Week 2 DNA Read, Write and Edit
Assignment 2
I created a Benchling account, loaded up the Lambda DNA, and then tried different combinations of the following restriction enzymes.
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
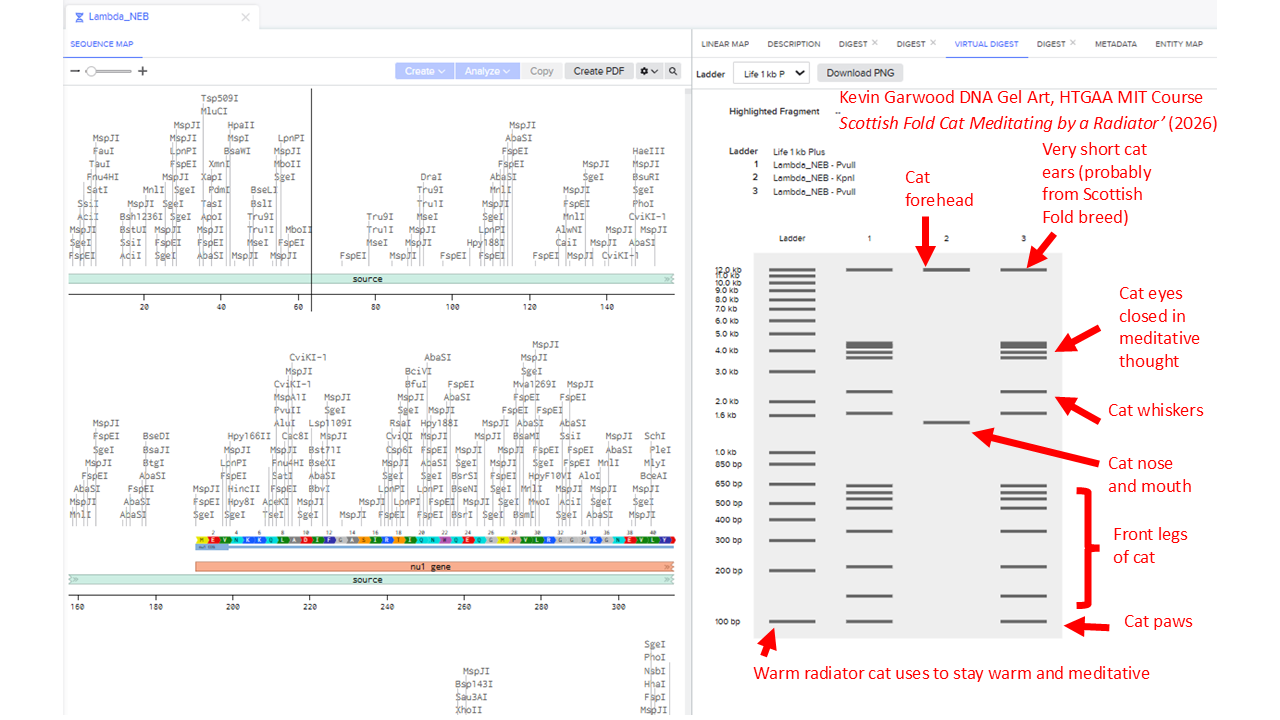
I note that the Automation Art tools produces randomly created electrophoresis ladders, but I excluded Ndel, Pvull and Xhol - because they were not in the list we were supposed to use.
I found it difficult to produce virtual digest ladders that could be combined into recognisable artistic shapes. To get an idea of the range of possible patterns, I systematically looked at choosing combinations of 1, 2, 3, 4 and 5 enzymes.
I’ll include some examples of this brute force way of assessing potential patterns. But that didn’t work well. So I widened my enzyme list to include the enzyme Pvull as well. I ended up with this pattern:
Part 3: DNA Design Challenge
3.1
I’ve chosen the sequence for staphyloxanthin, the protein found in the bacteria Staphylococcus aureus that creates a deep yellow colour that might be similar to the chrome yellow pigment van Gogh used to paint the centres of his sunflowers in the ‘Sunflowers’ painting hanging in the National Gallery.
