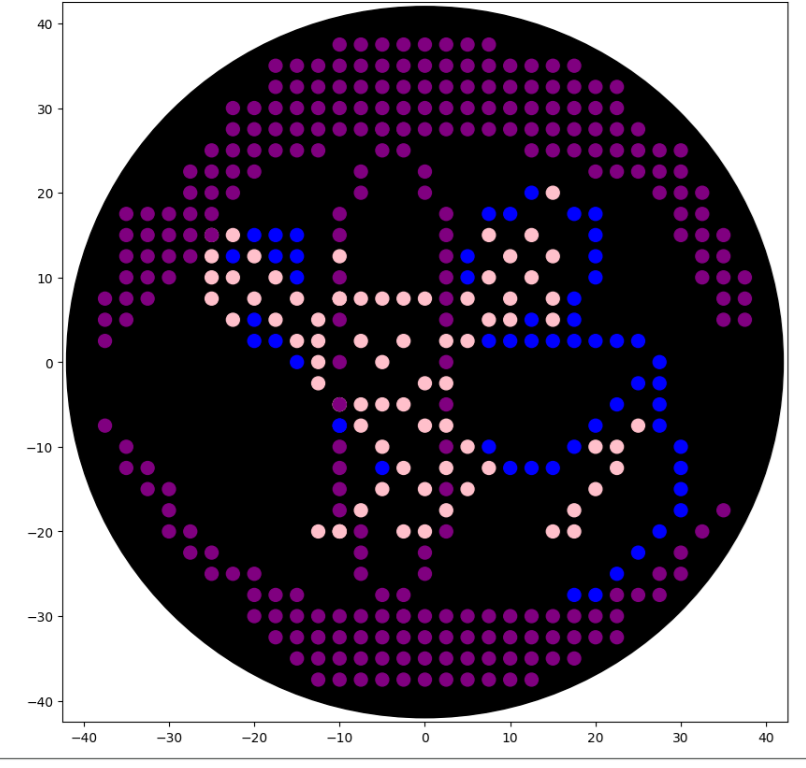
Note that because the lab only had colours for blue, pink and purple in stock, I had to rework my art. I call this one ‘Rattlesnake Dinner’ - it is meant to show how a snake might view a mouse through a thermal vision perspective.
Post Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The paper I chose was:
Taguchi S, Matsuzawa R, Suda Y, Irie K, Ozaki H. Investigating the effects of liquid handling robot pipetting speed on yeast growth and gene expression using growth assays and RNA-seq. Micropublication Biology. 2025 May 13;2025:10-7912.
Available here
The paper notes that: “.the influence of pipetting speed on biological experiments, —particularly when systematically varied using liquid-handling robots and evaluated through gene expression and cell growth—remains poorly investigated.” It conducted multiple experimental runs on an Opentrons to determine how the variation in pipetting speed influenced gene expression of Saccharomyces cerevisiae.
The authors write: “In conclusion, within the range of pipetting speeds investigated, variations in pipetting speed did not impact the maximum relative growth rate and the gene expression profiles of yeast.” The finding implies that if the Opentrons OT-2 were run at its top pipetting speed, there would not be much difference in gene expression of yeast colonies.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
For my final project, I plan to 3D print multiple objects based on models derived from various Green Man sculptures found in architecture (and particularly churches). I have already begun to use the application Polycam to take hundreds of pictures of Green Man figures in Ely Cathedral. I’ve been able to convert those into *.STL files which I hope to eventually print in the Lifefabs Lab. I will be experimenting with which sculpture provides the best surface properties to grow colonies of GFP-containing bacteria.
Week 4 Protein Design Part 1
Part A: Conceptual Questions
Choosing 9 of 11 questions to answer
Q1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
I’ve made some assumptions:
The meat mass does not include air or water.
The meat mass does not include organic materials that are not amino acids
A Dalton is a unit of mass used to express atomic and molecular weights. I used this converter to determine how many Daltons are in 500 grams of organic material. 500 g = 3.011086821E+26 daltons. If an amino acid is 100 daltons, then 500 g would contain 3.011086821E+24 molecules.
Q2 Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans digest beef, the process breaks the organic materials down into base materials that their bodies can use to build amino acids according to human DNA. The DNA found in the beef is not transferred and does not displace the DNA in our bodies.
Q3 Why are there only 20 natural amino acids?
Doig examines why the 20 standard amino acids were selected by Nature. 1 One theory is that by chance those 20 were selected and became established when there could have been alternative sets. But Doig seems to think their properties alone and in combination provided a kind of almost ideal set of amino acids. He writes:
“…they were selected to enable the formation of soluble structures with close-packed cores, allowing the presence of ordered binding pockets. Factors to take into account when assessing why a particular amino acid might be used include its component atoms, functional groups, biosynthetic cost, use in a protein core or on the surface, solubility and stability. Applying these criteria to the 20 standard amino acids, and considering some other simple alternatives that are not used, we find that there are excellent reasons for the selection of every amino acid. Rather than being a frozen accident, the set of amino acids selected appears to be near ideal.”
Q4 Can you make other non-natural amino acids? Design some new amino acids.
Q5 Where did amino acids come from before enzymes that make them, and before life started?
Cowing explains that amino acids could have formed in the early planetesimal bodies of the early solar system, far from the sun 2. Those bodies would contain large amounts of ice that could have been melted by the heat produced by radioactive materials. The melted water could have interacted with other volatile compounds to form amino acids. These could have been contained in meteorites which struck Earth, providing a large concentration of amino acids which could have led to life on Earth.
Amino acids could have also been created through chemical synthesis reactions happening on Earth. In 1953, Miller and Urey combined ammonia, hydrogen, methane and water vapour and in a flask and subjected it to electrical sparks. From this experiment, they were able to create eleven standard amino acids.3
Q6 If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect it would be left-handed. Novotny writes:
“a-Helices composed of L-amino acids are energetically more favourable in a right-handed conformation than in the left-handed mirror image of this arrangement due to steric hindrance between side-chain atoms and the main-chain carbonyl moiety. Conversely, D-amino acids will form more stable a-helices with a left-handed than with a righthanded conformation” 4
Q7 Can you discover additional helices in proteins?
So far in class we’ve discussed the alpha helix. In Tamar’s “Protein Structure Hierarchy”, the author describes the pi helix and the collagen helix. 5
Q8 Why are most molecular helices right-handed?
Because d-sugars direct the formation of right-handed helices and left hand versions don’t fit well.
MacDermott explains: “Biopolymer chirality is definitely determined by monomer chirality: l-amino acids can form only right-handed α-helices in protein secondary structures, and DNA naturally coils up into a right rather than left-hand B-form double helix because it is made of d-sugars. Why? Because of diastereomeric effects: d-sugars automatically direct the formation of a right-hand B-form double helix, because they simply do not fit well into a left-hand version. It is true that DNA does sometimes form a left-hand double helix, but this ‘Z-form’ helix is not a mirror image of the B-form, it has a totally different backbone conformation in order to be able to accommodate the d-sugars in a left-hand helix” 6
Eric Lindahl’s Youtube video explains that a helix such as the alpha helix is right-handed because it is due to L amino acids. 7
As a paper by Cole notes: “The remarkable predominance of right-handedness in beta-alpha-beta helical crossovers has been previously explained in terms of thermodynamic stability and kinetic accessibility.” 8
Q9a Why do beta sheets tend to aggregate?
Beta sheets are prone to intermolecular hydrogen bonding at the sheet edges with other beta sheets. Other parts of beta sheets are hydrophobic. They will tend to be attracted to each other and repelled by water, which will tend to bring them together.
Q9b What is the driving force for β-sheet aggregation?
Hydrophobic forces tend to repel beta sheets from water towards each other. Once they are close together, the attractive forces from intemolecular hydrogen bonding orders them together.
Q10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Q11. Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualisation
1. Briefly describe the protein you selected and why you selected it.
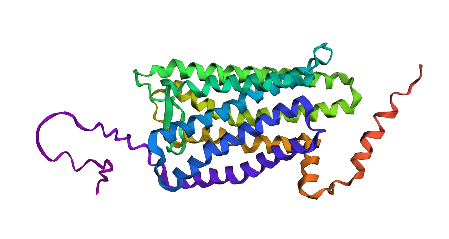
I’ve chosen rhodopsin, whic is a light-sensitive receptor protein found in the rod cells of the retina, and important for dim light vision. 9. The human protein for rhodopsin is described in P08100 · OPSD_HUMAN.
2. Identify the amino acid sequence of your protein.
The sequence is:
MNGTEGPNFYVPFSNATGVVRSPFEYPQYYLAEPWQFSMLAAYMFLLIVLGFPINFLTLYVTVQHKKLRTPLNYILLNLAVADLFMVLGGFTSTLYTSLHGYFVFGPTGCNLEGFFATLGGEIALWSLVVLAIERYVVVCKPMSNFRFGENHAIMGVAFTWVMALACAAPPLAGWSRYIPEGLQCSCGIDYYTLKPEVNNESFVIYMFVVHFTIPMIIIFFCYGQLVFTVKEAAAQQQESATTQKAEKEVTRMVIIMVIAFLICWVPYASVAFYIFTHQGSNFGPIFMTIPAFFAKSAAIYNPVIYIMMNKQFRNCMLTTICCGKNPLGDDEASATVSKTETSQVAPA
2a. How long is it?
The sequence is 348 amino acids long.
2b. What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The most frequent amino acid is ‘Alanine’, which appears 32 times.
2c. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
By using (Uniprot BLAST)[https://www.uniprot.org/blast] I identified 5 homologs belonging to the Chimpanzee, Northern white-cheeked gibbon, and the Western lowland gorilla.
2e. Does your protein belong to any protein family?
Rhodopsin belongs to the family of G protein–coupled receptors (GPCRs). 10
3. Identify the structure page of your protein in RCSB
I entered the Uniprot ID for rhodopsin, P08100, into the RCSB search and ran the query. I then
visualised the 3D representation through the page and obtained this result:
3a. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
It appears to have first been released between 2015 and 2016. Its resolution is 3.3 Å
3b. Are there any other molecules in the solved structure apart from protein?
The page is subtitled: “Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser”. Arrestin is another protein.
3c. Does your protein belong to any structure classification family?
