Week 4 Protein Design Part 1
Part A: Conceptual Questions
Choosing 9 of 11 questions to answer
Q1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) I’ve made some assumptions:
- The meat mass does not include air or water.
- The meat mass does not include organic materials that are not amino acids
A Dalton is a unit of mass used to express atomic and molecular weights. I used this converter to determine how many Daltons are in 500 grams of organic material. 500 g = 3.011086821E+26 daltons. If an amino acid is 100 daltons, then 500 g would contain 3.011086821E+24 molecules.
Q2 Why do humans eat beef but do not become a cow, eat fish but do not become fish? When humans digest beef, the process breaks the organic materials down into base materials that their bodies can use to build amino acids according to human DNA. The DNA found in the beef is not transferred and does not displace the DNA in our bodies.
Q3 Why are there only 20 natural amino acids? Doig examines why the 20 standard amino acids were selected by Nature. 1 One theory is that by chance those 20 were selected and became established when there could have been alternative sets. But Doig seems to think their properties alone and in combination provided a kind of almost ideal set of amino acids. He writes:
“…they were selected to enable the formation of soluble structures with close-packed cores, allowing the presence of ordered binding pockets. Factors to take into account when assessing why a particular amino acid might be used include its component atoms, functional groups, biosynthetic cost, use in a protein core or on the surface, solubility and stability. Applying these criteria to the 20 standard amino acids, and considering some other simple alternatives that are not used, we find that there are excellent reasons for the selection of every amino acid. Rather than being a frozen accident, the set of amino acids selected appears to be near ideal.”
Q4 Can you make other non-natural amino acids? Design some new amino acids.
Q5 Where did amino acids come from before enzymes that make them, and before life started? Cowing explains that amino acids could have formed in the early planetesimal bodies of the early solar system, far from the sun 2. Those bodies would contain large amounts of ice that could have been melted by the heat produced by radioactive materials. The melted water could have interacted with other volatile compounds to form amino acids. These could have been contained in meteorites which struck Earth, providing a large concentration of amino acids which could have led to life on Earth.
Amino acids could have also been created through chemical synthesis reactions happening on Earth. In 1953, Miller and Urey combined ammonia, hydrogen, methane and water vapour and in a flask and subjected it to electrical sparks. From this experiment, they were able to create eleven standard amino acids.3
Q6 If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? I would expect it would be left-handed. Novotny writes: “a-Helices composed of L-amino acids are energetically more favourable in a right-handed conformation than in the left-handed mirror image of this arrangement due to steric hindrance between side-chain atoms and the main-chain carbonyl moiety. Conversely, D-amino acids will form more stable a-helices with a left-handed than with a righthanded conformation” 4
Q7 Can you discover additional helices in proteins? So far in class we’ve discussed the alpha helix. In Tamar’s “Protein Structure Hierarchy”, the author describes the pi helix and the collagen helix. 5
Q8 Why are most molecular helices right-handed? Because d-sugars direct the formation of right-handed helices and left hand versions don’t fit well.
MacDermott explains: “Biopolymer chirality is definitely determined by monomer chirality: l-amino acids can form only right-handed α-helices in protein secondary structures, and DNA naturally coils up into a right rather than left-hand B-form double helix because it is made of d-sugars. Why? Because of diastereomeric effects: d-sugars automatically direct the formation of a right-hand B-form double helix, because they simply do not fit well into a left-hand version. It is true that DNA does sometimes form a left-hand double helix, but this ‘Z-form’ helix is not a mirror image of the B-form, it has a totally different backbone conformation in order to be able to accommodate the d-sugars in a left-hand helix” 6
Eric Lindahl’s Youtube video explains that a helix such as the alpha helix is right-handed because it is due to L amino acids. 7
As a paper by Cole notes: “The remarkable predominance of right-handedness in beta-alpha-beta helical crossovers has been previously explained in terms of thermodynamic stability and kinetic accessibility.” 8
Q9a Why do beta sheets tend to aggregate? Beta sheets are prone to intermolecular hydrogen bonding at the sheet edges with other beta sheets. Other parts of beta sheets are hydrophobic. They will tend to be attracted to each other and repelled by water, which will tend to bring them together.
Q9b What is the driving force for β-sheet aggregation? Hydrophobic forces tend to repel beta sheets from water towards each other. Once they are close together, the attractive forces from intemolecular hydrogen bonding orders them together.
