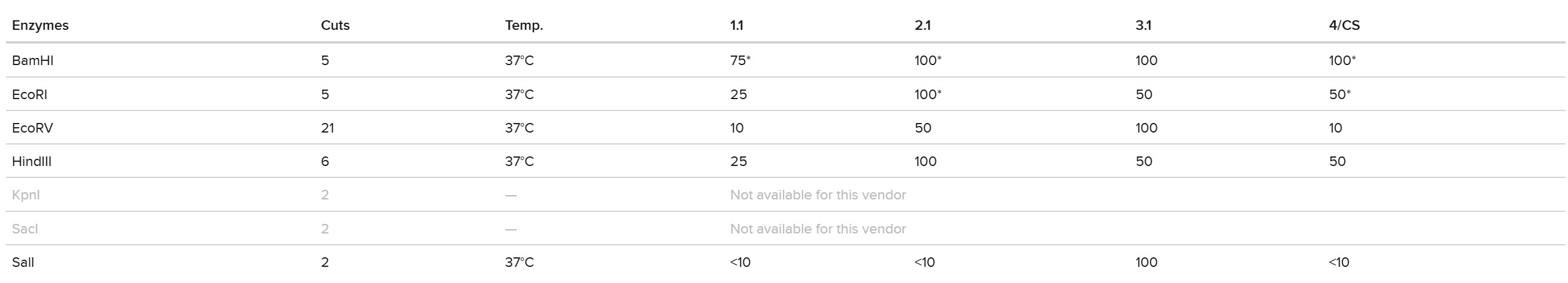
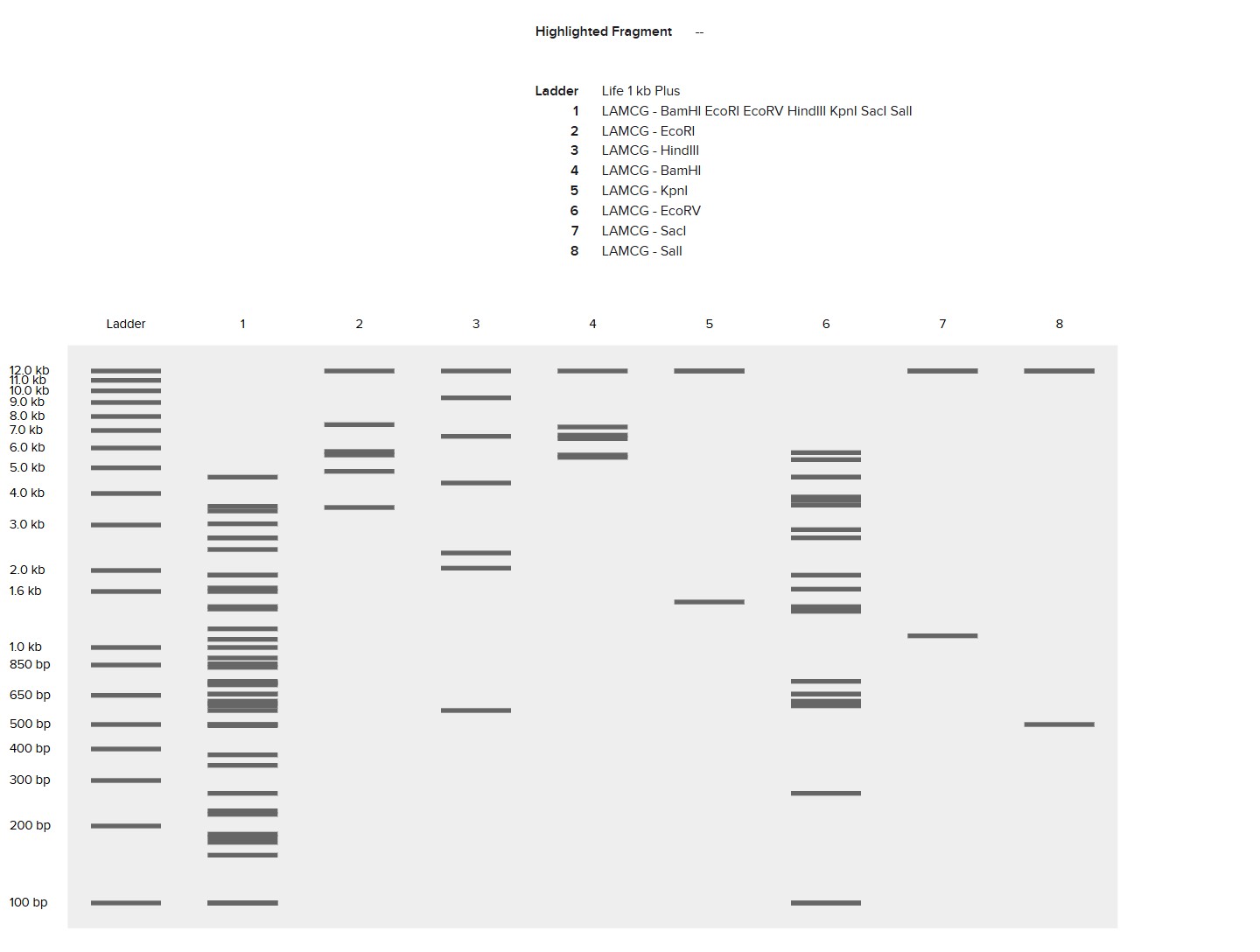
This week, I reviewed how gel electrophoresis turns a DNA “mixture” into an interpretable pattern. In an agarose gel, DNA fragments migrate toward the positive electrode because DNA is negatively charged, and smaller fragments travel farther through the gel matrix than larger ones. A DNA ladder provides a size reference so unknown bands can be estimated in base pairs. When a restriction enzyme digest is performed, the DNA sequence is converted into a predictable set of fragment lengths, and those fragments appear as bands at specific positions. Band brightness is roughly related to how much DNA mass is in that fragment (longer fragments can look brighter if molar amounts are similar). Overall, the key idea is that restriction digests plus gels let you “read out” a cutting pattern, validate identity, and compare designs or conditions in a simple visual way.
I created a “gel art” pattern inspired by the idea that restriction digests can produce recognizable visual signatures. The design uses symmetry and band density as the main visual elements: enzymes with few cuts generate sparse lanes (lighter), while enzymes with many cuts generate dense lanes (darker).
Lane plan (left → right): Ladder (Life 1 kb Plus), ApaI, EcoRI, HaeIII, EcoRI, ApaI.
HaeIII creates a high-density fragmentation pattern that acts as the “dark center,” while EcoRI and ApaI provide low-cut, high-molecular-weight bands that frame the pattern.
Part 3 — DNA Design Challenge
3.1 Protein choice
I chose sfGFP (superfolder GFP) as the target protein because it is a robust fluorescent reporter widely used to validate expression, folding, and cloning workflows. It provides an easy quantitative readout (fluorescence) and is a standard “sanity check” part in many synthetic biology builds.
3.2 Reverse translation (baseline CDS)
Starting from the sfGFP amino-acid sequence, I generated a DNA coding sequence (CDS) by back-translation using a codon-usage–matching approach (Benchling output). This produces a valid CDS encoding the same protein sequence.
