Week 4 HW: Protein Design Part I
Week 4 — Protein Design Part I
Part A — Conceptual Questions (9/11)
Selection note: The assignment allows answering 9 out of 11 questions.
I focused on questions most directly connected to protein design: size/constraints, chirality and secondary structure, and why β-structures tend to aggregate.
Q1) How many amino acids are in a typical protein? How large is it?
It depends on the organism and the protein family, but a practical rule of thumb is:
- Typical bacterial proteins: ~250–350 aa
- Typical eukaryotic proteins: ~350–600 aa (more domains and regulation)
- Real range: from microproteins <50 aa to very large proteins like titin (~30,000+ aa).
In terms of mass:
- A rough average is ~110 Da per amino acid.
- Therefore, a 300 aa protein is ~33 kDa (300 × 110 Da).
Key point: “typical size” is not a rule; it reflects tradeoffs among function, biosynthetic cost, folding constraints, and domain modularity.
Q2) Why can’t humans eat grass and become like cows? (i.e., why can’t we digest cellulose?)
Humans lack cellulases, the enzymes needed to hydrolyze the β(1→4) glycosidic bonds of cellulose.
- We can digest starch (α(1→4) and α(1→6)) using amylases.
- Cellulose is still glucose-based, but the bond stereochemistry changes polymer geometry and packing: it becomes crystalline and rigid, and our enzymes do not recognize/attack it effectively.
Cows are not “magical” either:
- They rely on a rumen microbiome (bacteria/protozoa/fungi) that produces cellulases.
- In practice, the cow hosts an internal bioreactor and absorbs the breakdown/fermentation products.
Q3) Why are there 20 amino acids (and not 10 or 50)?
The canonical set of 20 amino acids likely represents an evolutionary “sweet spot” balancing:
Sufficient chemical diversity
- charged (+/−), polar, hydrophobic, aromatic, nucleophilic, sulfur-containing side chains, etc.
- enough to build catalysis, recognition, and stable structures.
Translation cost and fidelity
- more amino acids ⇒ more tRNAs, aminoacyl-tRNA synthetases, quality control
- higher energetic cost and potentially higher error burden.
Genetic code robustness
- the code is redundant; point mutations often yield chemically similar substitutions
- supports robustness while still offering broad functional expressivity.
Also, biology already extends beyond 20 through:
- selenocysteine (Sec, U) and pyrrolysine (Pyl, O), and
- post-translational modifications (phosphorylation, glycosylation, etc.) that expand functional chemistry without rewriting the entire code.
Q4) What advantages would proteins with non-natural amino acids have?
Potential advantages include:
- New chemistry: functional groups not available in the canonical 20 (azides, alkynes, photoreactive groups, bioorthogonal handles).
- Greater stability: increased resistance to proteases, oxidation, or unfolding (context dependent).
- External control: photoactivatable or chemically switchable residues.
- Improved pharmacology: longer half-life, reduced degradation, potentially altered immunogenicity.
- Enhanced catalysis: introduce designed nucleophiles or metal-binding functionalities.
Main limitation: the cellular “stack” must support it (e.g., genetic code expansion with orthogonal tRNA/synthetase systems, and ribosomal compatibility).
Q5) Could amino acids form under prebiotic conditions? How?
Yes—there is classic experimental evidence:
- Miller–Urey-type chemistry produces simple amino acids (e.g., glycine, alanine) from small molecules plus energy inputs (e.g., electrical discharge).
- Plausible additional routes include meteoritic synthesis (amino acids detected in meteorites) and chemistry on mineral surfaces.
However, amino acids alone do not imply functional proteins. Key barriers include:
- Polymerization: long peptide formation in water is thermodynamically challenging.
- Chirality: abiotic synthesis yields racemic mixtures; life uses mostly L-amino acids.
- Functional folding: protein function requires information-rich sequences, not random polymers.
Q6) Can an α-helix form with D-amino acids?
Yes. The α-helix exists as a geometry; what changes is handedness.
- With L-amino acids, α-helices are typically right-handed.
- With D-amino acids, the corresponding helix tends to be left-handed.
Design relevance: D-peptides can preserve stable secondary structure while being highly protease-resistant, since most proteases are adapted to L-amino acid substrates.
Q8) Why are most α-helices in proteins right-handed?
Because proteins are made of L-amino acids, and for L-backbones the right-handed α-helix is energetically favored (reduced steric clashes in backbone and side-chain packing).
Left-handed helices can occur but are typically short, rare, and associated with specific constraints rather than being the default.
Q9) Why do β-sheets tend to aggregate?
β-structures are “sticky” because β-strands expose backbone hydrogen-bond donors/acceptors in a geometry that can pair with other β-strands.
If a β-prone region becomes exposed or partially unfolded, it can nucleate intermolecular β-pairing, leading to aggregation.
Additional contributors:
- β-prone sequences are often hydrophobic or have low net charge, enabling stacking.