Q10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Q11. Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualisation
1. Briefly describe the protein you selected and why you selected it. I’ve chosen rhodopsin, whic is a light-sensitive receptor protein found in the rod cells of the retina, and important for dim light vision. 9. The human protein for rhodopsin is described in P08100 · OPSD_HUMAN.
2. Identify the amino acid sequence of your protein. The sequence is: MNGTEGPNFYVPFSNATGVVRSPFEYPQYYLAEPWQFSMLAAYMFLLIVLGFPINFLTLYVTVQHKKLRTPLNYILLNLAVADLFMVLGGFTSTLYTSLHGYFVFGPTGCNLEGFFATLGGEIALWSLVVLAIERYVVVCKPMSNFRFGENHAIMGVAFTWVMALACAAPPLAGWSRYIPEGLQCSCGIDYYTLKPEVNNESFVIYMFVVHFTIPMIIIFFCYGQLVFTVKEAAAQQQESATTQKAEKEVTRMVIIMVIAFLICWVPYASVAFYIFTHQGSNFGPIFMTIPAFFAKSAAIYNPVIYIMMNKQFRNCMLTTICCGKNPLGDDEASATVSKTETSQVAPA
2a. How long is it? The sequence is 348 amino acids long.
2b. What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. The most frequent amino acid is ‘Alanine’, which appears 32 times.
2c. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. By using (Uniprot BLAST)[https://www.uniprot.org/blast] I identified 5 homologs belonging to the Chimpanzee, Northern white-cheeked gibbon, and the Western lowland gorilla.
2e. Does your protein belong to any protein family? Rhodopsin belongs to the family of G protein–coupled receptors (GPCRs). 10
3. Identify the structure page of your protein in RCSB I entered the Uniprot ID for rhodopsin, P08100, into the RCSB search and ran the query. I then visualised the 3D representation through the page and obtained this result:
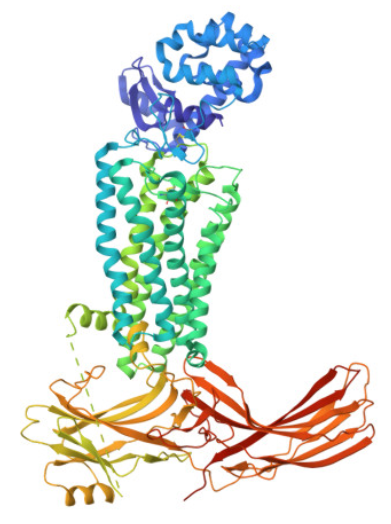
3a. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
 It appears to have first been released between 2015 and 2016. Its resolution is 3.3 Å
It appears to have first been released between 2015 and 2016. Its resolution is 3.3 Å
3b. Are there any other molecules in the solved structure apart from protein? The page is subtitled: “Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser”. Arrestin is another protein.
3c. Does your protein belong to any structure classification family? It belongs to a GPCR superfamily. A paper by Zhou indicates: “Rhodopsin is a member of class A of the GPCR superfamily, which is a large group of cell surface signaling receptors that transduce extracellular signals into intracellular pathways through the activation of heterotrimeric G proteins” 11 I tried typing entering ‘p08100’ into the EBI’s SCOP tool and used this query, yielded two domains. I clicked on 5W0P A:1-324 which indicated it was a member of the Class A (rhodopsin) G protein-coupled receptor-like’
4 Open the structure of your protein in any 3D molecule visualization software: PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands) Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
First I downloaded and installed PyMol. Then I searched for the PDB ID for ‘rhodopsin’ and obtained the ID 6QNO. I loaded the PDB structure for 6QNO and it provided me this:
Cartoon
 Ribbon
Ribbon
 Ball and stick
Ball and stick
Color the protein by secondary structure. Does it have more helices or sheets? I think it has more sheets. In this diagram, helices are coloured red and cover 3067 atoms. Beta sheets are coloured in yellow and cover 2845 atoms. If we count the distinct number of shapes rather than focus on atom coverage, I think there are more helices.