It belongs to a GPCR superfamily. A paper by Zhou indicates: “Rhodopsin is a member of class A of the GPCR superfamily, which is a large group of cell surface signaling receptors that transduce extracellular signals into intracellular pathways through the activation of heterotrimeric G proteins” 11 I tried typing entering ‘p08100’ into the EBI’s SCOP tool and used this query, yielded two domains. I clicked on 5W0P A:1-324 which indicated it was a member of the Class A (rhodopsin) G protein-coupled receptor-like’
4 Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
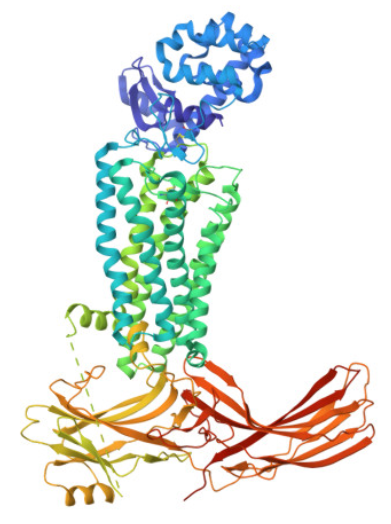
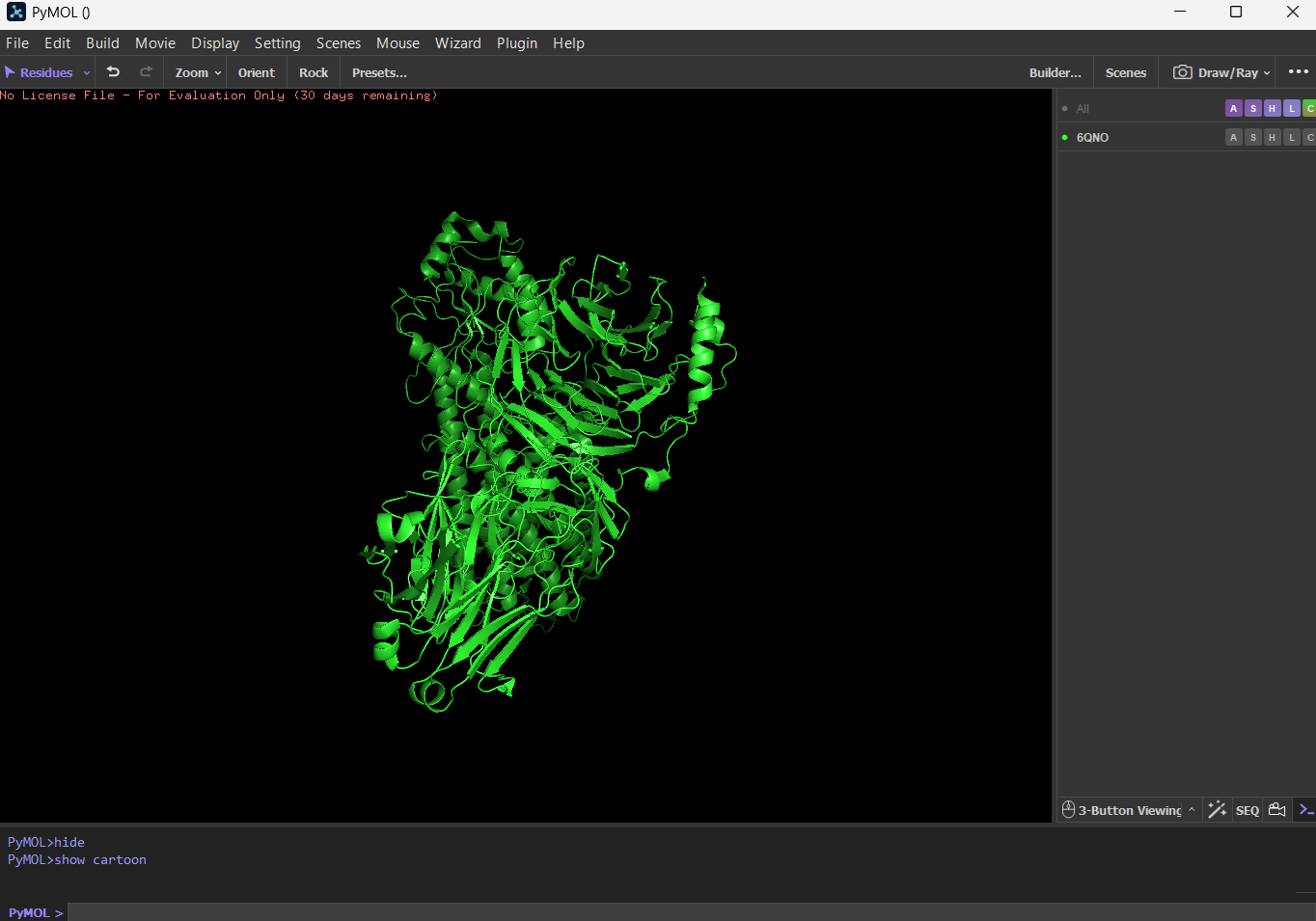
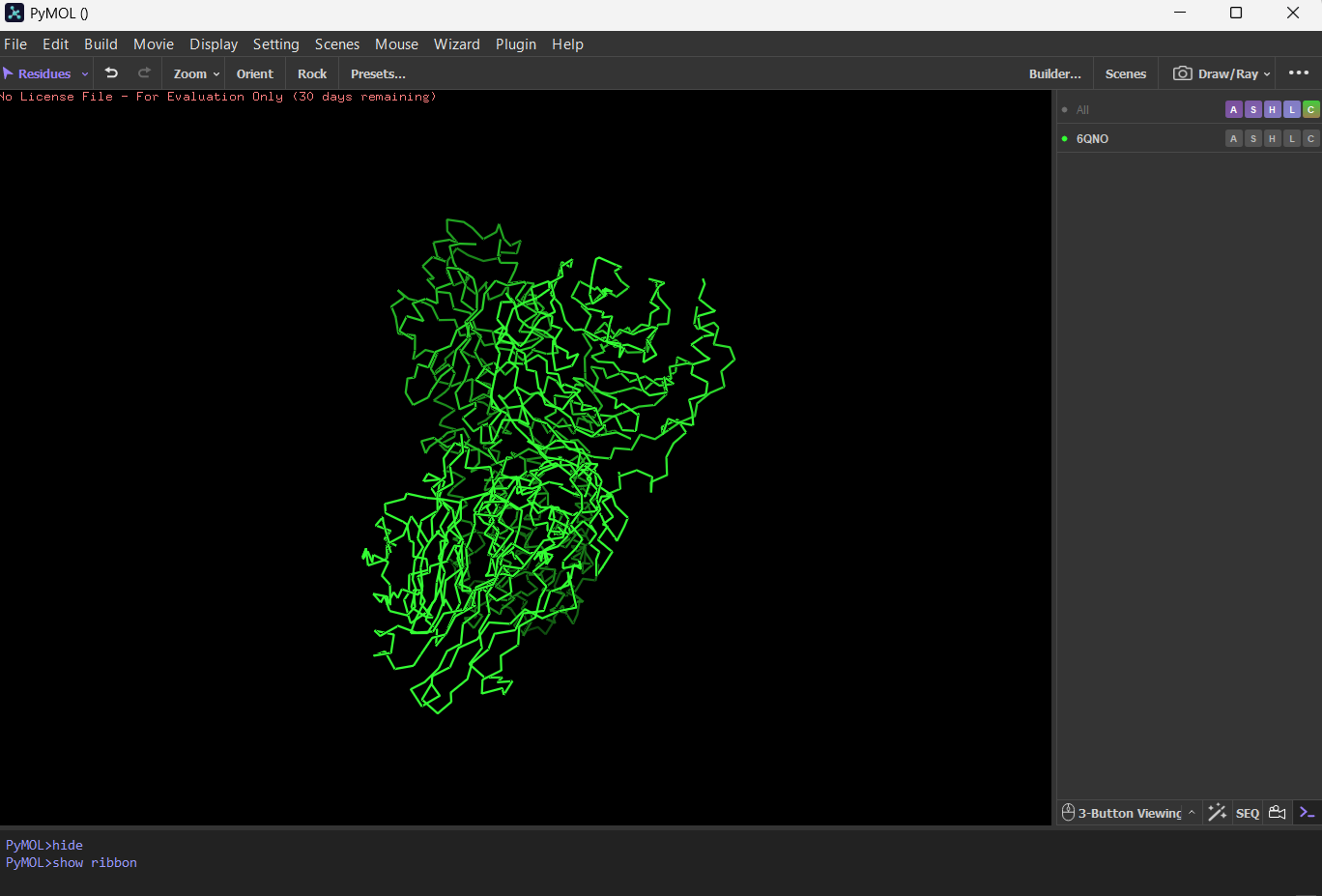
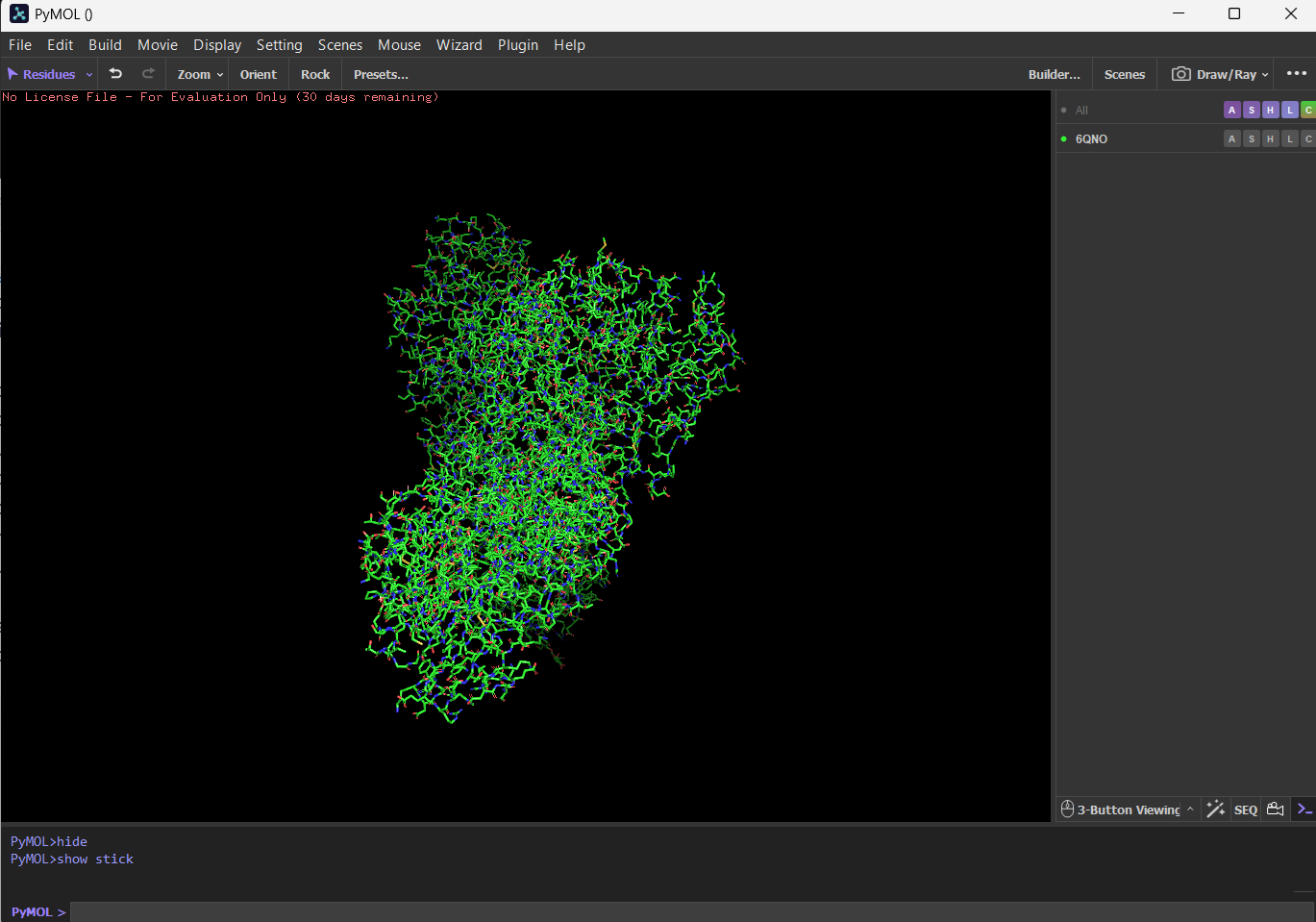
First I downloaded and installed PyMol. Then I searched for the PDB ID for ‘rhodopsin’ and obtained the ID 6QNO. I loaded the PDB structure for 6QNO and it provided me this:
CartoonRibbonBall and stick
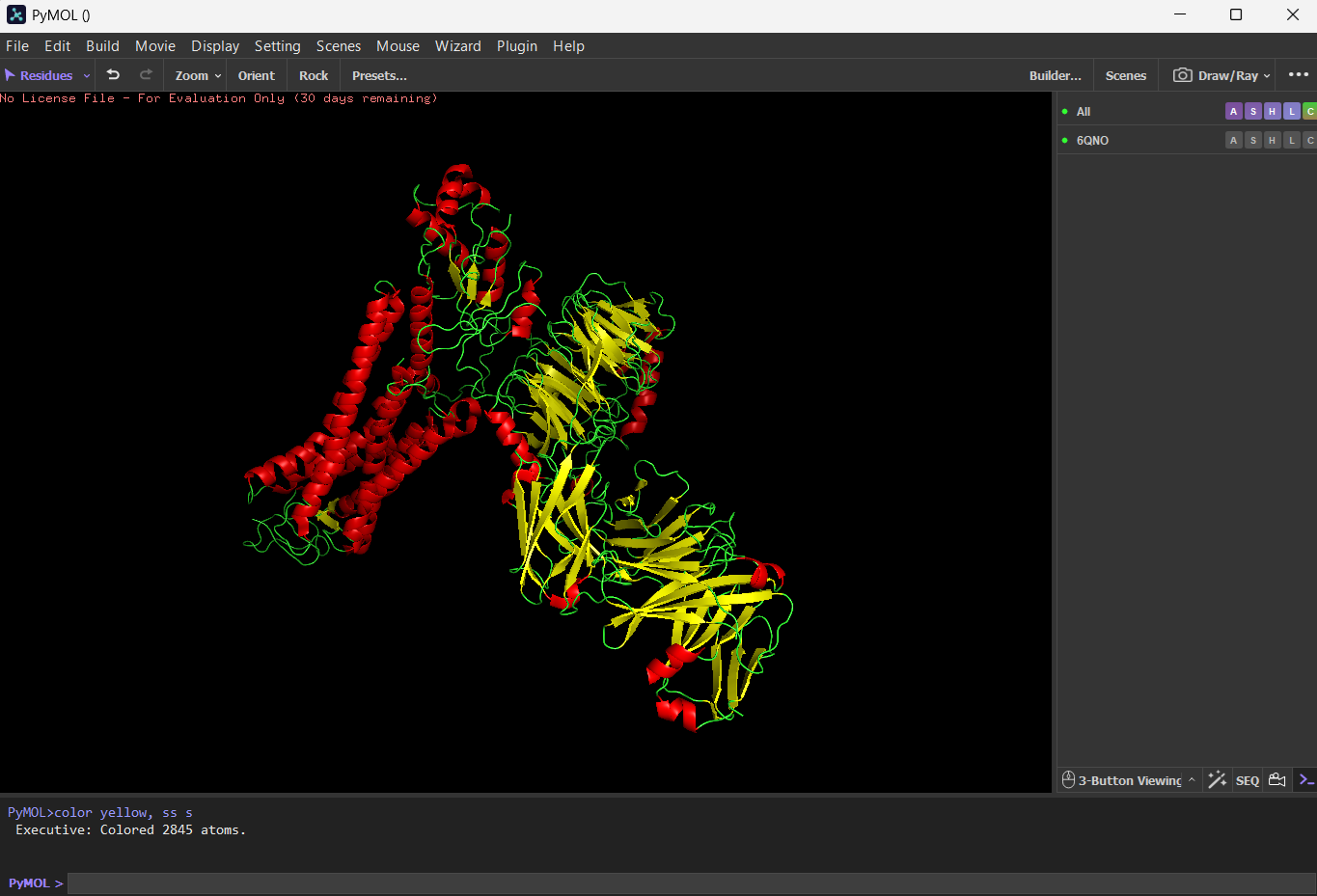
Color the protein by secondary structure. Does it have more helices or sheets?
I think it has more sheets. In this diagram, helices are coloured red and cover 3067 atoms. Beta sheets are coloured in yellow and cover 2845 atoms. If we count the distinct number of shapes rather than focus on atom coverage, I think there are more helices.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I coloured hydrophilic residues blue and hydrophobic residues yellow. I observe that one bunch of hydrophobic residues tend to concentrate together in yellow on the left.
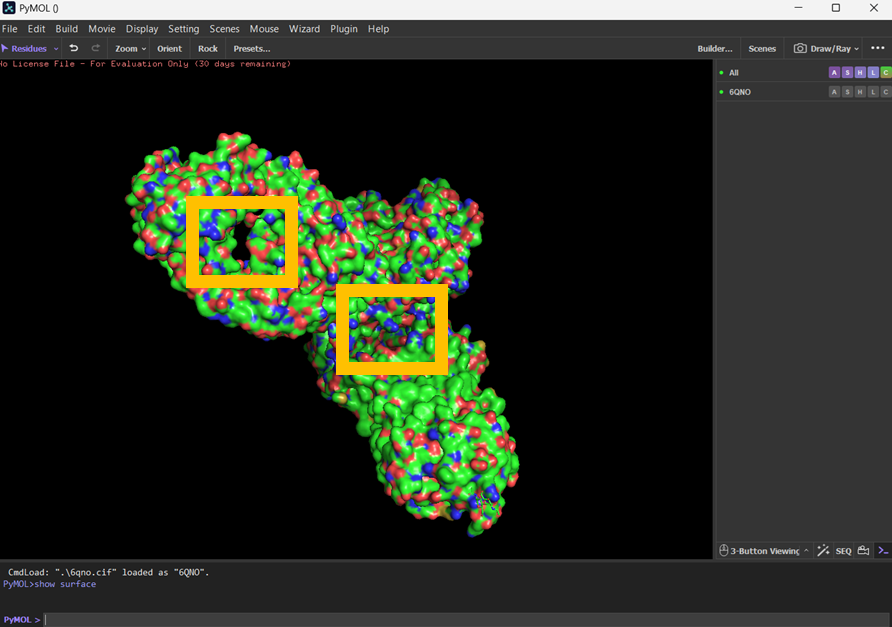
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I visualised the surface, rotated it and found two holes that I’ve highlighted in thick orange squares.