- Aggregation is thermodynamically favorable because it satisfies backbone H-bonds and buries hydrophobic surface area.
Q10) Why do amyloids form so easily?
Amyloids (cross-β architecture) form readily because this state is an accessible energetic minimum for many sequences:
- Stabilization comes from extensive backbone hydrogen-bond networks, not requiring very specific side-chain chemistry.
- Once a nucleus forms, growth proceeds by templating: monomers add like bricks.
In energy landscape terms, native states can be kinetically stable, but stress, mutations, high concentration, or impaired proteostasis can redirect proteins into this alternative “valley.” This is why cells invest heavily in chaperones and quality-control pathways.
(Optional) Reflection — Why this matters for protein design
- Many design failures come from confusing folding with function, especially for membrane-active or oligomeric systems.
- β-aggregation highlights the need for negative design (avoid exposed β-edges and aggregation-prone motifs).
- Language-model scoring can help rank mutations, but it may penalize sequences that are intentionally unusual (e.g., toxic or membrane-disruptive proteins).
Part B — Protein Analysis & Visualization (Cas12a)
Protein selected
-## Protein sequence and database metadata
For this analysis, I used the protein chain from the RCSB structure 8I54, corresponding to Lb2Cas12a from Lachnospiraceae bacterium MA2020.
- Protein: Lb2Cas12a / CRISPR-associated endonuclease Cas12a
- PDB ID: 8I54
- Chain analyzed: Chain A
- Protein length: 1206 amino acids
- Structure method: Cryo-EM
- Resolution: 3.95 Å
- Complex: Cas12a–crRNA–DNA ternary complex
- Other molecules present: crRNA, target DNA strand, non-target DNA strand
- Protein family: Type V CRISPR-associated nuclease / Cas12a family
- Functional class: RNA-guided DNA endonuclease
- Structure quality note: The 3.95 Å cryo-EM resolution is moderate. It is sufficient to interpret the global architecture, nucleic-acid binding channel, and domain organization, but local side-chain positions should be interpreted cautiously.
Because the full Cas12a sequence is long, I used the complete Chain A sequence for structural metadata and focused the ML-based analysis on a shorter subsequence, residues 450–800, to keep runtime practical.
For amino-acid composition, the sequence can be analyzed using the HTGAA Colab frequency tool or any FASTA parser. In the final interpretation, I treated charged, polar, and basic residues near the nucleic-acid channel as especially relevant because Cas12a binds RNA/DNA substrates.
Why I chose it: Cas12a is a programmable CRISPR nuclease used in genome editing and diagnostics. This structure includes both guide RNA and target DNA, which makes it ideal to visualize the binding channel (“pocket”), the protein–nucleic acid interface, and design constraints for activity.
PyMOL visualizations
Figure 1 — Global view (cartoon + nucleic acids).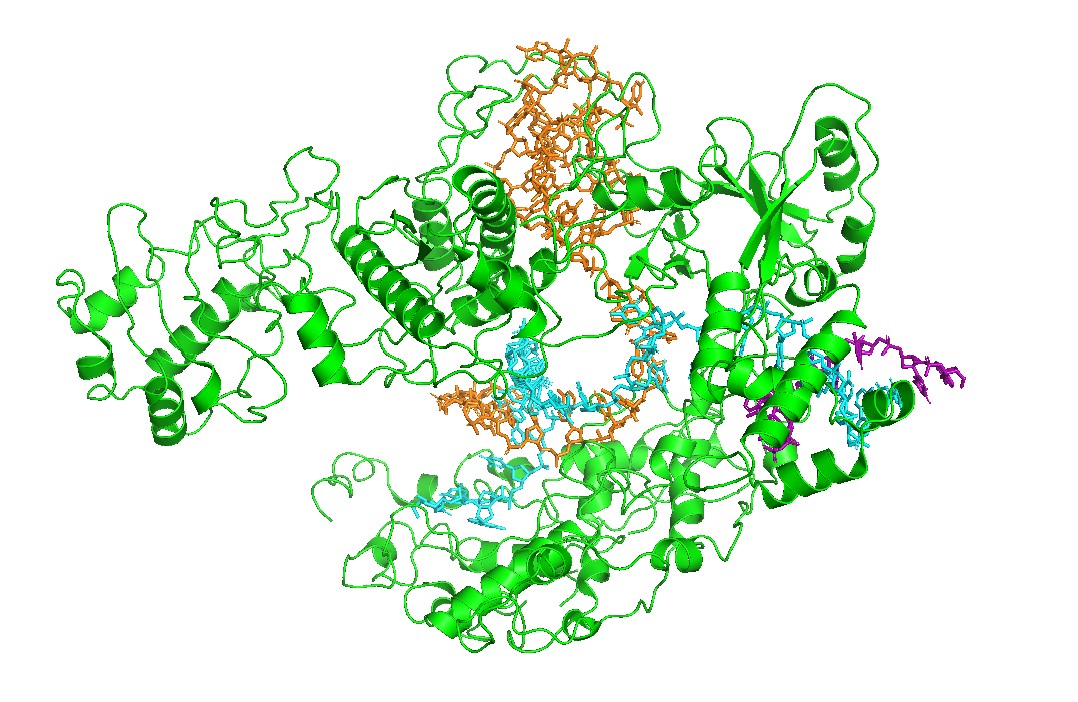
Cas12a is shown in cartoon representation and the RNA/DNA strands are shown as sticks. The nucleic acids sit inside a prominent groove formed by the protein, highlighting that substrate positioning is a primary structural constraint for function.