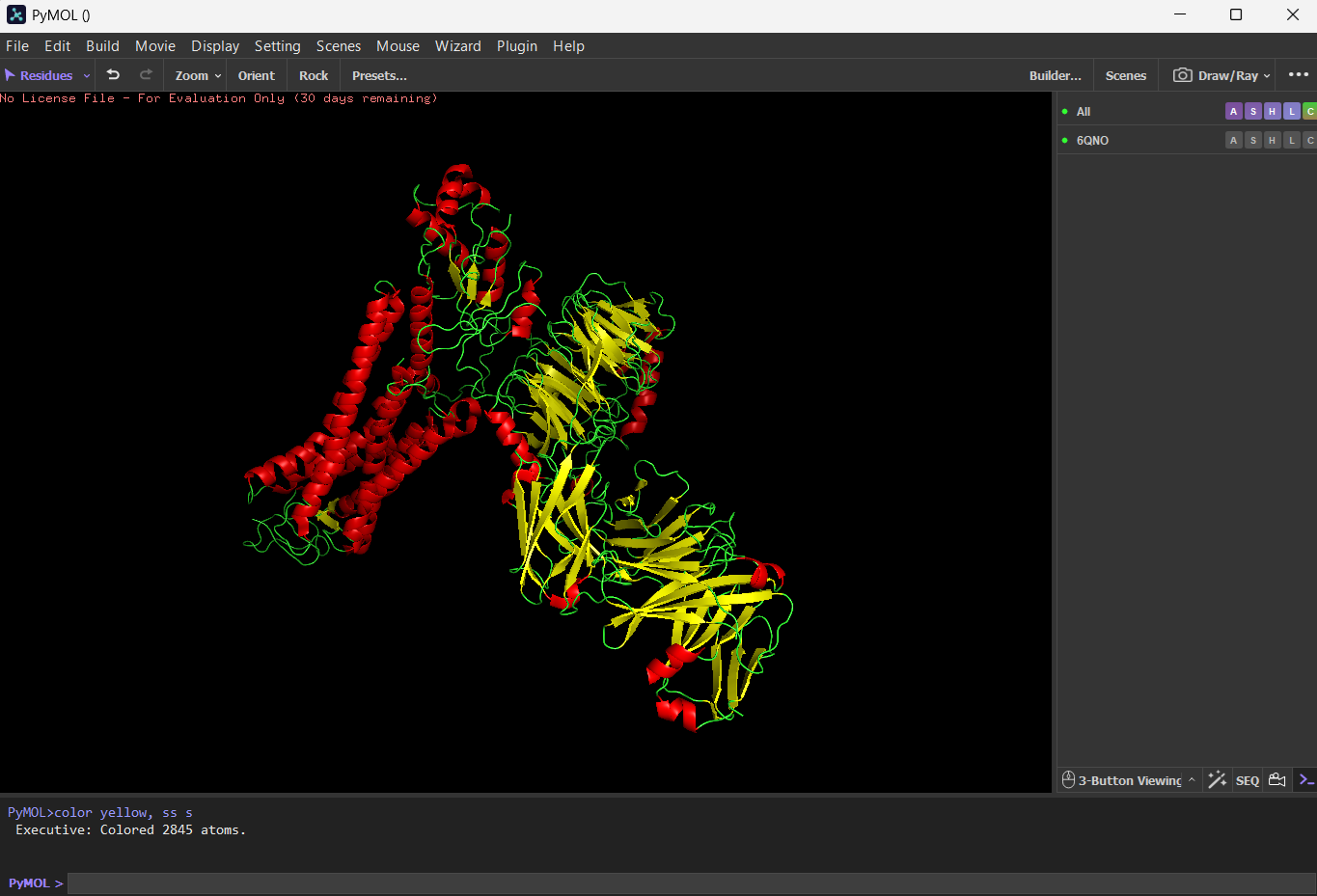
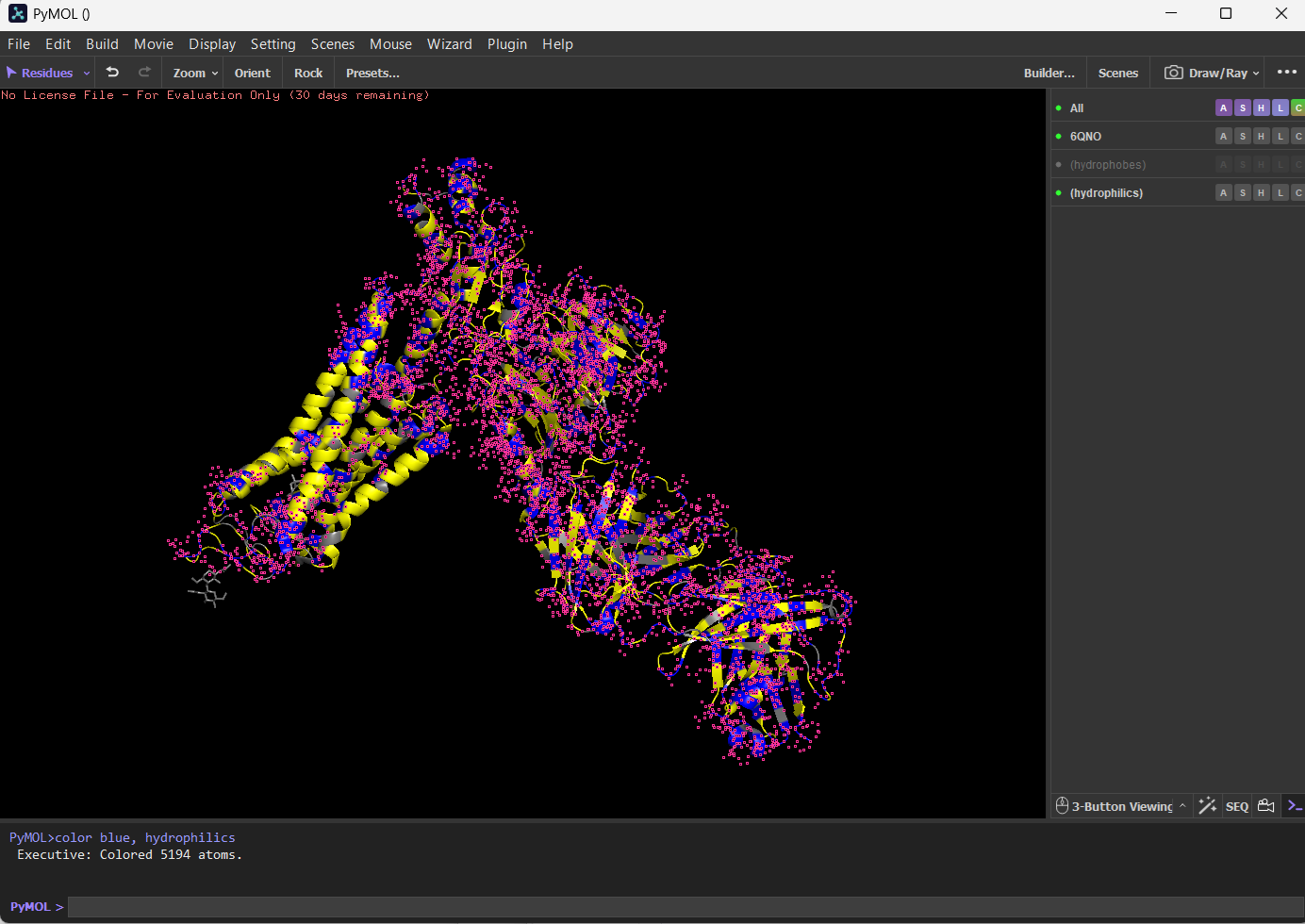
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I coloured hydrophilic residues blue and hydrophobic residues yellow. I observe that one bunch of hydrophobic residues