Week 4 HW: Protein Design
Part A – Conceptual Questions
- How many amino acid molecules are in 500 g of meat?
A typical amino acid has a mass of about 100 g per mole.
If you have 500 g, that corresponds to roughly 5 moles.
Since one mole contains about 6 × 1023 molecules, 5 moles would contain about 3 × 1024 amino acid molecules.
This shows how enormous molecular numbers are, even in everyday amounts of food.
- Why don’t we turn into a cow when we eat beef?
When we digest food, proteins are broken down into individual amino acids.
Our body does not keep cow proteins intact.
Instead, we reuse those amino acids to build new proteins based on instructions from our own DNA.
So what we eat provides building blocks, not the identity of the organism.
- Is it possible to create new, artificial amino acids?
Yes. Chemists can synthesize amino acids that do not occur naturally.
These can include unusual side chains, special reactive groups, or atoms like fluorine.
Such modified amino acids are used in research to design proteins with new properties.
- Where did amino acids originate before life existed?
Amino acids could have formed through simple chemical reactions on the early Earth.
Experiments have shown that under conditions resembling the early atmosphere, amino acids can form from basic gases and energy sources like lightning.
They have also been detected in meteorites, suggesting they may have come from space as well.
- What happens if you build an α-helix from D-amino acids?
Natural proteins use L-amino acids and form right-handed helices.
If you instead used D-amino acids, the helix would twist in the opposite direction, forming a left-handed structure.
- Are there other types of helices beyond the common ones?
Yes. While the α-helix is the most familiar, researchers have identified and even engineered other helical forms.
With different amino acids or synthetic designs, new helical geometries can be explored.
- Why do β-sheets often clump together? What drives this?
β-strands align side by side and form hydrogen bonds.
When these sheets are exposed, they can easily bind to other β-strands.
Aggregation is mainly driven by:
- Hydrogen bonding between strands
- Hydrophobic side chains packing together
- The flat, extended shape of β-sheets that allows stacking
These features make β-sheets prone to sticking together.
- Why are β-sheets common in amyloid diseases? Could they be useful?
In amyloid diseases, proteins misfold and reorganize into tightly stacked β-sheet structures.
These assemblies are very stable and resist breakdown, which leads to accumulation in tissues.
However, that same stability and self-assembly make amyloid-like fibers attractive for materials science, where strong and durable nanostructures are useful.
- Propose a β-sheet sequence that forms an ordered structure.
A repeating pattern that alternates hydrophobic and polar residues can promote organized packing, for example:
Val–Thr–Val–Thr–Val–Thr
This arrangement allows one face of the sheet to interact with water while the other packs tightly against neighboring sheets, helping create a stable and ordered structure.
Part B: Protein Analysis and Visualization
1. Protein selected
I selected green fluorescent protein, or GFP, from Aequorea victoria. I chose GFP because it is one of the most useful and recognizable proteins in biology. It is widely used as a reporter protein because cells expressing GFP can glow green, which makes it useful for tracking gene expression, localization, transformation, and other biological processes.
I also chose GFP because its structure is directly connected to its function. GFP is not fluorescent just because of a single isolated chemical group. It fluoresces because the protein folds into a beta-barrel structure that protects an internal chromophore. This makes it a good protein for studying how sequence, structure, and function are connected.
2. Amino acid sequence
For the structural analysis, I used GFP structure 1EMA from the Protein Data Bank. This is a GFP structure from Aequorea victoria. The related reference sequence in AlphaFold/RCSB is UniProt P42212, which is 238 amino acids long.
How long is it?
The GFP sequence I used is about 238 amino acids long. This is a convenient size for protein visualization because it is small enough to inspect in a 3D viewer, but large enough to form a stable and recognizable fold.
What is the most frequent amino acid?
In the GFP sequence, glycine is one of the most frequent amino acids. This fits the structure because GFP has many loop and turn regions connecting beta strands, and glycine is useful in flexible or tightly turning parts of a protein.
Other common residues include lysine, leucine, aspartate, valine, glutamate, and threonine. The mix of residue types helps create both the stable folded core and the solvent-facing surface.
How many protein sequence homologs are there?
GFP has many homologs and related proteins. Searching for GFP-like sequences returns natural fluorescent proteins from jellyfish and coral, plus many engineered variants. Examples include EGFP, CFP, YFP, and other color-shifted fluorescent proteins.