Figure 2 — Surface representation reveals the binding channel (“pocket”).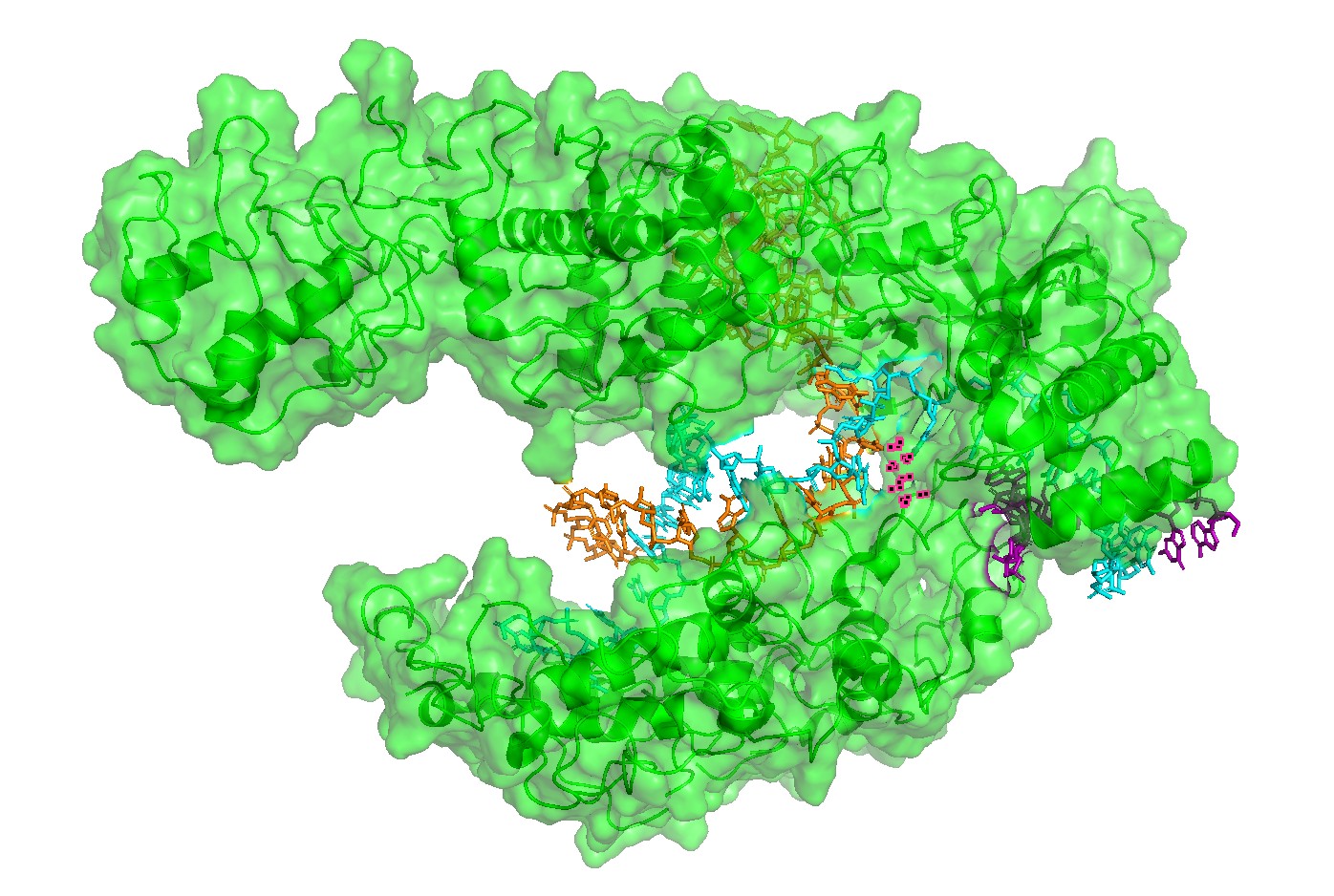 A semi-transparent surface view emphasizes a continuous channel accommodating the RNA–DNA duplex. This channel is the most obvious pocket-like feature in this complex and suggests that mutations lining the groove can strongly affect binding and activity.
A semi-transparent surface view emphasizes a continuous channel accommodating the RNA–DNA duplex. This channel is the most obvious pocket-like feature in this complex and suggests that mutations lining the groove can strongly affect binding and activity.