tend to concentrate together in yellow on the left.
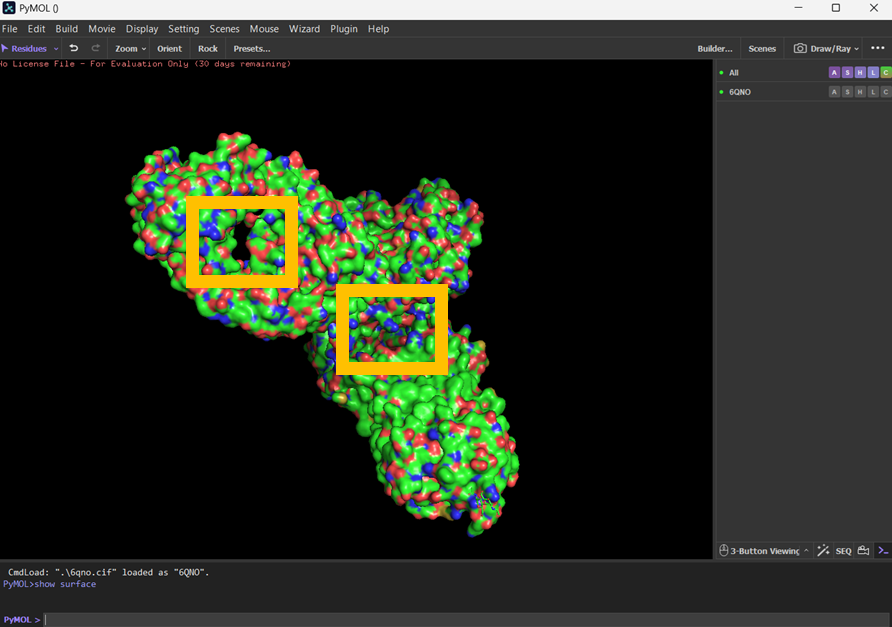
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I visualised the surface, rotated it and found two holes that I’ve highlighted in thick orange squares.