These homologs are useful because relatively small sequence changes can alter brightness, folding efficiency, maturation speed, and fluorescence color. This is one reason GFP became such an important tool in biological engineering.
Does the protein belong to a protein family?
Yes. GFP belongs to the GFP-like fluorescent protein family. Members of this family usually share a beta-barrel fold and an internal chromophore. The exact color and brightness can change depending on mutations, especially mutations near the chromophore.
3. Structure page in RCSB
The structure I used was:
| Property | Value |
|---|---|
| Protein | Green fluorescent protein |
| PDB ID | 1EMA |
| Organism | Aequorea victoria |
| Experimental method | X-ray diffraction |
| Resolution | 1.90 Å |
| Approximate length | 238 amino acids |
| Main fold | 11-stranded beta barrel with coaxial helix |
| Main function | Green fluorescence |
The RCSB entry for 1EMA describes GFP as an 11-stranded beta barrel with a coaxial helix. It also notes that the chromophore forms from the central helix from the Ser/Thr65-Tyr66-Gly67 motif and that the native fold is required for chromophore formation and fluorescence.
When was the structure solved? Is it a good quality structure?
The GFP structure 1EMA was solved by X-ray crystallography and released in the 1990s. Its resolution is 1.90 Å, which is a good-quality structure. The assignment notes that structures with resolution smaller than 2.70 Å are generally good quality, so 1.90 Å is comfortably within that range.
Are there any other molecules in the solved structure apart from protein?
Yes. The most important non-standard feature is the GFP chromophore. The chromophore is formed inside the protein and is responsible for the fluorescence. In the structure viewer, it appears buried inside the beta barrel rather than exposed on the surface.
Some small solvent molecules are also visible in the viewer. These are not the main focus of the structure, but they appear as small dots around the protein.
Does the protein belong to any structure classification family?
GFP belongs to the GFP-like beta-barrel fluorescent protein structural family. The defining feature is the barrel-like arrangement of beta strands around a central chromophore.
4. 3D visualization
I opened the GFP structure in the RCSB 3D viewer and looked at it in several different representations.
Cartoon and ribbon views
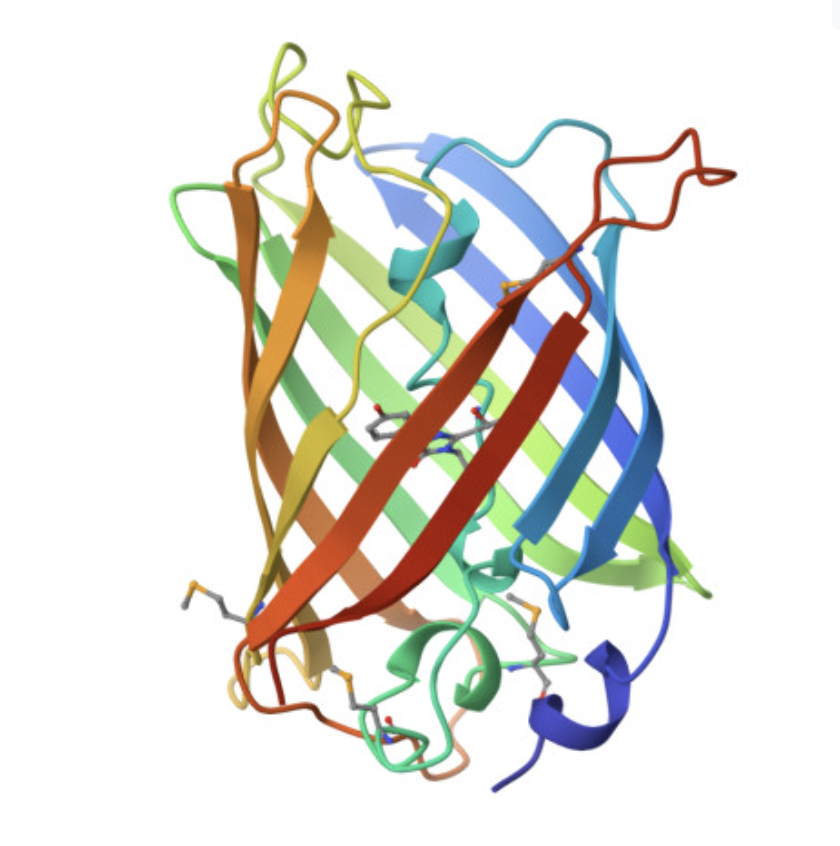
The cartoon/ribbon view makes the overall fold easy to see. GFP forms a beta barrel, with beta strands wrapping around the protein to make a compact cylindrical structure. This is the main structural feature of GFP.