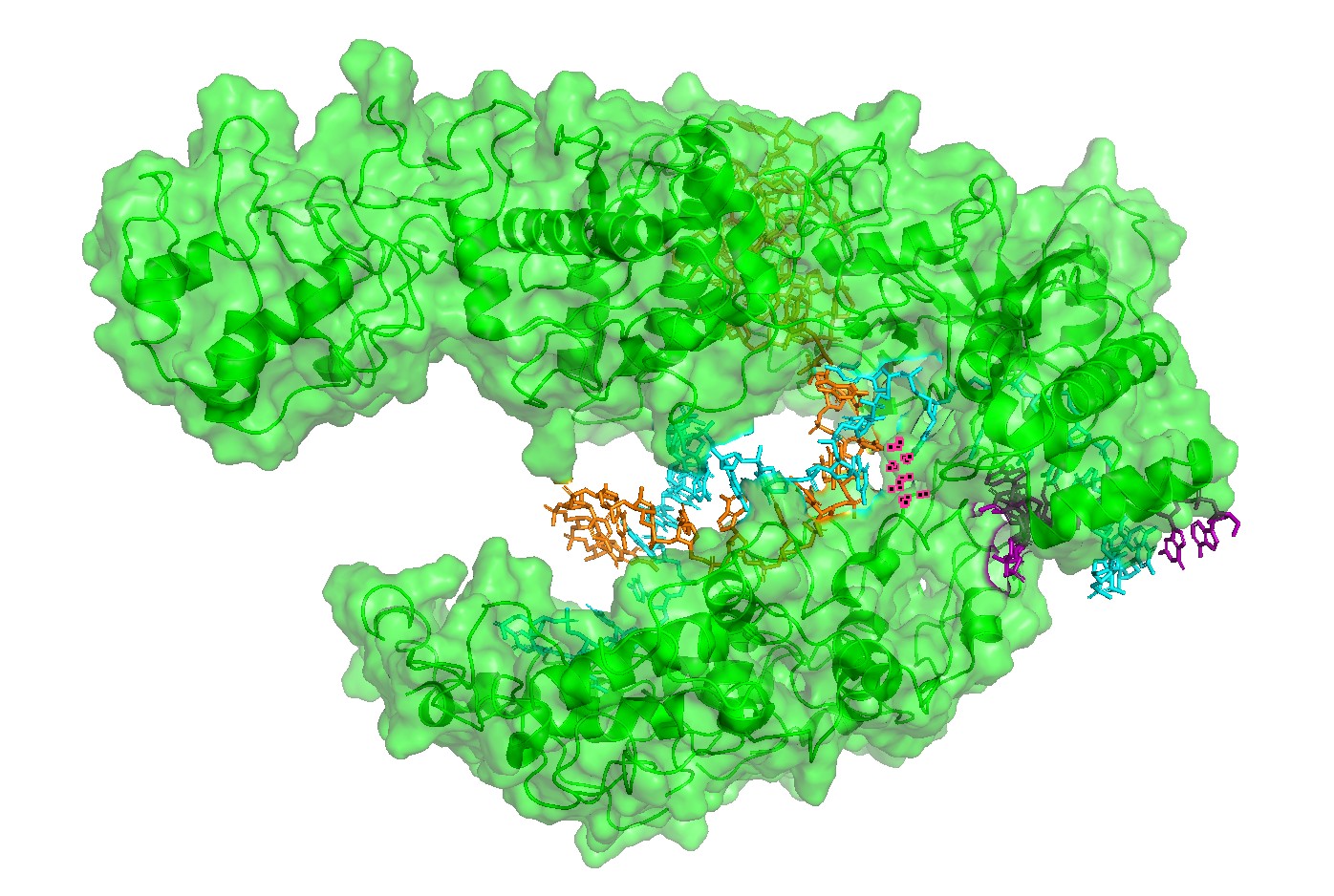
Figure 3. Alternative surface/channel view of Cas12a. This second viewpoint helps confirm that the nucleic acids traverse a defined channel rather than binding to a flat surface.