3.2 Reverse translate
I found the Uniprot entry for 4,4’-diapophytoene synthase, which is used in the biosynthesis of the yellow-orange carotenoid staphyloxanthin. In Benchling, I imported an AA sequence and specified ‘A9JQL9’ and ‘Uniprot’ to import from a database. I selected the entire protein sequence, right-clicked and clicked ‘Backtranslate’. From there, I obtained this DNA Sequence:
ATGACTATGATGGATATGAATTTCAAATATTGTCATAAAATAATGAAAAAACACAGTAAAAGTTTCTCTTATGCCTTTGATTTACTTCCAGAAGACCAAAGAAAGGCTGTATGGGCAATTTATGCAGTTTGTCGCAAAATTGATGACTCAATAGATGTTTATGGTGACATTCAATTTTTAAATCAAATAAAGGAAGATATTCAATCTATAGAAAAATATCCATACGAATATCATCATTTTCAAAGTGATAGAAGAATTATGATGGCACTACAGCACGTGGCTCAACATAAAAATATTGCTTTCCAGAGCTTTTATAATCTTATTGATACCGTCTATAAAGATCAACATTTTACAATGTTTGAAACTGATGCGGAGTTATTCGGATATTGCTATGGTGTTGCTGGTACAGTTGGTGAAGTCTTAACACCTATCTTATCAGATCATGAAACGCATCAAACATATGACGTGGCGCGTCGTCTTGGAGAATCATTGCAATTAATTAATATTTTAAGAGATGTAGGCGAGGATTTTGAAAATGAACGTATTTACTTTTCAAAACAACGACTAAAACAATATGAGGTAGATATTGCTGAAGTTTATCAAAATGGGGTAAACAACCATTATATTGATTTATGGGAATATTACGCAGCAATCGCAGAAAAAGATTTTCGAGATGTTATGGATCAAATTAAAGTATTTTCTATTGAAGCACAACCTATAATAGAACTCGCCGCACGTATCTATATCGAAATATTAGATGAAGTTAGACAAGCTAATTATACTTTGCACGAAAGAGTATTTGTGGAAAAACGTAAGAAAGCTAAGTTATTTCATGAGATTAATTCGAAATACCATAGGATT
3.3. Codon optimization.
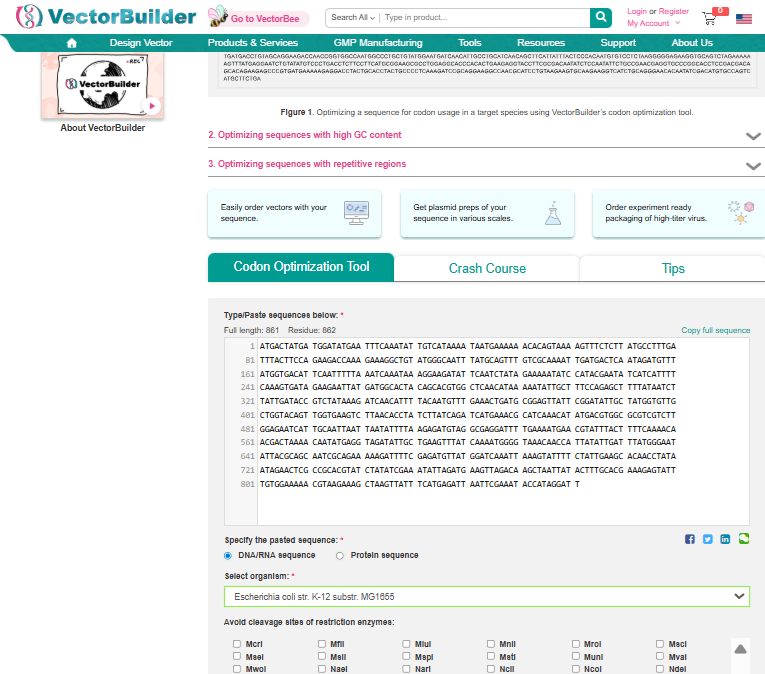
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
I used Vector Builder’s Codon Optimisation Tool and specified the sequence from 3.2 to be inserted into E coli strain K-12 substr: MG1655.