Protein length: 246 aa
DNA CDS length (no stop codon): 738 bp
sfGFP amino-acid sequence (246 aa):

MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTL
VTTLTYGVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLV
NRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKNGIKANFKIRHNVEDGSVQLAD
HYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYKGS
HHHHHH


Back-translated / codon-usage–matched CDS (low GC target):
ATGTCAAAAGGTGAGGAATTATTTACCGGAGTAGTACCAATACTGGTAGAATTAGATGGCG
ATGTTAATGGGCATAAGTTTTCAGTGCGTGGAGAAGGAGAAGGCGATGCTACAAATGGAAA
ATTAACGTTAAAATTTATTTGTACTACTGGGAAACTACCTGTACCTTGGCCAACTTTAGTT
ACAACCTTAACATATGGTGTACAATGTTTTTCTCGTTATCCAGATCATATGAAACGTCATG
ATTTTTTTAAAAGTGCGATGCCTGAAGGTTACGTTCAAGAAAGAACTATATCTTTTAAAGAT
GATGGTACATATAAAACACGAGCTGAAGTAAAATTTGAAGGTGATACTTTGGTTAATAGAAT
TGAACTTAAAGGGATTGATTTTAAGGAAGATGGAAATATTCTCGGACACAAATTAGAATACA
ATTTTAATTCACATAATGTTTACATAACAGCTGATAAACAAAAAAATGGCATAAAAGCAAAT
TTTAAAATAAGACATAATGTAGAAGATGGAAGTGTCCAATTAGCAGATCATTATCAGCAAAA
CACACCAATTGGTGATGGTCCTGTCCTTTTACCAGATAATCATTATTTATCAACCCAATCTG
TTTTGTCAAAAGATCCGAATGAAAAAAGAGATCATATGGTTTTATTGGAATTTGTAACAGCA
GCAGGTATTACTCATGGCATGGATGAATTATATAAAGGCTCTCATCATCATCATCATCAT
Codon optimization for E. coli
I then codon-optimized the CDS for Escherichia coli using a “use best codon” strategy. As expected, the amino-acid sequence is unchanged, but the nucleotide sequence changes due to synonymous codon choices that better match E. coli translation preferences.
Nucleotide identity (baseline vs optimized): 76.96%
GC content (baseline, codon-usage–matched): 33.0%
GC content (optimized, best-codon): 50.0%
Rare codons: 11 (baseline) vs 0 (optimized)
Hairpins (reported by the tool): 0 in both
Thymine fraction (reported by the tool): 0.30 (baseline) vs 0.21 (optimized)
ATGAGCAAAGGCGAAGAACTGTTTACCGGCGTGGTGCCGATTCTGGTGGAACTGGATGGCGAT
GTGAACGGCCATAAATTTAGCGTGCGCGGCGAAGGCGAAGGCGATGCGACCAACGGCAAACT
GACCCTGAAATTTATTTGCACCACCGGCAAACTGCCGGTGCCGTGGCCGACCCTGGTGACCA
CCCTGACCTATGGCGTGCAGTGCTTTAGCCGCTATCCGGATCATATGAAACGCCATGATTTT
TTTAAAAGCGCGATGCCGGAAGGCTATGTGCAGGAACGCACCATTAGCTTTAAAGATGATGG
CACCTATAAAACCCGCGCGGAAGTGAAATTTGAAGGCGATACCCTGGTGAACCGCATTGAAC
TGAAAGGCATTGATTTTAAAGAAGATGGCAACATTCTGGGCCATAAACTGGAATATAACTTT
AACAGCCATAACGTGTATATTACCGCGGATAAACAGAAAAACGGCATTAAAGCGAACTTTAA
AATTCGCCATAACGTGGAAGATGGCAGCGTGCAGCTGGCGGATCATTATCAGCAGAACACCC
CGATTGGCGATGGCCCGGTGCTGCTGCCGGATAACCATTATCTGAGCACCCAGAGCGTGCTG
AGCAAAGATCCGAACGAAAAACGCGATCATATGGTGCTGCTGGAATTTGTGACCGCGGCGGGC
ATTACCCATGGCATGGATGAACTGTATAAAGGCAGCCATCATCATCATCATCATCAT
Best way to obtain the DNA
For a ~0.74 kb CDS like sfGFP, the most straightforward approach is gene synthesis (ordering a dsDNA fragment). It is fast, accurate, and does not require an existing template. If a plasmid template is already available, an alternative is PCR amplification + cloning (e.g., restriction cloning or Gibson), but synthesis avoids PCR-introduced mutations and simplifies the workflow.