Part C: Using ML-Based Protein Design Tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU. Choose your favorite protein from the PDB. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
I’ve used the sequence for rhodopsin: MNGTEGPNFYVPFSNATGVVRSPFEYPQYYLAEPWQFSMLAAYMFLLIVLGFPINFLTLYVTVQHKKLRTPLNYILLNLAVADLFMVLGGFTSTLYTSLHGYFVFGPTGCNLEGFFATLGGEIALWSLVVLAIERYVVVCKPMSNFRFGENHAIMGVAFTWVMALACAAPPLAGWSRYIPEGLQCSCGIDYYTLKPEVNNESFVIYMFVVHFTIPMIIIFFCYGQLVFTVKEAAAQQQESATTQKAEKEVTRMVIIMVIAFLICWVPYASVAFYIFTHQGSNFGPIFMTIPAFFAKSAAIYNPVIYIMMNKQFRNCMLTTICCGKNPLGDDEASATVSKTETSQVAPA
C1. Protein Language Modeling
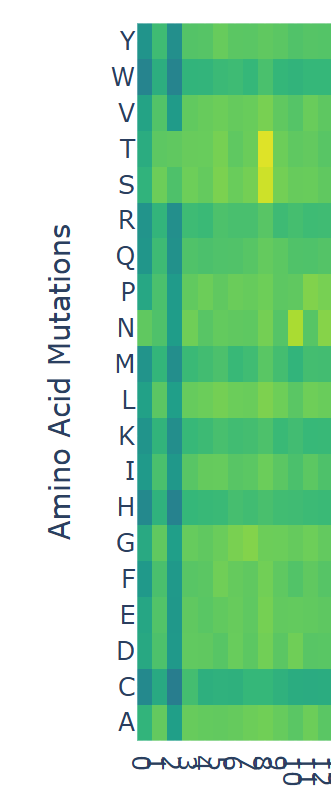
Deep Mutational Scans Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out) (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I looked at the scan and noticed that at position ‘8’ there were some yellow bars. Counting from 0 to 8 inclusively, the sequence ends in ‘F’, which has a model score of
model score = 0.5256555 012345678 MNGTEGPNF
model score = 2.980395 012345678 MNGTEGPNS
model score = 3.427129 012345678 MNGTEGPNT
If I assume that the higher the model score, the more likely an amino acid will occupy the position, it suggests that although ‘F’ appears in the rhodopsin sequence, in other proteins that begin with MNGTEGPN are more likely to have a ‘T’. For any of the dark blue squares (which tend to be very negative) I would assume their presence in the amino acid would be unstable or cause harm.
Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins? I think they do. I zoomed into the map and tried to pick two very close neighbours. My plan was to find the Uniprot IDs for neighbouring proteins and do a Protein blast sequence to determine how similar they were to one another. But I found it difficult to get IDs for neighbouring pairs.