Part C: Using ML-Based Protein Design Tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
I’ve used the sequence for rhodopsin: MNGTEGPNFYVPFSNATGVVRSPFEYPQYYLAEPWQFSMLAAYMFLLIVLGFPINFLTLYVTVQHKKLRTPLNYILLNLAVADLFMVLGGFTSTLYTSLHGYFVFGPTGCNLEGFFATLGGEIALWSLVVLAIERYVVVCKPMSNFRFGENHAIMGVAFTWVMALACAAPPLAGWSRYIPEGLQCSCGIDYYTLKPEVNNESFVIYMFVVHFTIPMIIIFFCYGQLVFTVKEAAAQQQESATTQKAEKEVTRMVIIMVIAFLICWVPYASVAFYIFTHQGSNFGPIFMTIPAFFAKSAAIYNPVIYIMMNKQFRNCMLTTICCGKNPLGDDEASATVSKTETSQVAPA
C1. Protein Language Modeling
Deep Mutational Scans
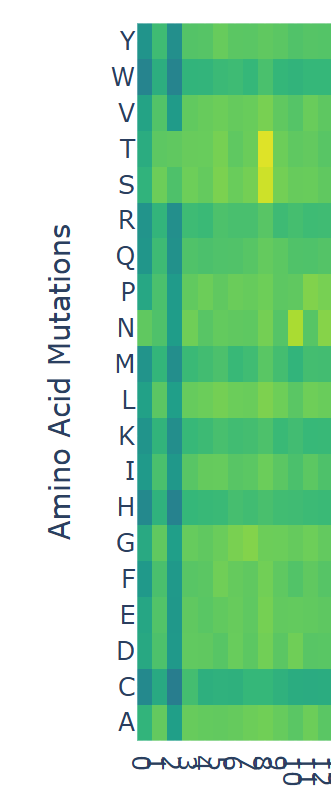
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I looked at the scan and noticed that at position ‘8’ there were some yellow bars. Counting from 0 to 8 inclusively, the sequence ends in ‘F’, which has a model score of
model score = 0.5256555
012345678
MNGTEGPNF
model score = 2.980395
012345678
MNGTEGPNS
model score = 3.427129
012345678
MNGTEGPNT
If I assume that the higher the model score, the more likely an amino acid will occupy the position, it suggests that although ‘F’ appears in the rhodopsin sequence, in other proteins that begin with MNGTEGPN are more likely to have a ‘T’. For any of the dark blue squares (which tend to be very negative) I would assume their presence in the amino acid would be unstable or cause harm.
Latent Space Analysis
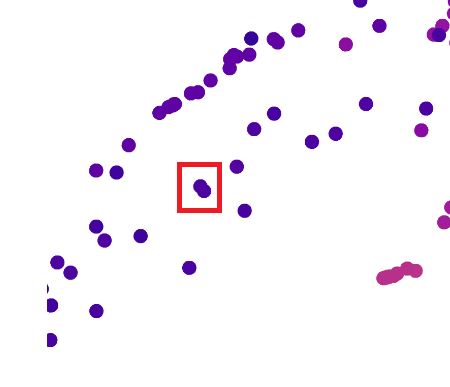
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
I think they do. I zoomed into the map and tried to pick two very close neighbours. My plan was to find the Uniprot IDs for neighbouring proteins and do a Protein blast sequence to determine how similar they were to one another. But I found it difficult to get IDs for neighbouring pairs.