Codon-optimized CDS (best codons, medium GC target)
## Part 4 — DNA Write (Ordering + Construct Design)
### 4.1 Expression cassette design (what I would build)
To express **sfGFP in *E. coli***, I would build a standard bacterial expression cassette:
- **Promoter:** T7 promoter (for high expression in BL21(DE3)-like strains) or a strong constitutive promoter if T7 is not desired
- **RBS:** strong bacterial RBS (e.g., a consensus Shine–Dalgarno / gene10-like RBS)
- **CDS:** sfGFP coding sequence, codon-optimized for *E. coli* (AA sequence unchanged)
- **Tag / stop:** optional **C-terminal 6xHis** tag for purification + **stop codon**
- **Terminator:** strong transcription terminator (e.g., T7 terminator / bacterial terminator)
This design is simple, robust, and makes fluorescence an immediate readout for “does expression work?”.
### 4.2 What I would order (DNA “write” step)
Because the sfGFP CDS is short (~0.7–0.8 kb), the most straightforward approach is **DNA synthesis** (a dsDNA fragment or a cloned gene). Concretely, I would order one of these:
**Option A — Gene fragment (fast + flexible)**
- Order the **sfGFP insert as dsDNA** with flanking overlaps for Gibson/HiFi assembly (or with restriction sites).
- Then clone into an expression plasmid in the lab.
**Option B — Cloned gene in a plasmid (one-step ready)**
- Order **sfGFP already cloned** into a high-copy plasmid backbone.
### 4.3 Twist Bioscience access limitation (Argentina) + workaround plan
From my location (Argentina), the Twist ordering portal is not accessible and prompts me to contact a local operator. In a real order scenario, I would do one of the following:

1) **Contact Twist local sales/support** (as requested) and place the order via email (sequence + vector + cloning format).
2) Use an **alternative synthesis provider** that ships to my region (e.g., ordering a dsDNA fragment from another vendor) and then perform the same assembly into an equivalent plasmid backbone.
For the purposes of this homework, I describe the intended order and construct as if placing a standard synthesis + cloning order.
### 4.4 Vector choice and final construct
If using Twist’s catalog, I would choose a standard **high-copy AmpR plasmid backbone** (e.g., a pTwist Amp high-copy–type vector), and insert the sfGFP expression cassette into it.
Final construct conceptually looks like:
**[T7 promoter] – [RBS] – [sfGFP CDS (E. coli optimized)] – [6xHis] – [STOP] – [Terminator]**
### 4.5 How I would obtain protein from this DNA (high-level workflow)
1) **Assemble** the insert into the plasmid (Gibson/HiFi or restriction cloning).
2) **Transform** into *E. coli* (expression strain if using T7).
3) **Verify** by sequencing (to confirm sfGFP is correct and in-frame).
4) **Express** and measure fluorescence as a fast functional readout.
5) (Optional) **Purify** via His-tag if purification is required.
This approach separates “DNA write” (ordering/synthesis) from “DNA read” (sequencing verification) and “DNA function” (fluorescence output).
## Part 5 — DNA Read / Write / Edit (Dengue focus: Argentina)
### 5.1 DNA Read
**(i) What DNA/RNA would I want to sequence and why?**
I would focus on **genomic surveillance of Dengue virus (DENV) in Argentina**, integrating **clinical** and **environmental** sequencing to support public health decisions in real time.
Concretely, I would sequence:
1) **Clinical DENV genomes (RNA → cDNA)** from a **representative subset** of confirmed cases:
- **Across regions** (e.g., AMBA vs. northern provinces where dengue burden can be higher).
- **Across time** (weekly/biweekly sampling during season peaks).
- **Across epidemiological contexts** (outbreak clusters, travel-associated cases, and sporadic detections).
**Why:**
- To track **serotype dynamics** (DENV-1/2/3/4) and detect shifts that may correlate with outbreak intensity.
- To monitor **lineage introductions** (new clades entering a province) and infer **transmission connectivity** between regions.
- To support **molecular epidemiology**: identify clusters, potential superspreading contexts, and genomic signatures associated with rapid spread (without overclaiming causality).
- To generate local datasets that strengthen **regional capacity** and reduce dependence on external sequencing pipelines.