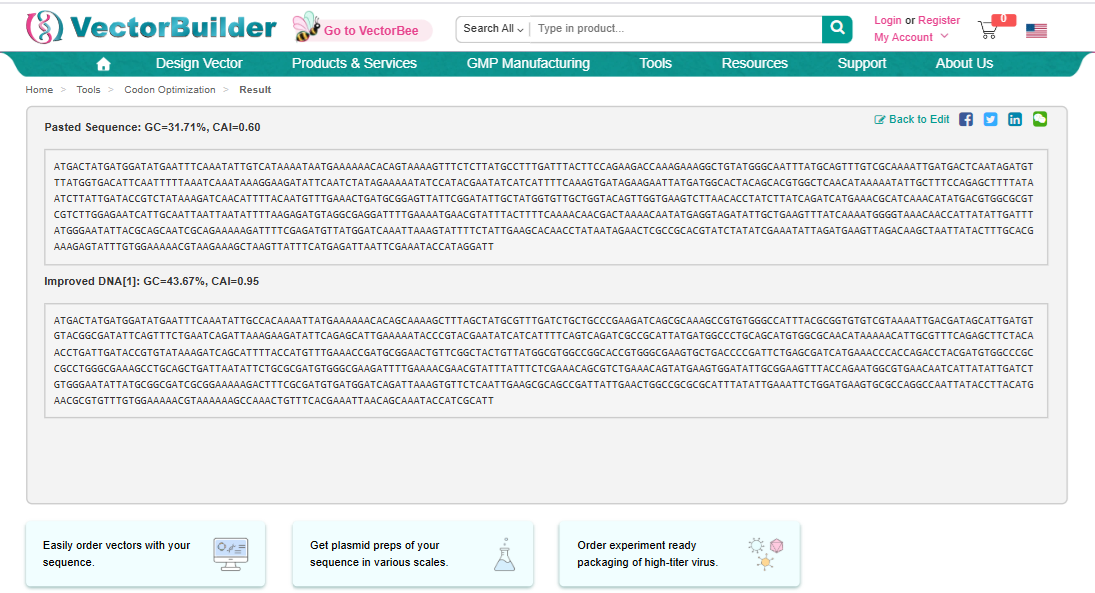
ATGACTATGATGGATATGAATTTCAAATATTGCCACAAAATTATGAAAAAACACAGCAAAAGCTTTAGCTATGCGTTTGATCTGCTGCCCGAAGATCAGCGCAAAGCCGTGTGGGCCATTTACGCGGTGTGTCGTAAAATTGACGATAGCATTGATGTGTACGGCGATATTCAGTTTCTGAATCAGATTAAAGAAGATATTCAGAGCATTGAAAAATACCCGTACGAATATCATCATTTTCAGTCAGATCGCCGCATTATGATGGCCCTGCAGCATGTGGCGCAACATAAAAACATTGCGTTTCAGAGCTTCTACAACCTGATTGATACCGTGTATAAAGATCAGCATTTTACCATGTTTGAAACCGATGCGGAACTGTTCGGCTACTGTTATGGCGTGGCCGGCACCGTGGGCGAAGTGCTGACCCCGATTCTGAGCGATCATGAAACCCACCAGACCTACGATGTGGCCCGCCGCCTGGGCGAAAGCCTGCAGCTGATTAATATTCTGCGCGATGTGGGCGAAGATTTTGAAAACGAACGTATTTATTTCTCGAAACAGCGTCTGAAACAGTATGAAGTGGATATTGCGGAAGTTTACCAGAATGGCGTGAACAATCATTATATTGATCTGTGGGAATATTATGCGGCGATCGCGGAAAAAGACTTTCGCGATGTGATGGATCAGATTAAAGTGTTCTCAATTGAAGCGCAGCCGATTATTGAACTGGCCGCGCGCATTTATATTGAAATTCTGGATGAAGTGCGCCAGGCCAATTATACCTTACATGAACGCGTGTTTGTGGAAAAACGTAAAAAAGCCAAACTGTTTCACGAAATTAACAGCAAATACCATCGCATT
3.4. You have a sequence! Now what?
I’d follow the instructions in Homework Week 2 ‘Prepare a Twist DNA Synthesis Order’ to create an expression cassette, which would be designed to drop into a plasmid vector. This would involve specifying a sequence comprising: a promoter, a Ribosome Binding Site (RBS), a start codon, the optimised codon sequence that will be designed to make the staphyloxanthin work best with E. coli, the 7x His Tag, a stop codon and a terminator. I’d carefully annotate each of these components and export a Fasta file that I would then upload to Twist. The order from twist would presumably arrive in London as the freeze-dried sequence for the cassette.