I zoomed in on these two data points:
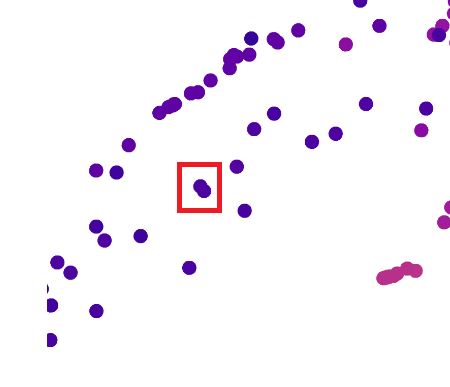
Up close, this is what those two dots represented:
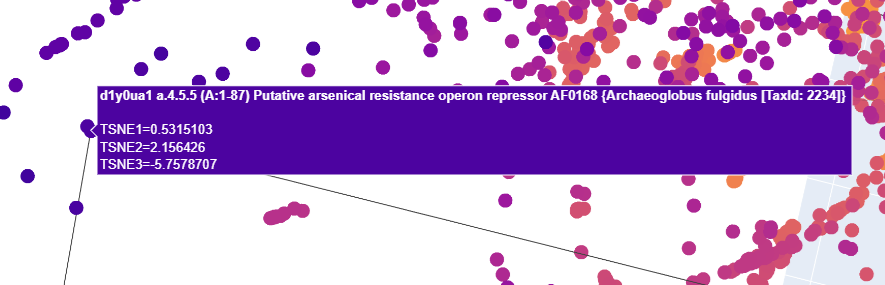
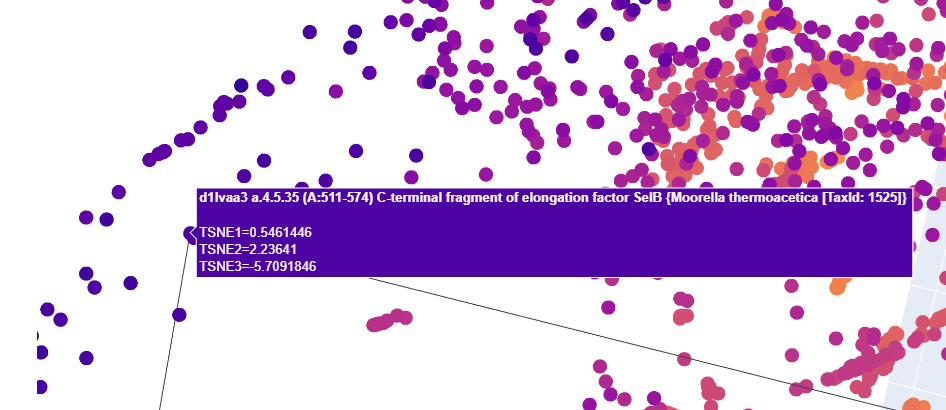
I tried to find Uniprot entries for both of these for each
- Putative arsenical resistance operon repressor AF0168 Archaeoglobus fulgidus
- C-terminal fragment of elongation factor SelB Moorella thermoacetica
I downloaded FASTA files for each. I then went to the NCBI’s BLASTP facility:
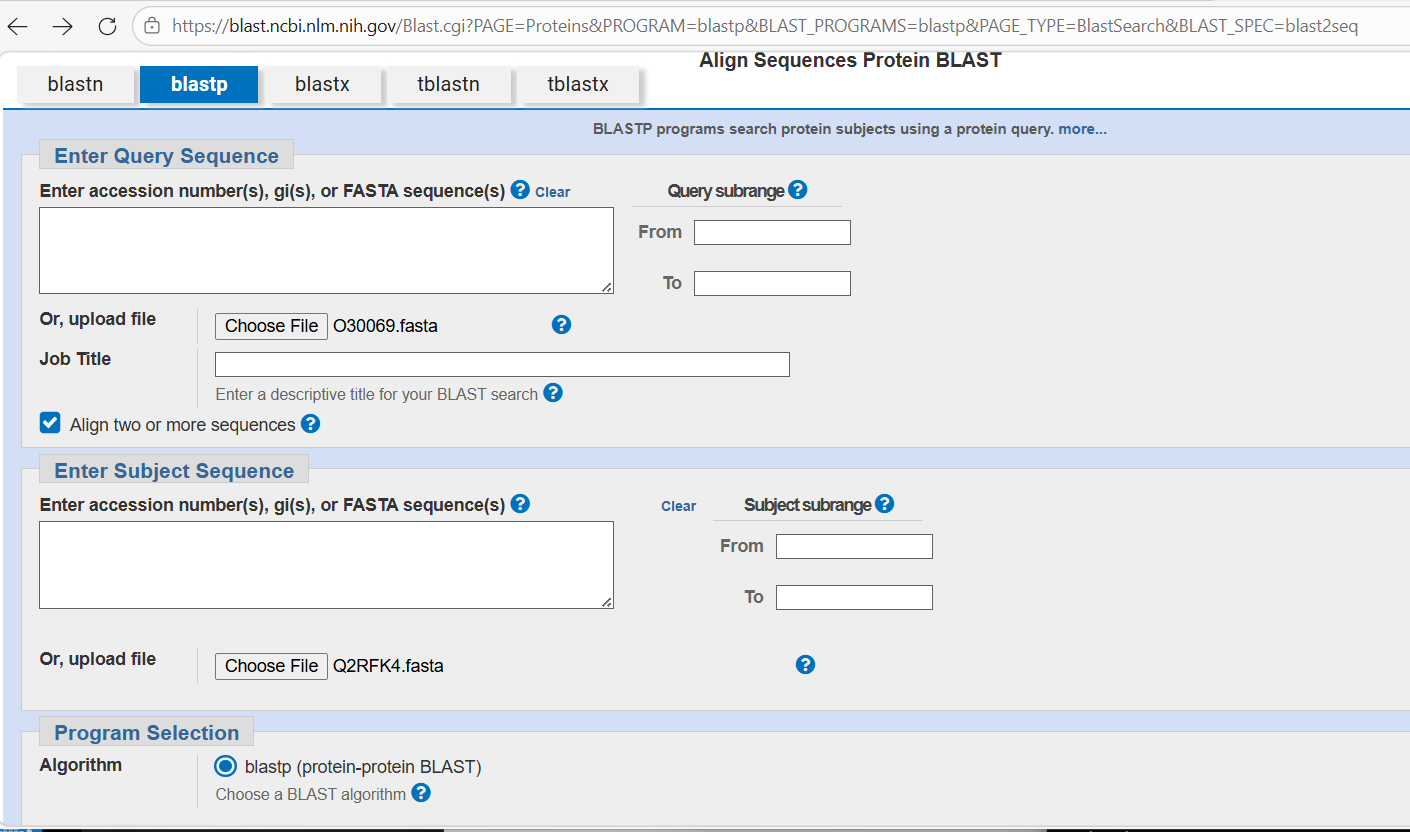
I loaded the FASTA files and then waited for the blast to complete. The results indicated they don’t appear to have much similarity with one another and I’m not