I zoomed in on these two data points:
Up close, this is what those two dots represented:
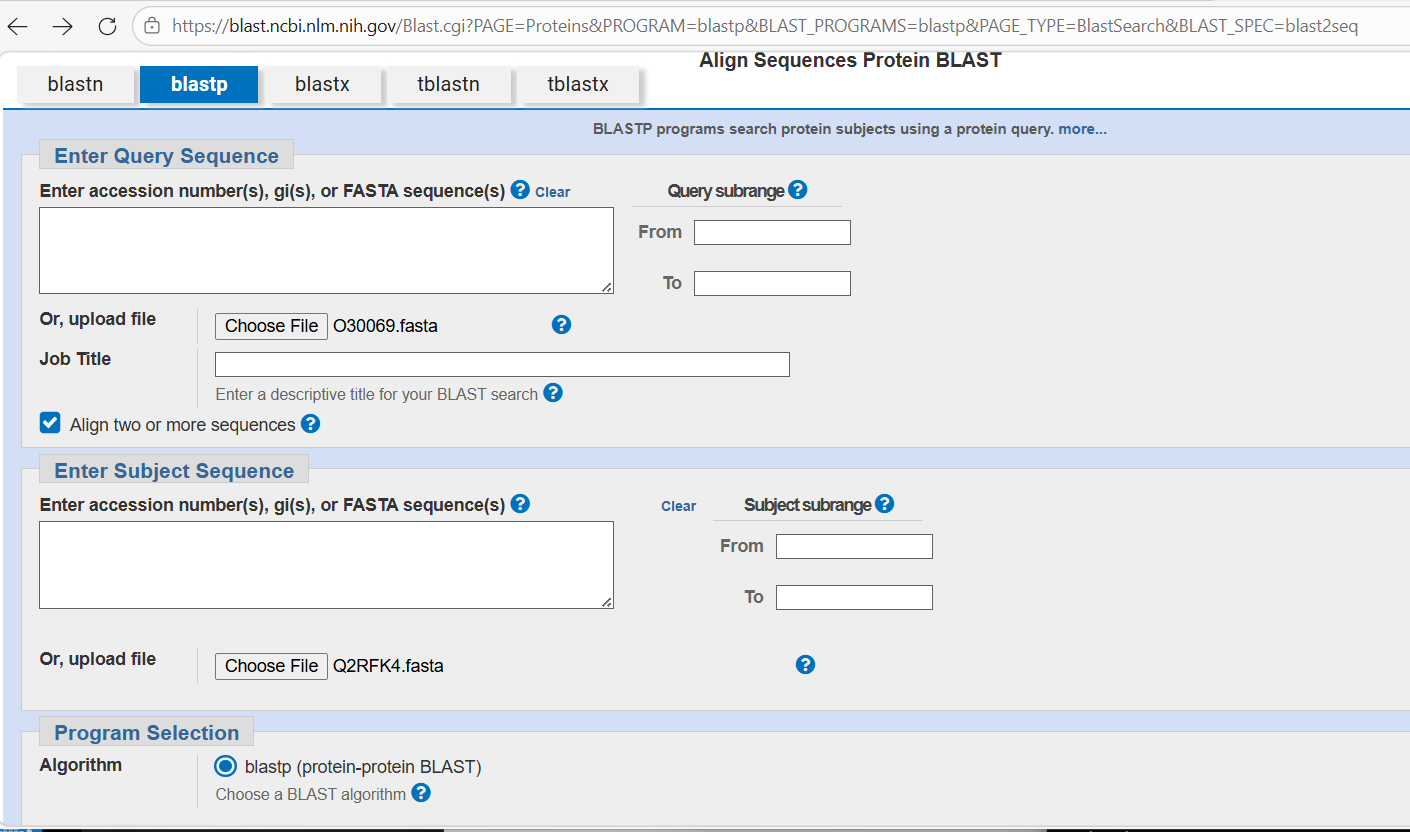
I tried to find Uniprot entries for both of these for each
I downloaded FASTA files for each. I then went to the NCBI’s BLASTP facility:
I loaded the FASTA files and then waited for the blast to complete. The results indicated they don’t appear to have much similarity with one another and I’m not sure why - that doesn’t fit well with the apparent similarity suggested by the close collocation of the proteins in the visualisation.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?**
First I tried to fold the rhodopsin protein with no changes, and then in the third position where I substituted a ‘F’ for a ‘T’. There wasn’t much change. But then I made the sequence include a lot of Cs and Qs and I started to notice the left end started to distort a lot.
Rhodopsin protein with no changes.
Rhodopsin protein with many alterations.
Part D. Group Brainstorm on Bacteriophage Engineering
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
For my review of the Bacteriophage Final Projects Goals for engineering the L Protein, I focused on the goal of designing bacteriophages that showed increased stability. Initially I prompted ChatGPT to help determine how researchers might stabilise a lysis protein that is created by bacteriophages used to kill e coli12
According to the response, the challenges for making stable bacteriophages include the risk of denaturation at higher temperatures or broad ranges of pH, degradation by bacterial enzymes, and the risk of aggregation or precipitation when the bacteriophage is purified for therapeutic use.
It recommended four strategies to consider:
rational design, which is: “…a molecular method aimed at altering the genetic makeup of existing enzymes to improve their structural and functional properties in a predictable manner, relying on prior knowledge of the enzyme’s molecular details”13
fusion proteins, which are: “…A fusion protein is a single polypeptide composed of two or more distinct protein domains encoded initially by separate genes but artificially linked to function as one unit.” They are often incorporate tags into a protein which help improve the solubility, purification and detection of the main protein of interest.14
directed evolution, which involves “… creating mutants by random mutagenesis and recombination followed by the screening of mutants for desired characteristics such as increase stability and changed catalytic specificity”15
expression optimisation, which: “…focuses on maximizing the yield of proteins or other biological molecules in various expression systems” 16
I’d focus on directed evolution, and perhaps randomly introducing random mutations in an amino acid sequence for the L protein, and using ESM2 to help identify which ones would be likely to form. I could then try to use tools that tested how well the resulting L proteins would bind to mutated proteins that act as the chaperone sites in the host such as DnaJ. I think the ESM2 model would help me make some intelligent guesses about mutations that would result in a viable protein.
Doig, Andrew J. “Frozen, but no accident–why the 20 standard amino acids were selected.” The FEBS journal 284.9 (2017): 1296-1305. ↩︎
Gutiérrez-Preciado, Ana, Hector Romero, and Mariana Peimbert. “An evolutionary perspective on amino acids.” Nature Education 3.9 (2010): 29. ↩︎
Novotny, Marian, and Gerard J. Kleywegt. “A survey of left-handed helices in protein structures.” Journal of molecular biology 347.2 (2005): 231-241. ↩︎
Schlick, Tamar. “Protein structure hierarchy.” Molecular Modeling and Simulation: An Interdisciplinary Guide: An Interdisciplinary Guide. New York, NY: Springer New York, 2010. 105-128. ↩︎
MacDermott, A. J. “8.2 Perspective and concepts: Biomolecular significance of homochirality: The origin of the homochiral signature of life.” Comprehensive chirality. 2012. 11-38. ↩︎
Cole, Benjamin J., and Christopher Bystroff. “Alpha helical crossovers favor right‐handed supersecondary structures by kinetic trapping: The phone cord effect in protein folding.” Protein Science 18.8 (2009): 1602-1608. ↩︎
Zhou, X. Edward, Karsten Melcher, and H. Eric Xu. “Structure and activation of rhodopsin.” Acta Pharmacologica Sinica 33.3 (2012): 291-299. ↩︎
“How would researchers try to stabilise the lysis protein that is created by bacteriophages used to kill e coli? Please include sources” prompt. ChatGPT, 29 March version, OpenAI, 29 March 2026, chatgpt.com. ↩︎
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
I visited the Uniprot page for (P00441)[https://www.uniprot.org/uniprotkb/P00441/entry#sequences] and the normal sequence is:
A4V represents one mutation where the ‘A’ changes to a ‘V’ at position 4:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
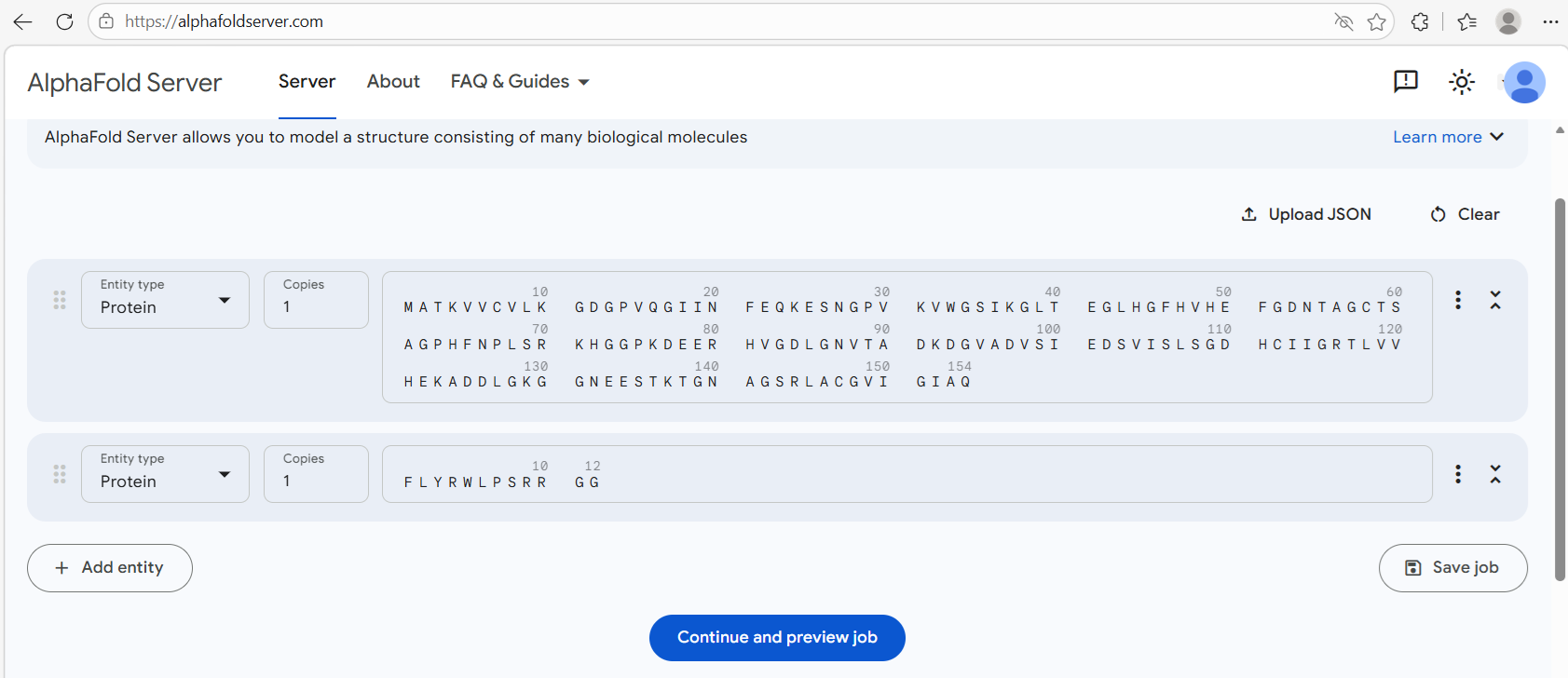
I opened my copy of the PepMLM script and entered the sequence. I tried adjusting the slider to change the peptide length from 15 to 12, but the slider appears fixed. So I opened the code and just changed the peptide length to ‘12’. I set the number of peptides to 4.
![Adjusting the PepMLM script]](sod1_a4v_pepMLM1.png)
I then ran the code for ‘Load Model’. Next, I modified ‘Generate Peptides’ so that it would include the known binder FLYRWLPSRRGG I obtained one binder result:
Navigate to the AlphaFold Server: alphafoldserver.com. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind.