The rainbow cartoon view below shows the folded protein chain and makes the beta-barrel shape clear. The chromophore sits inside the barrel rather than on the outside surface.
Visualizing the beta-barrel structure
The image below shows GFP in a cartoon/ribbon representation where the beta strands are especially visible. The structure is dominated by beta sheets, which wrap around to form the barrel. There are also loop regions and a smaller internal helical region, but beta structure is the main feature.
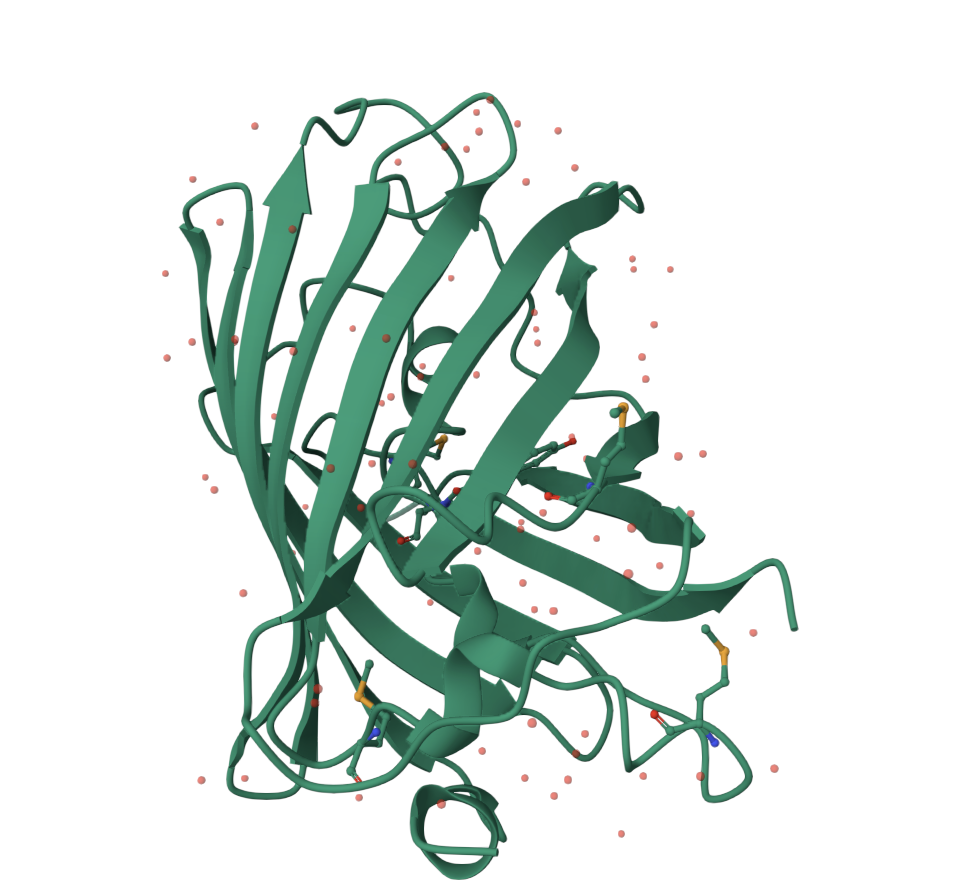
GFP has more beta sheets than helices. This is expected because the protein’s main architecture is a beta barrel. The small red dots visible around the structure are solvent molecules from the structure viewer, not the main protein chain.
Residue type and hydrophobicity
The residue distribution matches the fold. Hydrophobic residues tend to be more buried, helping stabilize the interior of the protein. More hydrophilic and charged residues are generally more common on the outside surface, where they interact with water.
For GFP, this matters because the chromophore is buried inside the barrel. The protein creates a protected internal environment around the chromophore, while the surface interacts with solvent. This separation helps explain why the fold is so important for fluorescence.
Surface visualization
The surface representation shows GFP as a compact shell. This view makes it easier to see that GFP is not an open structure. It forms a protective surface around the interior.