sure why - that doesn’t fit well with the apparent similarity suggested by the close collocation of the proteins in the visualisation.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?** First I tried to fold the rhodopsin protein with no changes, and then in the third position where I substituted a ‘F’ for a ‘T’. There wasn’t much change. But then I made the sequence include a lot of Cs and Qs and I started to notice the left end started to distort a lot.
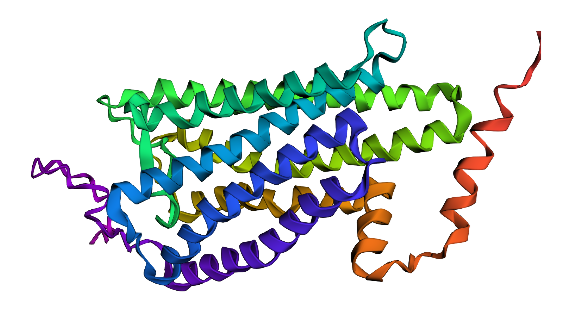 Rhodopsin protein with no changes.
Rhodopsin protein with no changes.
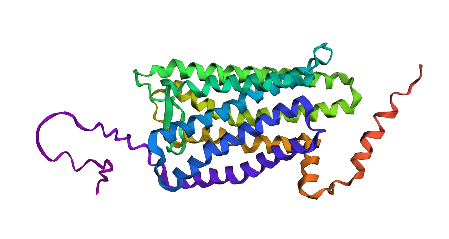 Rhodopsin protein with many alterations.
Rhodopsin protein with many alterations.