For each peptide sequence, I used the alphafold query form as shown:
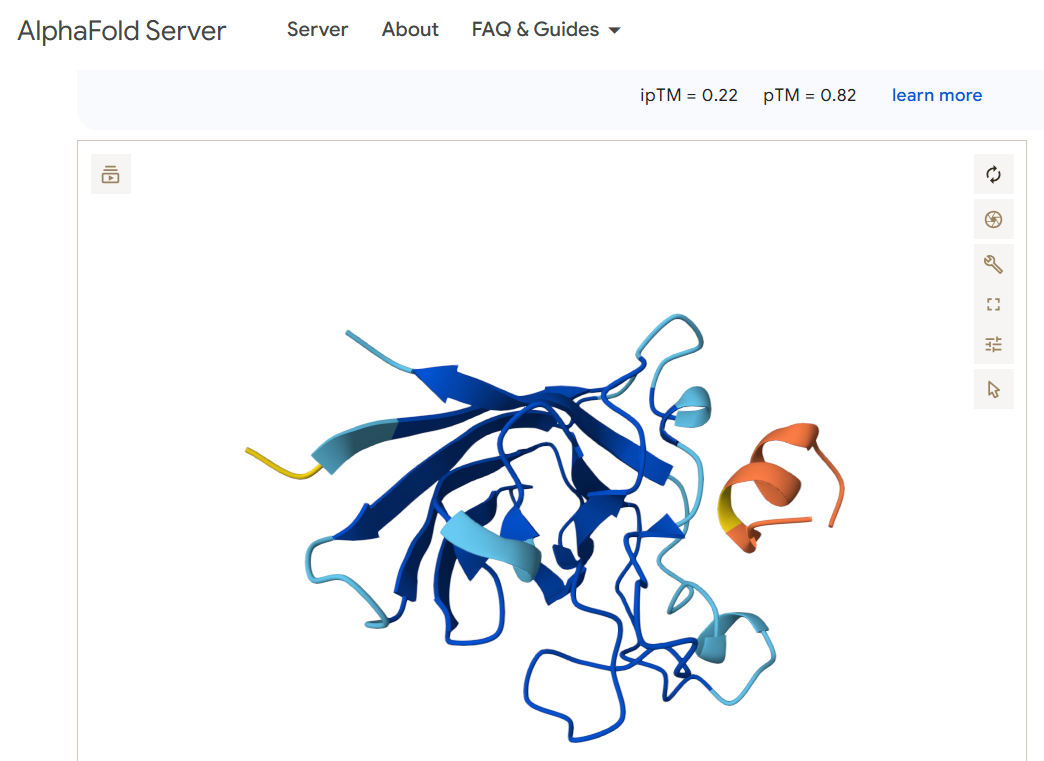
For the first candidate binder FLYRWLPSRRGG, I obtained this image:
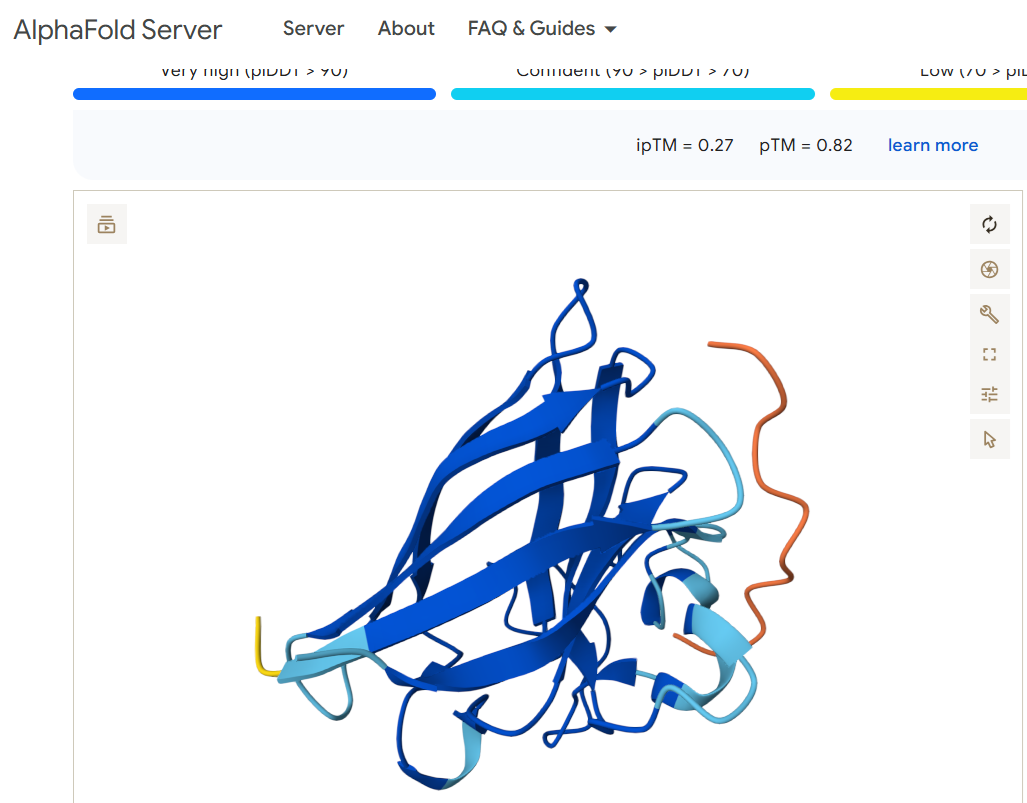
For the binder WRYPVAAAEWGE, I obtained this image:
For the other three binder candidates, I got an error:
Does it localize near the N-terminus where A4V sits?
Neither of the two viable binding candidates FLYRWLPSRRGG or WRYPVAAAEWGE appears to bind anywhere near the N terminus (shown in the left part of the diagrams)
Does it engage the β-barrel region or approach the dimer interface?
The peptides appear to engage the beginning of the beta sheet.
Does it appear surface-bound or partially buried?
Both of the peptides appear to hover over the other protein and seem to me like they may be more surface bound than becoming buried or entangled.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The two binder candidates that are able to work have a similar pTM score. Both are above 0.5 which suggests the overall predicted fold for the complex might be similar to the true structure. Both also have a very low ipTM, which suggests they are low-confidence predictions of relative positions of subunits within the complex. The candidate WRYPVAAAEWGE seems slightly better a candidate than FLYRWLPSRRGG, but not by much. Both seem like poor candidates as they don’t appear to bind anywhere near the N terminus where the mutation exists.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
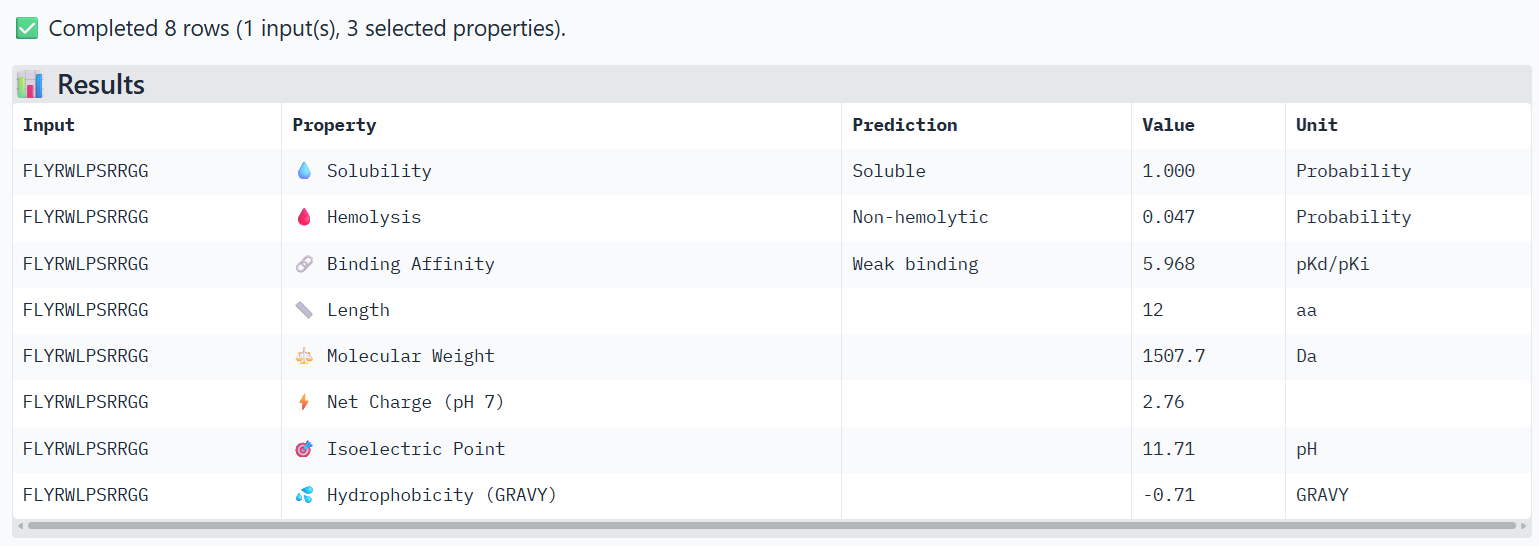
For the main suggested candidate FLYRWLPSRRGG, I obtained these results:
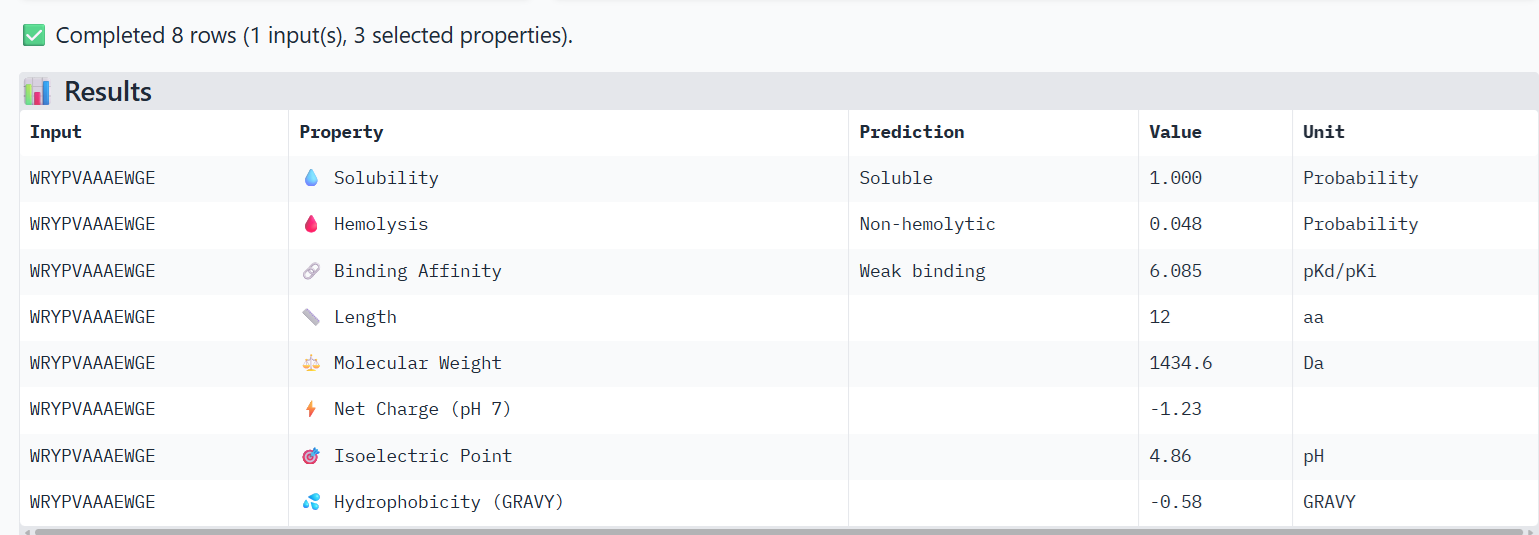
For the other binder candidate WRYPVAAAEWGE, I obtained these results:
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
I observe that WRYPVAAAEWGE, which has a slightly higher ipTM value than FLYRWLPSRRGG, also shows very slightly higher hemolysis and binding affinity properties, but still not much. Both are considered soluble, non-hemolytic and weak binding.