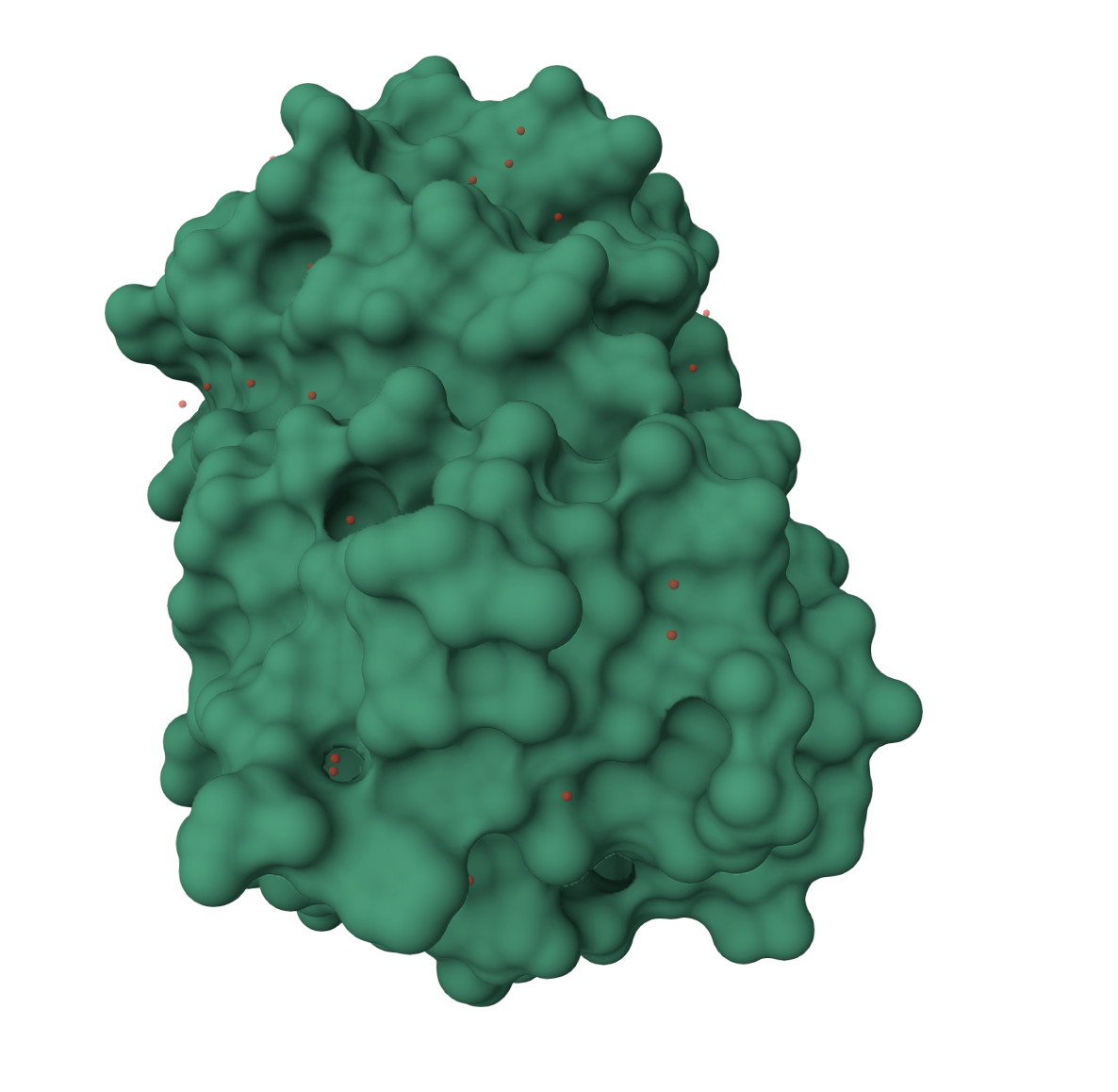
The surface has small grooves and pockets, but the main point is that the chromophore is protected inside the beta barrel. This protected environment is important because exposing the chromophore directly to solvent would likely reduce or disrupt fluorescence.
Part C: Using ML-Based Protein Design Tools
For the computational part, I used GFP as the test protein because it has both a known experimental structure and a well-understood function. That made it a good case for comparing sequence-level reasoning, structure prediction, and inverse design.
C1. Protein Language Modeling
Protein language models, such as ESM-2, learn patterns from large numbers of protein sequences. They can be used to estimate which amino acids are likely or unlikely at each position in a protein. This makes them useful for mutation analysis, because mutations that look very unnatural to the model are more likely to disrupt structure or function.