Next, I’d use PCR to amplify the insert sequence (comprising all the components I specified in Benchling) and then add it to a tube that contains the three enzymes used in Gibson Assembly cloning. First, the T5 exonuclease would cause the 5’ ends of the insert sequence and linearised vector (plasmid backbone) to be chewed back to create overlaps. Polymerase would begin to fill in at the overhangs to prevent excessive enzyme chewing. When the overhangs become stable, taq ligase would complete the fusion of the inserted staphyloxanthin sequence into the backbone sequence of the plasmid. The recommended temperature for this would be about 50’C.
In this process, the enzymes would cut at specific places on the plasmid, which would create a gap for the inserted sequence. I’d need to select appropriate primers: a forward primer that would create an overlap between the sequence leading to the beginning of the gap and the beginning of the inserted sequence. Then I’d need to pick a reverse primer that would create an overlap between the end of the inserted sequence and the beginning of the sequence that starts at the other end of the gap. The end product would be a plasmid where the sequence had been inserted into the gap.
The plasmids would be then be inserted into E. coli by applying temporary heat shock to the cells. The shock would create temporary pores in the membrane of the bacteria which would allow the plasmid to pass inside. Once inside, the E. coli’s own machinery would express the genes encoded into the plasmids as if it were its own. The promoter encoded into the insert sequence would signal the optimised staphyloxanthin sequence to be first transcribed into RNA and then later translated into a protein that would then produce the yellow colour associated with the staphyloxanthin gene.
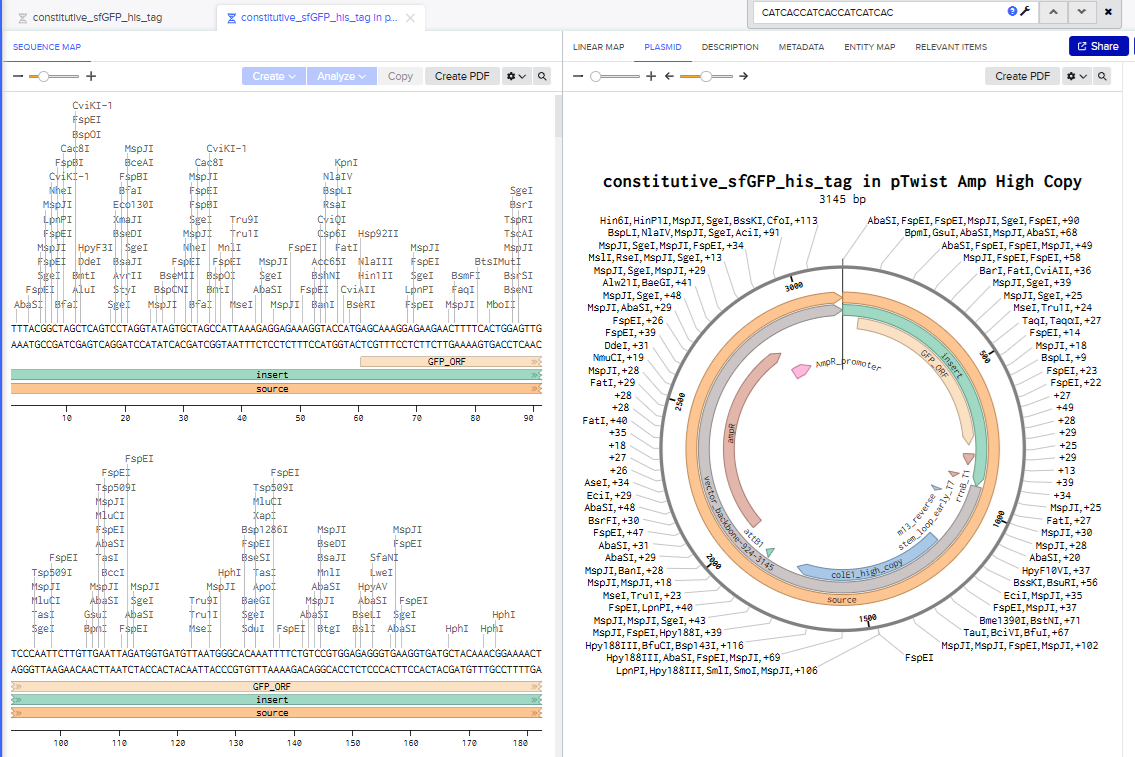
Part 4: Prepare a Twist DNA Synthesis Order
I went through all the instructions for this part using Benchling and Twist, and the final plasmid looked like this in Benchling:
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would want to sequence the protein Proteorhodopsin. When I was working on my Masters dissertation in Art History, I came across this paper: “Frangipane, Giacomo, et al. “Dynamic density shaping of photokinetic E. coli.” Elife 7 (2018): e36608.”