2) **Environmental DENV surveillance in Aedes aegypti pools** (and optionally wastewater as exploratory):
- **Mosquito pools** (RT-PCR confirmed) from vector surveillance programs: this can provide early hints of circulating serotypes/lineages even before clinical case counts surge.
- **Wastewater** is less standard for DENV than for enteric viruses, but could be explored as a research add-on; vector-based sampling is usually more direct for arboviruses.
**Why:**
- To get **earlier warning signals** and a broader picture of circulation beyond who shows up at clinics.
- To link **vector circulation** with **human cases**, improving outbreak models.
---
**(ii) What sequencing technology would I use and why?**
I would use a **two-tier strategy**:
- **Illumina short-read sequencing (2nd generation)** for routine surveillance:
- High per-base accuracy, scalable multiplexing, strong variant calling.
- Great for producing reliable consensus genomes and phylogenies.
- **Oxford Nanopore sequencing (3rd generation)** for rapid, field-forward situations:
- Faster turnaround when you need same-week answers (e.g., suspected new introduction or unusual outbreak).
- Useful for decentralized labs or mobile workflows, at the cost of higher raw read error (mitigated by coverage + consensus polishing).
This hybrid approach fits a realistic public health workflow: Illumina as the “gold standard backbone”, Nanopore as the “rapid response tool”.
---
**1) Is it first-, second-, or third-generation? How so?**
- **Illumina = second-generation**: massively parallel short reads (sequencing-by-synthesis).
- **Nanopore = third-generation**: single-molecule sequencing, long reads, electrical signal through nanopores.
---
**2) What is the input? How do you prepare your input? Essential steps.**
**Input:** Dengue is an **RNA virus**, so the primary input is **viral RNA** extracted from samples, then converted to **cDNA**.
A practical pipeline:
**Clinical samples (serum/plasma/whole blood, depending on stage):**
1. **Sample + metadata collection** (date, location, Ct value, suspected serotype if known, etc.).
2. **RNA extraction**.
3. **RT step → cDNA**.
4. **Target enrichment strategy** (choose one):
- **Amplicon tiling PCR** (common for viral genomes; efficient and cheap).
- OR **capture-based enrichment** (more flexible but more expensive).
5. **Library preparation**:
- Illumina: adapter ligation + indexes (multiplexing), optional PCR.
- Nanopore: end-repair + adapter ligation, optional barcoding.
6. **Sequencing run**.
7. **Bioinformatics**: QC → mapping → consensus → variants → phylogeny.
**Mosquito pool samples:**
1. **Pool preparation** (Aedes aegypti pools, ideally with RT-qPCR confirmation).
2. **RNA extraction** (often with inhibitors → extra QC).
3. RT → cDNA, then same as above.
**Key practical note:** For DENV, sampling time matters: early infection tends to have higher viremia (better genome recovery). Also, using Ct thresholds to select samples improves success rate.
---
**3) How does it decode the bases (base calling)?**
- **Illumina**: fluorescent signals from nucleotide incorporation per cycle → base calls + quality scores.
- **Nanopore**: ionic current shifts as molecules pass through the pore → signal-to-sequence base calling (model-based), then consensus polishing.
---
**4) What is the output?**
- **FASTQ** reads (with quality scores).
- **BAM/CRAM** alignments to a reference genome.
- **Consensus genome FASTA** per sample.
- **Variant calls (VCF)** (when appropriate).
- **QC reports** (coverage depth, % genome recovered, contamination checks).
- Downstream: **phylogenetic trees** and **lineage/cluster summaries** for epidemiological interpretation.
---
### 5.2 DNA Write
**(i) What DNA would I want to synthesize and why? (Dengue-focused)**
I would “write” DNA that enables **faster and more deployable dengue diagnostics** and/or supports local R&D.
Three concrete synthesis targets:
1) **DENV diagnostic standards and controls** (safe, non-infectious):
- Synthetic **gene fragments** (e.g., conserved regions of DENV genome used in RT-qPCR/CRISPR assays).
- **Positive control templates** for assay development and QA/QC.