Figure 4 — Interface residues within ~4 Å of RNA/DNA.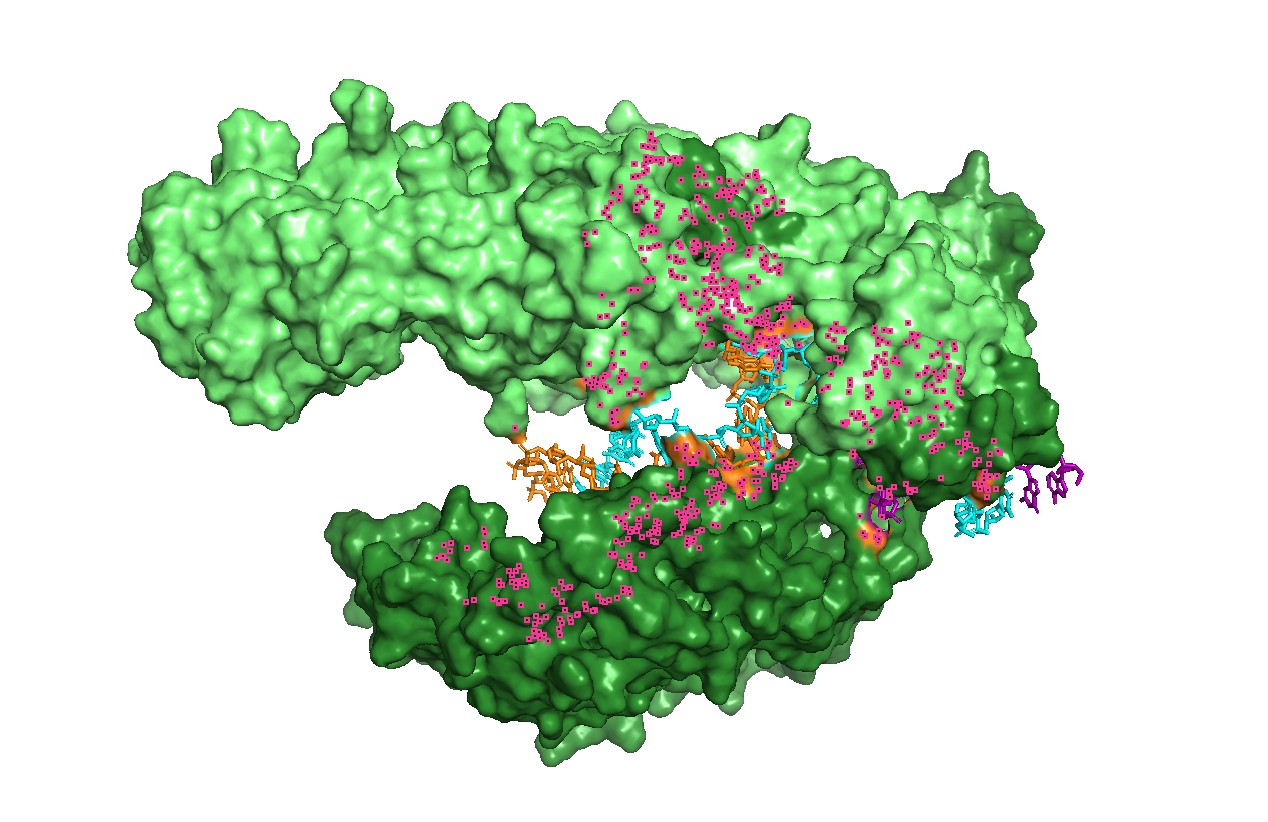
Residues located within ~4 Å of nucleic acids highlight the likely functional interface. This provides a rational set of positions expected to be more constrained in mutational scans (interface mutations can disrupt function even if the global fold remains stable).
Figure 5 — Qualitative “electrostatics-like” surface coloring (charged patches).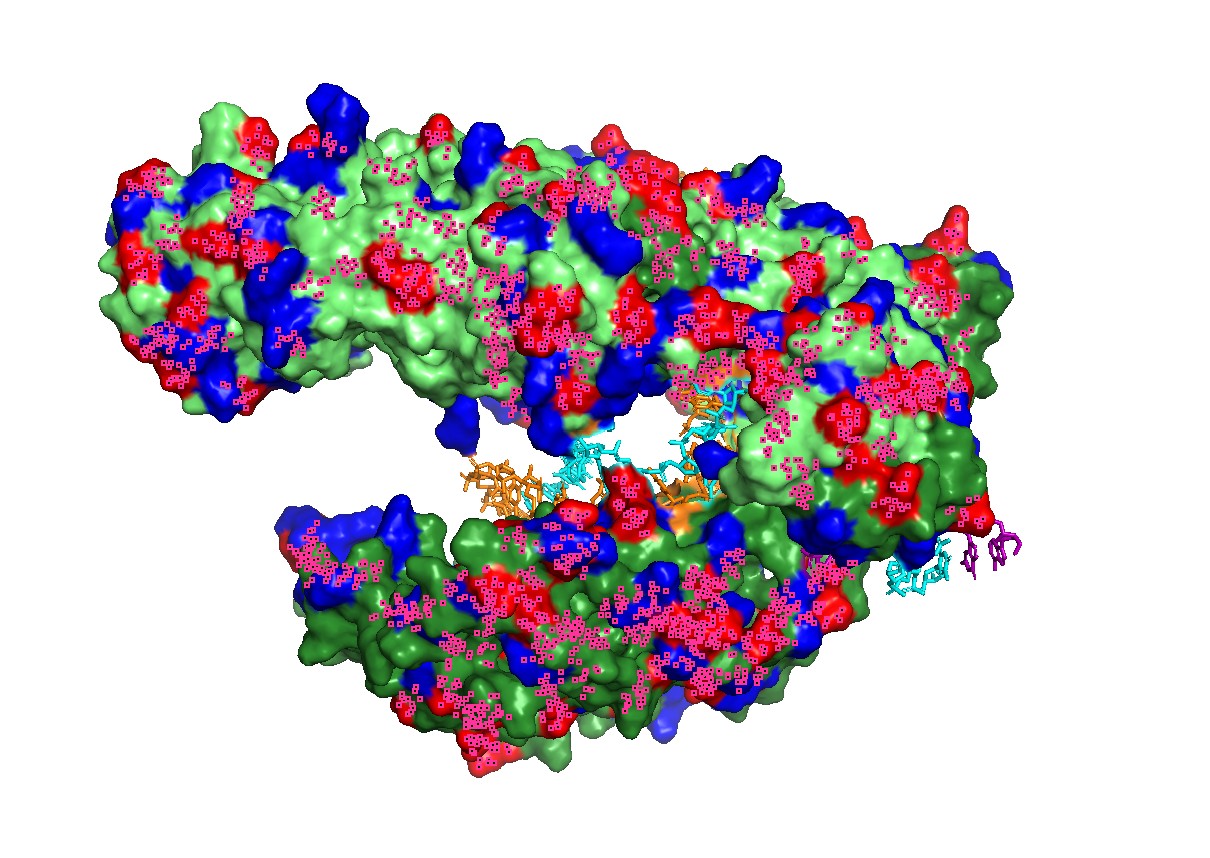
A qualitative mapping of charged residues on the surface shows patches consistent with nucleic-acid