The authors had created E. coli that used proteorhodopsin to make them respond to light. When light was shone on them, it provided energy that propelled the bacteria. They were able to manipulate combinations of light and darkness to help them clump together in a way that looked like a photographic negative.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions: I would use Illumina Sequencing (Short-Read), because they have often been used for metagenomic and transcriptomic sequencing of the genes for Photorhodopsin.
This paper about microbial rhodopsin dynamics by Laura Gómez-Consarnau makes use of the Illumina PE150 platform: Gómez-Consarnau, Laura, et al. “Unexpected microbial rhodopsin dynamics in sync with phytoplankton blooms.” Nature Communications (2025).
Is your method first-, second- or third-generation or other? How so? The method uses short reads and is an example of Next Generation (Second generation) sequencing.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
I read this paper by Dahui Qin to learn more about how NGS worked. Its main steps include:
DNA Fragmentation. Involves breaking up DNA into many short segements using various methods such as enzymatic digestion. The relevant sequences of interest are isolated using complementary probes.
Library preparation. This involves preparing the segments in a uniform way that makes it easier for sequencing primers to bind to fragments.
Sequencing. Allows massive parallel sequencing of all the fragments at the same time.
Analysis. Bioinformatics analysis tools are used to support base calling, read alignment, variant identification and variant annotation. The fragments are compared with a reference sequence to identify mutations and then all the fragment sequences are stitched together to create a complete sequence.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
As part of my final project, I’m looking at doing something similar to Part 4, but using a plasmid where sfGFP can be expressed in the presence of high levels of carbon dioxide. From what I’ve been gathering, the easiest way of doing this is to make a less specific function that would make it fluoresce green in the presence of (carbonic) acid. The main drawback is that it would then react to any acid, not just acid resulting from carbon dioxide dissolved in water. Another approach involves having the bacteria detect HC03 but I’m not yet sure how to do that.
I would be looking at a whole-cell biosensing bacteria that would likely use a cadBA promoter, which is supposed to be active as an acid stress response in E. coli. The promoter would act as a kind of switch to turn the GFP on.
See some famous examples of DNA design
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions:
I would be using Twist to synethesise the plasmid-containing bacteria that would fluoresce green in the presence of CO2, probably indirectly triggered by a response to an acid. So perhaps I’d be building a biosensor that was more about detecting acid than CO2.
What are the essential steps of your chosen sequencing methods? What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I’m not sure if this is relevant to what I’d like to do. I’m not wanting to synthesize DNA based on something that hasn’t been sequenced before. I’m actually more interested in using a documented promoter to turn the GFP on. I’d be doing something really similar to Part 4.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I’d want to edit the E. coli sequence so I can stuff the circuit I want into it.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions: I’d be using Twist to synthesise a genetically altered E. coli that would glow green in the presence of CO2 or perhaps a strong acid stress instead.
How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? I would be editing a plasmid in the way Part 4 outlines and getting Twist to do it.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?