Choose one peptide you would advance and justify your decision briefly.
I’m not sure I’d advance either, because their ipTM scores were so low. Yet, it appears that WRYPVAAAEWGE has slightly better properties than FLYRWLPSRRGG.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
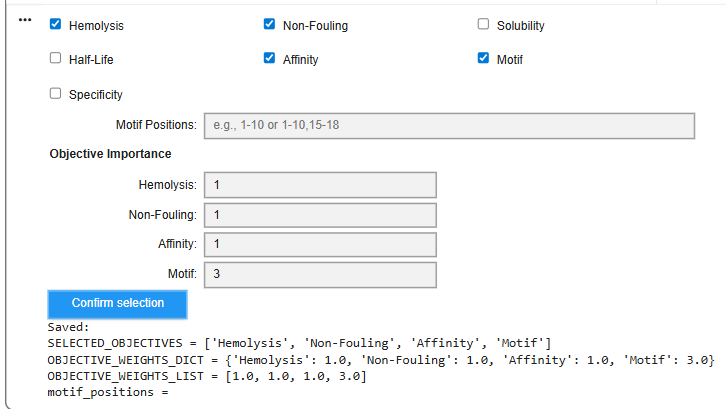
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
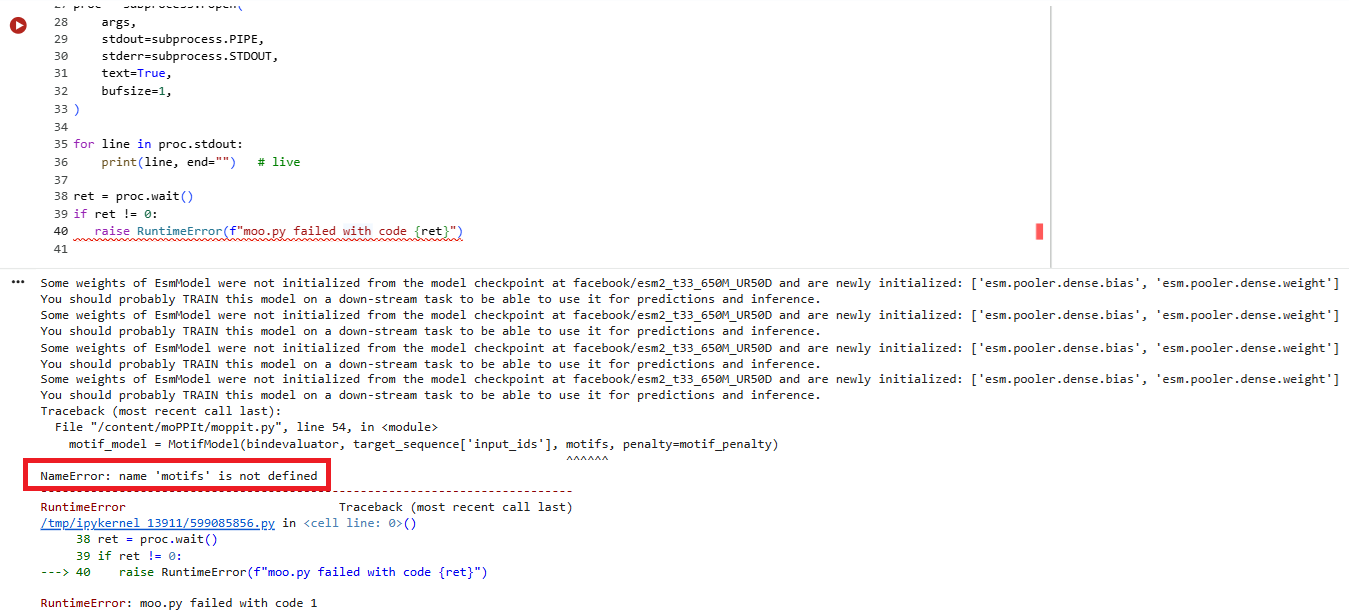
I ran the script and tried to specify a motif value of ‘2’ or ‘3’ - but the next script which generated the peptides complained of an error.
I then unselected ‘motif’ and tried to run it. I got the following sets of results:
I would try to insert A4V mutant SOD1 into animal and then human cell lines and see whether using the peptides reduced the problems associated with A4V.
Week 6 Genetic Circuits Part I: Assembly Technologies
Genetic Circuits Part I: Assembly Technologies
1 What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
A product page for Fisher Scientific indicate:
“Phusion High-Fidelity PCR Master Mix is convenient 2X mix containing Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2. Two master mix formulations are available: with HF Buffer (F-531S and F-531L) and with GC Buffer (F-532S and F-532L).”1
The optimised reaction buffer is a mix of chemicals that support the best environment to allow DNA polymerase to work during PCR amplification.2 The DNA polymerase are enzymes that are used to duplicate the genetic information stored in DNA, generating a faithful copy.3
Phusion DNA Polymerase
nucleotides
optimised reaction buffer
2 What are some factors that determine primer annealing temperature during PCR?
The annealing temperature, which is the optimal temperature at which the primers can bind to the DNA, is heavily related to the primer melting temperature and the guanine citosine (GC) content of the primer. The melting temperature of the primer is the temperature at which 50% of the DNA duplex separates and becomes single stranded. The melting temperature in turn is dependent on the length of the oligonucleotide sequence and the composition of the DNA molecule. Generally, the annealing temperature should be no more than 5 degrees lower than the melting temperature of the primers. If the annealing temperature is set too low, the the result will feature partial annealing with mismatched bases that produces non-specific amplifications. If it is set too high, it will reduce the PCR yield.
3 There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR focuses on a specific region of DNA defined by primers, whereas restriction enzymes focus on a specific sequence that may occur multiple times. Whereas PCR is used for DNA sequencing and gene isolation, restriction digests are used in cloning. Generally you would use PCR to multiply the number of sequences of interest to replicate and then use restriction digestion to prepare those sequences for cloning and to insert DNA fragments into plasmids.
4 How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The main consideration should focus on the overlap length the of the forward and reverse primers. The longer the insert and the larger the number of insert fragments, the longer the primers need to ensure stable annealing. According to a SnapGene video -for a single insert sequence of 0.1 to 0.5kb, the overlapping tails of between 15 to 30 nucleotides should be sufficient. For an insert sequence between 8 and 10 kb, the tail overlap should increase to about 40 bp in length.4
5 How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters the E. coli cells by passing through temporary pores that appear in the bacteria when it is stressed (typically by heat)
6 Describe another assembly method in detail (such as Golden Gate Assembly)
6.1 Explain the other method in 5 – 7 sentences plus diagrams (either handmade or online).
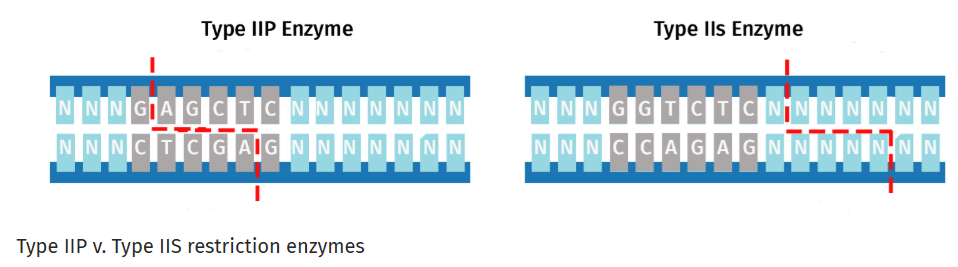
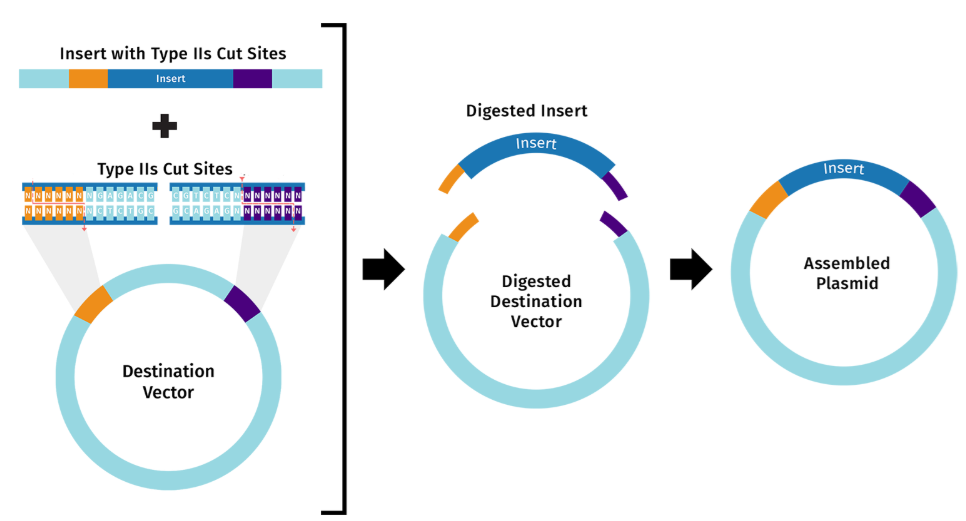
Golden Gate Assembly is a DNA assembly method that relies on Type IIS restriction enzymes such as Bsal that cleave DNA outside their recognition sequences. It has two steps that occur within the same reaction:
Type IIS restriction enzyme digestion
DNA ligation
Whereas Gibson Assembly relies on melting overlaps and filling gaps, Golden Gate uses Type IIS restriction enzymes to cleave outside their recognition sequences to produce unique overhangs. Those unique overhangs help make Golden Gate better able to assemble DNA that involves many many insert fragments.
These two diagrams borrowed from the SnapGene web site best describe the key parts of this approach.5
6.2 Model this assembly method with Benchling or Asimov Kernel!
Week 7 Genetic Circuits Part II: Neuromorphic Circuits
Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Traditional genetic circuits tend to have a more restricted set of states they can use to respond to stimuli. IANNs support analog signal processing, which allows them to continuously adapt and respond to varying conditions instead of set of those that only register as ‘1’s or ‘0’s. This property can make them better suited to cope with the environmental bioprocesses that can exhibit different metabolic properties depending on their location in space and time 1. IANNs are also better able to respond to noise in signals than traditional genetic circuits.