binding, supporting the idea that electrostatics contributes to substrate recruitment and stabilization in the binding groove.
Figure 6 — Charged patches + channel view (combined).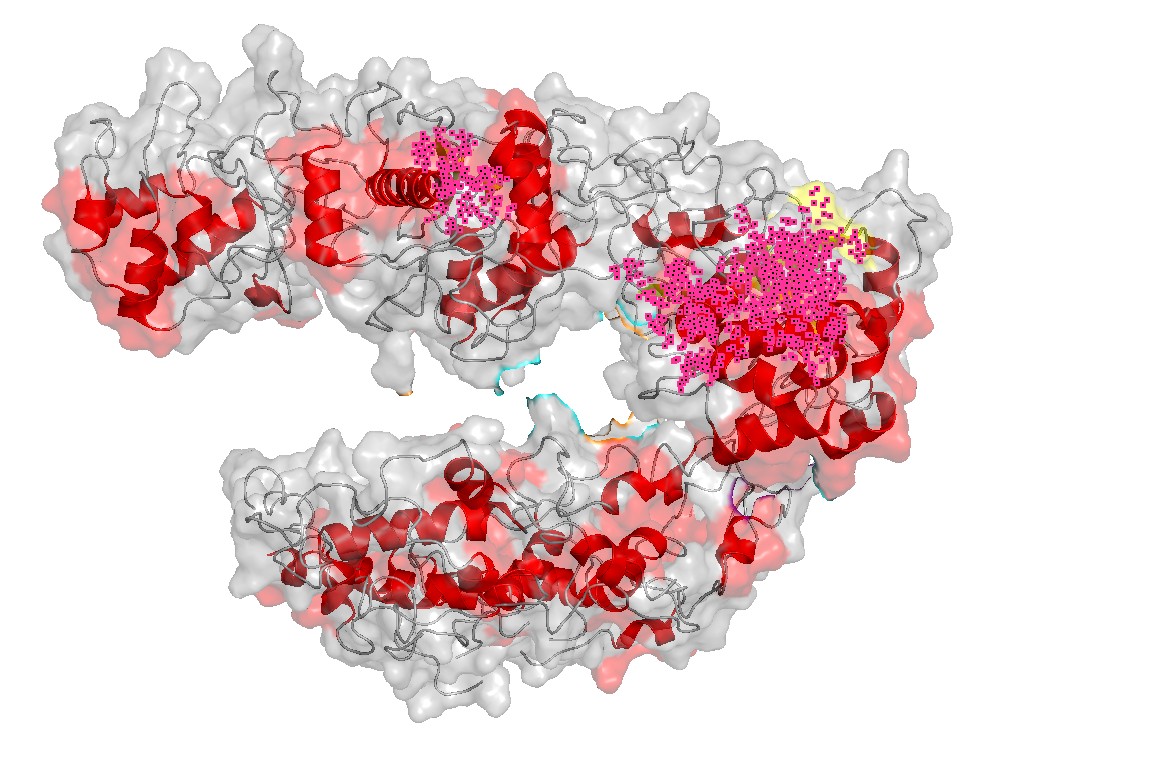 This combined view links charge distribution with geometry: charged surface regions are positioned near the nucleic-acid channel, consistent with a binding-and-positioning role.
This combined view links charge distribution with geometry: charged surface regions are positioned near the nucleic-acid channel, consistent with a binding-and-positioning role.