Part D. Group Brainstorm on Bacteriophage Engineering
Brainstorm Session Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”). Write a 1-page proposal (bullet points or short paragraphs) describing: Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”). Why do you think those tools might help solve your chosen sub-problem? Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”). Include a schematic of your pipeline. This resource may be useful: HTGAA Protein Engineering Tools Each individually put your plan on your HTGAA website Include your group’s short plan for engineering a bacteriophage
For my review of the Bacteriophage Final Projects Goals for engineering the L Protein, I focused on the goal of designing bacteriophages that showed increased stability. Initially I prompted ChatGPT to help determine how researchers might stabilise a lysis protein that is created by bacteriophages used to kill e coli12 According to the response, the challenges for making stable bacteriophages include the risk of denaturation at higher temperatures or broad ranges of pH, degradation by bacterial enzymes, and the risk of aggregation or precipitation when the bacteriophage is purified for therapeutic use. It recommended four strategies to consider:
- rational design, which is: “…a molecular method aimed at altering the genetic makeup of existing enzymes to improve their structural and functional properties in a predictable manner, relying on prior knowledge of the enzyme’s molecular details”13
- fusion proteins, which are: “…A fusion protein is a single polypeptide composed of two or more distinct protein domains encoded initially by separate genes but artificially linked to function as one unit.” They are often incorporate tags into a protein which help improve the solubility, purification and detection of the main protein of interest.14
- directed evolution, which involves “… creating mutants by random mutagenesis and recombination followed by the screening of mutants for desired characteristics such as increase stability and changed catalytic specificity”15
- expression optimisation, which: “…focuses on maximizing the yield of proteins or other biological molecules in various expression systems” 16
I’d focus on directed evolution, and perhaps randomly introducing random mutations in an amino acid sequence for the L protein, and using ESM2 to help identify which ones would be likely to form. I could then try to use tools that tested how well the resulting L proteins would bind to mutated proteins that act as the chaperone sites in the host such as DnaJ. I think the ESM2 model would help me make some intelligent guesses about mutations that would result in a viable protein.
Doig, Andrew J. “Frozen, but no accident–why the 20 standard amino acids were selected.” The FEBS journal 284.9 (2017): 1296-1305. ↩︎
Cowing, Keith. “How Were Amino Acids Formed Before The Origin Of Life On Earth?” Astrobiology, April 5, 2023. URL: https://astrobiology.com/2023/04/how-were-amino-acids-formed-before-the-origin-of-life-on-earth.html ↩︎
Gutiérrez-Preciado, Ana, Hector Romero, and Mariana Peimbert. “An evolutionary perspective on amino acids.” Nature Education 3.9 (2010): 29. ↩︎
Novotny, Marian, and Gerard J. Kleywegt. “A survey of left-handed helices in protein structures.” Journal of molecular biology 347.2 (2005): 231-241. ↩︎
Schlick, Tamar. “Protein structure hierarchy.” Molecular Modeling and Simulation: An Interdisciplinary Guide: An Interdisciplinary Guide. New York, NY: Springer New York, 2010. 105-128. ↩︎
MacDermott, A. J. “8.2 Perspective and concepts: Biomolecular significance of homochirality: The origin of the homochiral signature of life.” Comprehensive chirality. 2012. 11-38. ↩︎
Lindahl E., ‘Lecture 05, concept 08: The alpha helix is right-handed due to L amino acides.’, URL: https://www.youtube.com/watch?v=rdkXOxLHDws ↩︎
Cole, Benjamin J., and Christopher Bystroff. “Alpha helical crossovers favor right‐handed supersecondary structures by kinetic trapping: The phone cord effect in protein folding.” Protein Science 18.8 (2009): 1602-1608. ↩︎
Rhodopsin. URL: https://www.britannica.com/science/rhodopsin ↩︎
Palczewski, Krzysztof. “G protein–coupled receptor rhodopsin.” Annu. Rev. Biochem. 75.1 (2006): 743-767. ↩︎
Zhou, X. Edward, Karsten Melcher, and H. Eric Xu. “Structure and activation of rhodopsin.” Acta Pharmacologica Sinica 33.3 (2012): 291-299. ↩︎
“How would researchers try to stabilise the lysis protein that is created by bacteriophages used to kill e coli? Please include sources” prompt. ChatGPT, 29 March version, OpenAI, 29 March 2026, chatgpt.com. ↩︎
Rational Design, ScienceDirect, https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/rational-design ↩︎
Fusion Proteins Overview, SinoBiological, https://www.sinobiological.com/resource/protein-review/fusion-protein ↩︎
Directed Evolution, ScienceDirect, https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/directed-evolution ↩︎
Understanding Expression Optimization: Enhancing Protein Production Efficiently, InfinixBio, https://www.infinixbio.com/glossary/understanding-expression-optimization-enhancing-protein-production-efficiently/ ↩︎