I think it would be interesting to create an IANN to support people who have hemochromatosis, a disorder that causes the body to absorb too much iron. An IANN would rely on measuring a analogue signnal of free iron concentration. The processing part of the circuit would involve summing the input signal of Fe concentration to detect when it passed a certain level. When the threshold was reached, the circuit could encourage further ferritin synthesis, perhaps such that the amount produced matched the amount of free iron.
I don’t know if it’s a weakness but I’d wonder how long engineered cells would last in the body. I’d also wonder whether parts of the circuit would have a risk of changing - for example somehow as the circuit ages, the threshold of when it starts producing ferritin gets lower and lower (producing too much response) or higher and higher (triggering too little response)
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
I’m having trouble understanding this and spent half an hour on it before giving up to get more help on it.
I understand that the perceptron f1 would take as input the x1 - the DNA encoding for the Csy4 endoribonuclease and something else that could turn that ‘on’ or ‘off’. That output would then feed into the diagram that’s shown, and the presence of endoribonuclease would turn ‘off’ the production of fluorescent proteins. Beyond that I’m not sure.
Part II
1 What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium (the vegetative part of fungi) can be used to produce insulation, packaging, or construction materials. Mycelium is also used to make a leather-like substance to make fashion items 2. Research has also found that mycelium can be made to create a self-repairing fabric.3
Mycelium material can provide a biodegradable alternative to synthetic materials.4 There are claims that it can be more environmentally friendly, but I think the costs of growing it and reusing it at large scale need to happen before I can evaluate that claim.
The disadvantages of mycelium materials is that growing it for large-scale activities may be challenging. Its properties may not be as consistent as those of synthetic materials and its biodegradability means it would likely not be as durable.5
Also note fungal materials are also used to detect food pathogens, provide antibiotics, and alternative food materials to meat.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi can be genetically engineered to support bioremediation applications (e.g. sequestering heavy metals).6 For example, Jo reports that:
"Compared with bacteria, which are widely deployed for bioremediation and typically require a continuous water phase to grow, filamentous fungi can extend their mycelia across the air-liquid interface into air-filled pores in the soil. Fungal mycelia can thus reach areas inaccessible to bacterial biofilms and form bridges transporting nutrients and chemicals across discontinuous microhabitats. In these environments, fungal cells absorb molecules from enzymatic degradation together with other chemicals, such as heavy metals and metalloids, and convert them into less toxic forms intracellularly."
Roquet, Nathaniel, and Timothy K. Lu. “Digital and analog gene circuits for biotechnology.” Biotechnology journal 9.5 (2014): 597-608. ↩︎
Jo, Charles, et al. “Unlocking the magic in mycelium: Using synthetic biology to optimize filamentous fungi for biomanufacturing and sustainability.” Materials Today Bio 19 (2023): 100560. ↩︎
Week 9: Cell free systems
Week 9 — Cell-Free Systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The greatest advantage the cell-free protein synthesis has over in vivo methods is that the viability of the cell does not have to be maintained in order to maintain the viability of the protein synthesis. By allowing proteins to be grown in an open environment, greater control can be exercised over the key factors that produce the protein. Materials can be added to help protein production which may otherwise be toxic to cell populations.
Scientists can control ion concentrations, cofactors and energy sources. Enhancing and prohibiting materials can also be added. Growing proteins in a cell-free environment also means protein production does not have to accommodate cloning of cells.
2. Describe the main components of a cell-free expression system and explain the role of each component.
I asked ChatGPT with a main prompt and then found more specific references for parts of it.1
The main components of a cell-free expression system are:
Genetic Template. which contains the genetic information encoding the target protein. An example of a template could be a genetic circuit. The typical parts of the template would include:
a gene promoter, which is: “…a region of DNA upstream of a gene where relevant proteins (such as RNA polymerase and transcription factors) bind to initiate transcription of that gene. The resulting transcription produces an RNA molecule (such as mRNA)”2
a ribosome binding site (RBS), which is a RNA sequence found in the messager RNA to which ribosomes can bind and initiate translation 3
coding sequence, which could be something like the sequence to code for the green fluorescent protein GFP.
transcription terminator, which marks the end of a gene and cause transcription to stop.
Cell Extract., which contains all the machinery that will be used to support transcription and translation in the cell-free system. The machinery includes things such as:
ribosomes, which read messenger RNA (mRNA) and translates the genetic code into a sequence of amino acids 4
tRNAs, which ensure that the correct amino acid is inserted into the protein being created 5
translation factors, which are proteins that “…help control when and how genes are turned on or off in a cell by binding to nearby DNA and to other DNA-associated proteins” 6
metabolic enzymes which support energy procuction
aminoacyl-tRNA synthetases, which help ensure accurate translation of genetic code 7
RNA polymerase. Synthesis messenger RNA (mRNA) from the DNA template during transcription. Examples include T7 RNA and SP6 RNA polymerases.
amino acids, which provide the raw materials for building proteins
Energy source. This is needed to power protein synthesis and generally comes from ATP and GTP
Amino acids. These provide the raw materials to help build proteins.
Nucleotides. Needed for RNA synthesis during transcription and as energy carriers during translation. ATP and GTP are important for ribosomal functions and to charge transfer RNA (tRNA).
Buffers and salts. These help maintain the best conditions for supporting enzymatic activity and for stabilising ribosomes.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Transcription, translation and some enzymatic reactions require great amounts of ATP, an energy-carrying molecule that resides in cells.8 Without them, the reaction would stop. According to Dunn, “…cells in the human body depend on the hydrolysis of 100-150 moles of ATP per day to ensure proper functioning.”9.
It is easier for cell-based systems to generate ATP for transcription and translation because the cells already have various metabolic networks that produce it. In cell-free systems, you have to supply that machinery.
One way of sustaining the ATP it so use a regeneration reaction such as:
ADP + Creatine phosphate –> ATP + Creatine
When transcription and translation happen, the processes get their energy from molecules such as ATP. ATP releases energy by losing a phosphate group, which turns ATP into ADP. Creatine kinase can transfer a phosphate group to help the ADP molecule go back to being ATP.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Whereas prokaryotic systems typically emphasise extracts from bacteria, eukaryotic systems typically come from wheat germ, insects, yeast or mammalian cells. Prokaryotic cell free platforms are typically cheaper, simpler, faster, produce higher yields. However, eukaryotic systems are better at producing membrane proteins and are better suited for therapeutic and complex proteins.10.
In a prokaryotic system I would produce sfGFP because years of work have been spent adapting and refining GFP to be produced by E. coli bacteria. I would use a prokaryotic cell free system to produce Erythropoietin (EPO), a protein used to produce red blood cells. The protein’s requirement for glycosylation isn’t well supported by eukaryotic systems.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
First I would determine whether the protein of interest required post-translational modifications (PTMs).
Prokaryotes such as E. coli bacteria do not support PTMs that well because they lack the extra specialised cell compartments which can support modifying the protein after it is first translated. Therefore, if the protein did not need them I would focus on the simpler eukaryotic system. Otherwise I would focus on using a prokaryotic-based cell-free system.
Membrane proteins can be difficult to synthesise in cell-free systems because there is no membrane that could influence how they well they fold or their solubility. Therefore, the systems need to support materials that can mimic a membrane. Zemella lists nanodiscs, liposomes and certain detergents that have been developed to mimic the missing membrane. 10
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each
Three causes of low yield for target proteins in a cell-free system include:
inefficient transcription or translation.
protein misfolding
energy depletion
Inefficient transcription or translation may be caused by different causes, which include:
weak promoter that doesn’t activate translation of downstream genes as well as it could
the ribosome has difficulty binding to the RBS
Choosing a stronger promoter can mean the promoter switches transcription ‘on’ more often. Inserting filler base pairs round the RBS sequence can make it easier for the RBS to stand out and be grasped properly by the ribosome (like I put into my final project Twist sequence).
Misfolded proteins could be caused by various factors such as the proteins containing hydrophobic regions. In this case, the use of detergents, nanodiscs or liposomes could help. As Puthenveetil notes: “Nanodiscs provide an excellent system for the structure-function investigation of membrane proteins. Its direct advantage lies in presenting a water soluble form of an otherwise hydrophobic molecule, making it amenable to a plethora of solution techniques.” 11
The system could run out of supplies materials such as ATP, GTP, amino acids and magnesium that help provide the cell-free system with energy. It could also develop byproducts which could stall translation prematurely. One solution is to try to reduce these byproducts and another is to increase inputs such as ATP, amino acids and magnesium.
Homework question from Kate Adamala
1. Pick a function and describe it.
1a. What would your synthetic cell do? What is the input and what is the output?
Consider a synthetic cell that is capable of fluorescing when it detects the presence of lactose.12. The system’s input would be a lactose molecule that resides outside the synthetic cell and the output would be the Green Fluorescent Protein (GFP).
1b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, it could be done in a cell-free transcription/translation environment. It may even be preferable to using a solution with encapsulation, unless a project called for the biosensor cell to support other functions besides just sensing lactose. Then there may be a need for a synthetic cell to support different functions in different compartments.
1c. Could this function be realized by genetically modified natural cell?
Yes.
1d. Describe the desired outcome of your synthetic cell operation.
When a lactose molecule passes through the membrane pores of the synthetic cell, it can bind to a regulatory protein such as Lac and signal that transcription of the GFP protein should begin.
2. Design all components that would need to be part of your synthetic cell.
The cell would comprise three modules:
a sensing module, which could be a plasmid genetic circuit
a gene expression module that uses PURE (Protein synthesis using Recombinant Elements) system
an output module that transcribes and translates the Green Flourescent Protein inside a vesicle
2a. What would be the membrane made of?
I would use phospholipids because they are closest chemically to real biological membranes.
2b. What would you encapsulate inside? Enzymes, small molecules.
I would include the following systems:
the transcription-translation system made from PURE components
an energy system to sustain transcription and translation
small molecules used for protein synthesis
enzymes
2c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?need mammalian)
Bacteria is OK to use because lactose sensing is a prokaryotic regulatory feature. For example, the lactose operon exists in E. coli and contains genes involved with metabolising lactose 13. You could use a mammalian system but it would add more complexity without adding much benefit.
2d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
In order for the circuit to work, the GFP protein would need to be able to pass out of the membrane of the synthetic cell. One way to help that happen is to ensure the synthetic cell has generic pores that could transport it.
3. Experimental details.3a. List all lipids and genes.
A common set of lipids includes:
Phosphatidylcholine (PC)
Phosphatidylethanolamine (PE)
Phosphatidylglycerol (PG)
The lactose repressor gene would be the prominent gene in this system.
3b. How will you measure the function of your system?
I would measure the amount of green fluorescent protein (GFP) produced by using flow cytometry.