Figure 7 — Secondary structure emphasis (helices).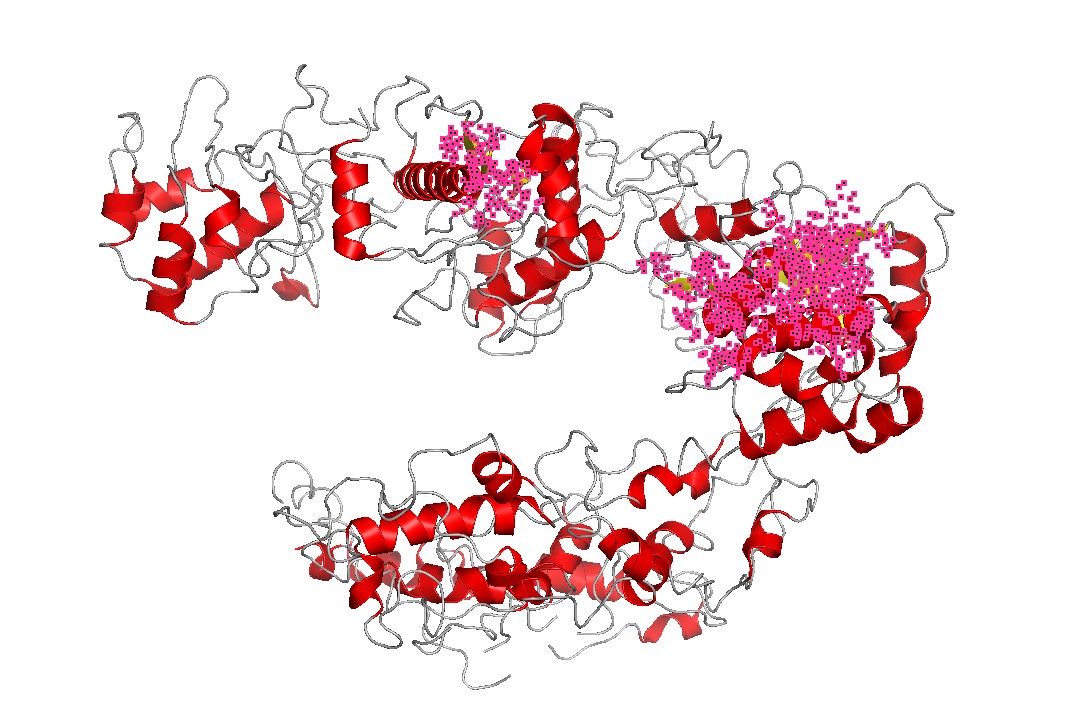
Cas12a is strongly helix-rich, consistent with many large nucleic-acid binding proteins that use extended helical scaffolds to shape binding channels and mediate conformational changes upon substrate binding.
Figure 8 — Coarse lobe/domain segmentation (REC vs NUC).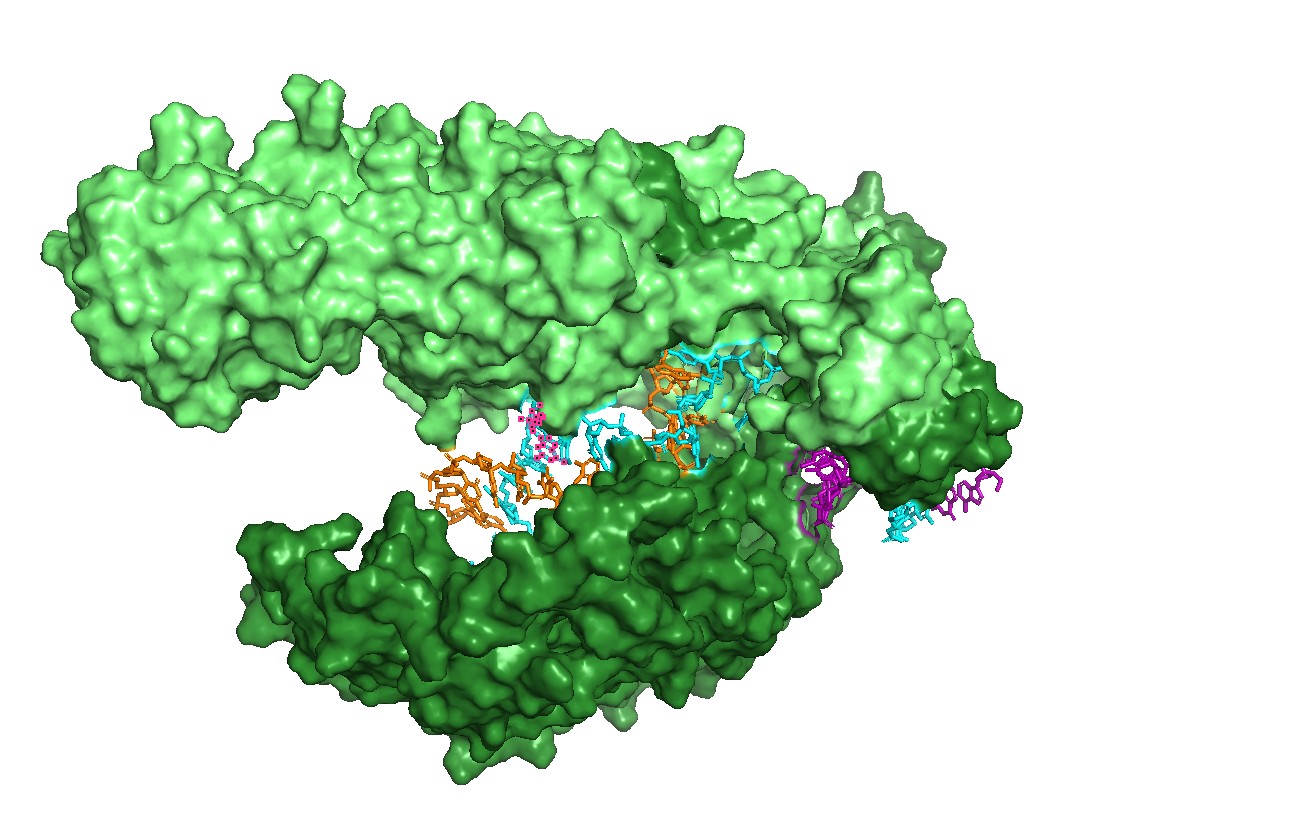
A coarse two-color segmentation illustrates Cas12a’s modular architecture: a recognition lobe (REC-like region) and a nuclease lobe (NUC-like region) together shape the binding channel and position substrates for cleavage.
Visualization modes used
I visualized the Cas12a complex in several molecular representations:
- Cartoon representation: used to inspect the global fold, domain organization, and secondary structure.
- Ribbon/cartoon-like representation: used to emphasize the overall path of the protein backbone and the helical architecture.
- Stick representation: used mainly for RNA and DNA strands to highlight the nucleic-acid binding channel.
- Surface representation: used to identify the main binding groove or pocket-like channel.
- Residue/interface selection: residues within approximately 4 Å of RNA/DNA were highlighted to identify likely functional interface positions.
The most informative representation was the semi-transparent surface with RNA/DNA shown as sticks, because it directly revealed the continuous nucleic-acid binding channel.
Key structural takeaways (summary)
- The RNA–DNA duplex runs through a clear binding channel, which can be treated as the main “pocket” in the complex.
- The ~4 Å interface highlights the most likely constrained region for function and provides candidate sites for mutational sensitivity (Part C).
- Surface charge patches near the groove suggest electrostatics is important for nucleic-acid binding, emphasizing that function depends on local chemistry, not only global folding.
Part C — ML-Based Protein Design Tools
To keep runtime practical, I analyzed a subsequence of Cas12a from the 8I54 structure (chain A, residues 450–800; 351 aa).
C1 — ESM2: in silico mutational scan
Example mutation interpretation