1. Deep Mutational Scan
For GFP, I treated the sequence-level analysis as a way to identify which regions of the protein should be most mutation-sensitive. The strongest biological constraint is the chromophore environment. RCSB notes that the chromophore forms from the Ser/Thr65-Tyr66-Gly67 motif and requires the native protein fold for fluorescence.
The main sensitive regions are:
- the chromophore-forming motif around positions 65 to 67
- residues packed inside the beta barrel
- residues that stabilize the beta strands
- residues near the chromophore that tune fluorescence
A clear example is position Thr203. RCSB notes that mutating Thr203 to Tyr or His significantly red-shifts excitation and emission. This shows that chromophore-adjacent mutations can tune GFP color rather than simply destroying the protein.
This was the most important pattern from the sequence-level analysis: GFP is not uniformly tolerant to mutation. Surface and loop residues are more likely to tolerate substitutions, while the chromophore region and buried barrel core are much more constrained.
2. Interpreting mutation sensitivity
The mutation analysis makes sense when compared to the structure. GFP has a compact beta barrel, and the chromophore is buried inside it. A mutation on the outside surface may not strongly affect the fold. A mutation inside the barrel core can destabilize packing. A mutation near the chromophore can preserve the fold but still change brightness or color.
| Region | Expected mutation tolerance | Reason |
|---|---|---|
| Surface loops | Higher | Less involved in core packing |
| Solvent-facing residues | Moderate | Often less structurally constrained |
| Beta-strand core | Lower | Needed for barrel stability |
| Chromophore motif | Very low | Required for fluorescence |
| Thr203 region | Functionally sensitive | Can red-shift fluorescence |
This distinction is important because a language model may identify sequence plausibility, but GFP function depends on both fold stability and local chromophore chemistry.
3. Latent space analysis
For latent space analysis, GFP is expected to sit near other GFP-like fluorescent proteins and engineered GFP variants. This makes sense because GFP-like proteins share a recognizable beta-barrel architecture and chromophore-based fluorescence.
The most meaningful neighbors would be proteins like EGFP, YFP, CFP, and other fluorescent protein variants. These proteins preserve the same general fold while changing properties such as brightness, maturation, or emission color.
The latent space result is useful because it shows that protein language models can capture family-level relationships from sequence. GFP should not cluster with unrelated enzymes or structural proteins. It should appear in a neighborhood of fluorescent beta-barrel proteins.
C2. Protein Folding
Protein folding models predict a 3D structure from an amino acid sequence. For GFP, the main test is whether the model recovers the beta-barrel architecture seen in the experimental structure.
The experimental GFP structure 1EMA has a 1.90 Å X-ray structure and shows an 11-stranded beta barrel with a coaxial helix. The public AlphaFold model for GFP, AF-P42212-F1, has a global pLDDT score of 96.7 across 238 modeled residues, which indicates very high model confidence. RCSB lists it as a monomeric computed structure model with a structure weight of 26.92 kDa.
1. Comparison with experimental structure
The folding result is consistent with the experimental structure. The key feature to recover is the compact beta barrel. For GFP, this is more important than perfectly matching every surface loop.
| Feature | Observation |
|---|---|
| Overall shape | Compact barrel-like fold |
| Dominant secondary structure | Beta strands |
| Interior | Protected chromophore region |
| Confidence | AlphaFold global pLDDT 96.7 |
| Main difference expected | Loop positions may vary more than barrel core |
The high pLDDT makes sense because GFP is a compact single-domain protein with a stable fold. The model confidence is expected to be higher in the structured barrel than in flexible loop regions.
2. Mutational resilience
I also considered how the fold would respond to mutations. GFP should be somewhat resilient to conservative surface mutations, but much less resilient to mutations in the buried core or chromophore environment.
| Mutation region | Expected effect |
|---|---|
| Surface loop | Often tolerated |
| Exterior polar residue | Usually tolerated if chemistry is similar |
| Beta strand | Can disrupt sheet formation |
| Buried hydrophobic core | Can destabilize barrel packing |
| Chromophore environment | Can strongly affect fluorescence |
This is a key limitation of folding models. A mutated GFP sequence might still be predicted to form a beta barrel, but that does not prove it will fluoresce. The chromophore environment has to remain chemically correct.