Homework question from Peter Nguyen
**Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch.
1. Write a one-sentence summary pitch sentence describing your concept.
My idea would be to develop a bite-proof body suit that would fluoresce with colour if it detected the presence of saliva from Ixodes ricinus, the tick that carries the Lyme disease bacteria in Europe.14.
2. How will the idea work, in more detail? Write 3-4 sentences or more.
Community health specialists who are assessing the risk of getting bitten by Lyme disease ticks on walking trails and fields could use the body suit to measure realistic exposure without getting bitten. A cell-free system would be integrated into the outer layer of clothing designed to prevent tick bites from penetrating the skin. The cell-free system would detect salivary protein IrAV422, which would be activated when the dried cell-free system encountered saliva from the bug. The system would then manufacture a fluorescent protein in the area of the bite attempt. The system would include peptides that could bind IrAV422 and trigger the production of the fluorescent protein.
3. What societal challenge or market need will this address?
As weather patterns change, Lyme ticks are able to live in a wider range for longer during the year. Lyme disease isn’t always obvious to detect and it would be a good way to show people where ticks were detected.
4. How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
I do wonder how effective such a system would be in sensing from such a small amount of saliva from the bug. The clothing may wear out and I would imagine putting it through a washing machine could severely degrade it. Perhaps it would be designed as a cheap, biodegradable fabric that is meant to be used only a few times until it needed to be washed.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space! For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/.
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
How do tardigrades respond physiologically to extreme environment stressors such as radiation, extreme temperature and a lack of oxygen? This topic could be important for understanding genes that could be useful in making microorganisms that could accomplish important tasks in inhospitable environments. I would be designing cell-free systems which could help monitor expression of proteins that helped it survive. This could be important for transporting biological materials when there is limited payload space available for systems to keep living organisms alive.
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
I would focus on genes that code for Dsup, a protein that helps tardigrades survive severe radiation.15
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
I would focus on the damage suppressor protein Dsup, which helps tardigrades survive extreme conditions.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis would be that if you integrated a cell-free system that detected Dup inside a microfluidics channel that housed the tardigrade, it would be expressed more in extreme conditions.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would create clones of tardigrades and then establish various experiments that used a control and an extreme condition. The control would correspond to conditions it enjoyed on Earth that would also be favourable to humans. For the extreme condition (e.g. extreme heat, cold, vacuum, radiation), I would use the cell-free system to help detect the presence of the Dsup protein (e.g. fluoresence, electrical signal). I would then measure each indicator between control and extreme condition. I would expect the extreme condition to reflect higher Dsup production.
Homework Part B: Individual Final Projec
Done
“Describe the main components of a cell-free expression system. Please include sources. Please include sources.” ChatGPT, OpenAI, 17 May 2026, https://chatgpt.com. ↩︎
Dunn, Jacob, and Michael H. Grider. “Physiology, adenosine triphosphate.” (2020). ↩︎
Zemella, Anne, et al. “Cell‐free protein synthesis: pros and cons of prokaryotic and eukaryotic systems.” ChemBioChem 16.17 (2015): 2420-2431. ↩︎↩︎
Puthenveetil, Robbins, Khiem Nguyen, and Olga Vinogradova. “Nanodiscs and solution NMR: preparation, application and challenges.” Nanotechnology reviews 6.1 (2017): 111-125. ↩︎
“Provide an example of how a synthetic cell system could perform a common function and describe its inputs and outputs”, ChatGPT, OpenAI, 17 May 2026, https://chatgpt.com/. ↩︎
“Describe how a cell-free system would be integrated into a textile that would fluoresce if it detected the presence of the Ixodes ricinus, the tick that carries the Lyme disease bacteria in Europe. Please include sources”, ChatGPT, OpenAI, 17 May 2026, https://chatgpt.com/↩︎
Chavez, Carolina, Cruz-Becerra, Grisel, Fei, Jia, Kassavetis, George, Kadonaga, James, “The tardigrade damage suppressor protein binds to nucleosomes and protects DNA from hydroxyl radicals”, eLife, https://elifesciences.org/articles/47682↩︎
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail
I’ve written most of these up in my Final Project Protocol. My project requires a genetic switch to show an off/on fluorescence effect so I’m not measuring gradual trends in fluorescence. My main measurements will relate to:
preparing proportions for the Agar LB mix with chloramphenicol. Measuring millilitres of water, grams of agar and grams of chloramphenicol.
bacterial transformation measurements. Temperature to measure the critical range for where competent E. coli are shocked and made receptive to accepting plasmids. Measuring time to wait for competent bacteria to absorb plasmids.
preparation of acetic acid solutions. Volumetric calculations to create separate solutions with pH 5.5 and pH 7.0. A pH meter will be needed to verify pH of solutions.
measuring dimensions of the 3D printed Green man sculptures I plan to create. I need to be careful measuring the dimensions of printed sculptures because I’ll need to contain the printed shape within a slightly larger bioreactor.
Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and Determine for each adjacent pair of peaks using the prescribed formula ()
Here is the table I produced through a spreadsheet:
To calculate the weight of the molecule, I would look at the highest peak (Peak 11), and calculate:
molecular weight = m/z ratio x z
= 875.9758 x 32.07761568
= 28099.22
The theoretical weight provided by Expasy is 28006.60.
**2.1 Determine for each adjacent pair of peaks (n, n+1) using the formula:
Z = M(Zn+1) / ()
Waters III - Peptide Mapping - primary structure
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid)
There are 20 Lysines (K) and 6 Arginines (R) in this sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEK****RDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
**2.How many peptides will be generated from tryptic digestion of eGFP?
Based on the count shown in the table of peptides, there are 19 peptides.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
First, I interpret >10% relative abundance as meaning >10% of 1.2e7 which would be > 1.2e6 - between the second and third tick marks on the y axis. I counted 18
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
There are fewer peaks than predicted. The difference may owe to limitations interpreting the graph. The point at 4.30 minutes just touches the line but is not above it. Other people may think of including that point in the list of peaks if they think it is greater than 10% of relative abundance.
5.Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its (m/z) and z.
The two successive peaks showing the most abundance are 525.76712 and 526.25918. Using the formula for z mentioned earlier,
Calculating the mass of that peptide it would be
= (m/z) x z
= 525.76712 x 1055.7996
= 555104.71 Daltons - which is wrong.
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
My answer in 5 is clearly wrong and don’t think it’s useful to calculate the accuracy as it is just too far away from any of the predicted peptide masses. However, based on the recitation video, you would calculate the ppm error by multiplying the accuracy by 1000000.
7. What is the percentage of the sequence that is confirmed by peptide mapping?
88%.
Waters Part V - Did I make GFP?
Thoeretical weight: 28006.6 Daltons (28.0066 kDa)
Observed weight on the Intact LC-MS: 28099.22 Daltons (28.09922kDa)
PPM mass error:
= accuracy x 1000000
= 0.0033070776 x 1000000 = 3,307 ppm.
Week 11: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
This is the pixel I contributed:
I saw that students appeared to be trying to create a person in the lower right corner and I added part of a left arm. I liked the fact that everyone could contribute a small instruction to a program that could drive an Opentrons robot remotely. Encourage art subjects to relate to the creatures that produce the fluorescent proteins.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1.Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase). contains the substances that are left after the E. coli’s membrane has been broken. It provides the machinery that is necessary to support translation activities in the cell-free system. The star lysate is designed to limit activity of RNase, which are enzymes that can degrade the mRNA that influence protein yield.
Salts/Buffer
Potassium Glutamate. Provides high concentrations of potassium ions and glutamate, which help to mimic the intracellular environment that would exist in cell-based systems.
HEPES-KOH pH 7.5. Helps provide optimal pH to support enzymes involved with transcription and translation.
Magnesium Glutamate. Magnesium ions help stabilise the function and structure of the ribosome, which helps translate messenger RNA into functional proteins.
Potassium phosphate monobasic. Helps maintain a stable pH by resisting a pH increase.
Potassium phosphate dibasic. Helps maintain a stable pH by resisting a pH decrease.
Energy / Nucleotide System
Ribose. It is a sugar that forms the backbone of ribonucleotides such as ATP, GTP, CTP, UTP, which are used for transcription and energy transfer.
Glucose. It is a sugar which can be used by enzymes in the lysate to create ATP, which is essential for translation and transcription.
AMP. Adenosine monophosphate can be converted into ADP and ATP, which allows it to help recycle nucleotides that accumulate in long-running cell-free protein synthesis reactions.
CMP. Cytidine monophosphate is one of the four monophosphates that are used to form the building blocks of RNA.
GMP. Guanosine monophosphate serves as a precursor for synthesising GTP, which is one of the four ribonucleotide triphosphates required for mRNA polymerisation.1
UMP.Uridine monophosphate is a precursor for UTP, which is required for mRNA synthesis.
Guanine. It is a precursor for guanosine nucleotides that are essential for translation and transcription activities.
Translation Mix (Amino Acids)
17 Amino Acid Mix. Provide the building blocks of the proteins that are produced and allow translation to occur.
Tyrosine. One of the 20 standard amino acids that cells can synthesise but which must be supplied externally in cell-free protein synthesis systems. The ribosome includes tyrosine into building polypeptide chains whenever it encounters a UAC or UAU codon.
Cysteine. One of the 20 standard amino acids that can help form disufide bonds that are critical for protein structures.
Additives
Nicotinamide. It is a precursor of NAD+/NADP+, which are essential cofactors for proteins that work in energy and redox reactions.
Backfill
Nuclease Free Water. A solvent for CFPS reactions that is designed to prevent degradation of nucleic acids.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences) The 1 hour optimised master mix is supports fast, high energy reactions that are good for short experiments with small-scale expression. The 20 hour mix supports slower reactions that have more sustainable nucleotide regeneration that is best for prolonged, high-yield protein synthesis at lower cost.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1.Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP. It has a higher fluorescence intensity and greater photostability than GFP, making sfGFP the better protein for imaging applications. [^22]
mRFP1. The fact that it emits in the red spectrum means it is useful for experiments that make use of multiple colours of fluorescent proteins. Red light experiences less autofluoresence in many biological systems.
mKO2. It is suitable for long-term expression monitoring because of its ability to resist degradation when it is exposed to light (photostability).
mTurquoise2. It has rapid chromophore formation which supports early fluorescence readout. It is ideal for short CFPS reactions.
mScarlet_I. High expression of the protein may lead to crowding effects that reduce effective fluorescence. The crowding effects reduce the volume that is occupied by macromolecules, which can influence how well they diffuse, react or support protein folding.
Electra2. Although it has a rapid maturation time, it still requires post-translational chromophore maturation that means for short CFPS reactions, some of the protein may not fluoresce before an experiment ends.
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Consider how we might maximise fluorescence of mTurquoise2, which experiences rapid chromophore formation and supports early flourescenct readout. It is good at supporting short cell-free protein syntheses, but 36 hours of incubation is a long time and could present some problems. Adding a high concentration of amino acids will ensure that translation continues and won’t stall. ATP and GTP levels may need to be kept high to support the energy demands required for such a prolonged period of translation.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
I’m not sure we did this for our cell-free lab.
4. The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
I’m not sure we did this for our cell-free lab. I remember we were using tiny magnetic beads that would allow us to help extract proteins we wanted to make.