**Why:** robust controls are crucial for reliable diagnostics, especially across multiple labs and seasons.
2) **CRISPR-based dengue detection components** (research prototype):
- Synthetic DNA templates to generate **RNA targets** (IVT) or **reporter constructs** for assay benchmarking.
- If building cell-free or isothermal detection workflows, you can synthesize the necessary templates without needing infectious material.
**Why:** safer, faster iteration.
3) **Aedes-related biosensor modules** (optional):
- DNA parts for sensor chassis optimization (e.g., expression cassettes for reporters in E. coli cell-free systems).
**Why:** create modular “plug-and-play” parts to accelerate prototyping.
---
**(ii) What technology would I use for DNA synthesis and why?**
- For ~0.3–3 kb fragments: **commercial gene synthesis** (dsDNA fragments or cloned gene in a plasmid).
- For many variants: **oligo pools** (array-based synthesis) + assembly.
**Why:** speed + reliability, avoids PCR errors, and supports rapid iteration (especially when you want multiple versions: different primers, target regions, or assay designs).
---
**1) Essential steps (high-level)**
- Design sequence (include constraints: avoid repeats/extreme GC, include needed cloning sites/overlaps).
- Order as dsDNA fragment (or oligos + assembly).
- If needed: clone into plasmid backbone (Gibson/HiFi or restriction cloning).
- Verify by sequencing (at least Sanger for inserts, or NGS for pools).
- Use as template/control in downstream assays.
---
**2) Limitations (speed, accuracy, scalability)**
- **Length & complexity**: longer sequences or high repeat content may fail or take longer.
- **Error rate**: increases with length; sometimes error correction or clone screening is needed.
- **Sequence constraints**: extreme GC, hairpins, homopolymers can reduce success.
- **Regulatory/shipping**: international access can be limited; some vendors require regional sales contact.
- **Cost**: scales with length and number of variants.
---
### 5.3 DNA Edit
**(i) What DNA would I want to edit and why? (Dengue context)**
I would focus on edits that are **ethically appropriate, feasible, and beneficial**, avoiding speculative or high-risk human germline scenarios.
Two realistic editing directions:
1) **Editing lab strains (E. coli or cell-free chassis) to improve dengue diagnostic prototyping**
Examples (conceptual):
- Reduce background nuclease activity that can degrade reporters.
- Improve expression stability of reporter proteins or enzymes used in readouts.
**Why:** more robust, reproducible diagnostics and faster prototyping cycles.
2) **Vector biology research (Aedes aegypti) — in controlled research settings**
Examples (high-level):
- Knock-in/knock-out genes to study **vector competence** or immune pathways relevant to arbovirus replication.
**Why:** better understanding of transmission biology can support long-term control strategies (with strong oversight and biosafety/ethics review).
---
**(ii) What technology would I use and why?**
- **CRISPR-Cas9** for knock-outs and knock-ins in model systems.
- **Base editing** for precise point mutations (when you want to avoid double-strand breaks).
- **Prime editing** for flexible small edits (insertions/deletions/substitutions) with less HDR dependence.
Choice depends on the edit:
- Big insertions → Cas9 + HDR (or targeted integration strategies).
- Single base changes → base editor.
- Small flexible edits → prime editor.
---
**1) How does it edit DNA? (conceptual steps)**
- Guide RNA targets a specific locus.
- Editor performs cut or base conversion.
- Cellular repair/processing results in the desired change.
- Screen and validate clones/lines.
---
**2) What preparation is needed and what is the input?**
- Target selection + guide design + off-target risk assessment.
- Editor delivery strategy (plasmid, mRNA, RNP).
- Optional donor template for HDR edits.
- Validation plan:
- PCR across the locus, Sanger/NGS confirmation,
- phenotype/functional assay relevant to the edit,
- off-target screening where appropriate.
---
**3) Limitations (efficiency/precision)**
- **Delivery** limitations (some cell types/organisms are difficult).
- **Off-targets** and unintended edits (varies with editor/guide).
- **HDR efficiency** can be low; requires careful design and screening.
- Need for **strong controls**, replication, and transparent reporting.