One mutation I selected for closer inspection was L706D. This substitution replaces a hydrophobic leucine with a negatively charged aspartate. In a folded protein core or hydrophobic structural region, this type of mutation is expected to be disruptive because it introduces charge and changes side-chain chemistry dramatically.
In the ESM2 mutational scan, strongly negative Δ log-probability values are interpreted as substitutions that are poorly compatible with the learned sequence context. Therefore, a mutation such as L706D is a useful example of a sequence-level warning: even before folding prediction, the language model suggests that this position may be chemically constrained.
In contrast, K518R is a conservative substitution because lysine and arginine are both positively charged basic residues. Such mutations are usually more tolerated, especially if the position mainly requires positive charge rather than a specific lysine geometry. I performed an in silico deep mutational scan (DMS-like) using ESM2 by masking each position and scoring all 20 substitutions (Δ log-prob = mutant − WT). More negative values indicate substitutions that are less compatible with the sequence context (more constrained positions), whereas values closer to zero indicate more tolerated substitutions.
Interpretation: The tolerance map shows heterogeneous constraint across the fragment, consistent with a folded scaffold containing both structurally constrained positions and more permissive regions. This provides a rational way to choose mutation sites (avoid strongly constrained positions; target tolerant ones) before structural screening.
C1b — Latent Space Analysis
To complement the ESM2 mutational scan, I performed a latent-space analysis using protein sequence embeddings. The goal was to project protein sequences into a reduced-dimensionality space where proteins with similar sequence features, evolutionary constraints, or functional properties tend to appear closer together.
Because the original SCOPe/ASTRAL dataset download failed in my Colab session, I built a smaller self-contained comparison set. This dataset included overlapping fragments from the same Lb2Cas12a protein, several unrelated protein structures downloaded from RCSB/PDB, and my query fragment: Lb2Cas12a chain A residues 450–800 from PDB 8I54.
I embedded the protein sequences using ESM2-derived mean sequence embeddings and then reduced the embedding space using PCA followed by t-SNE.
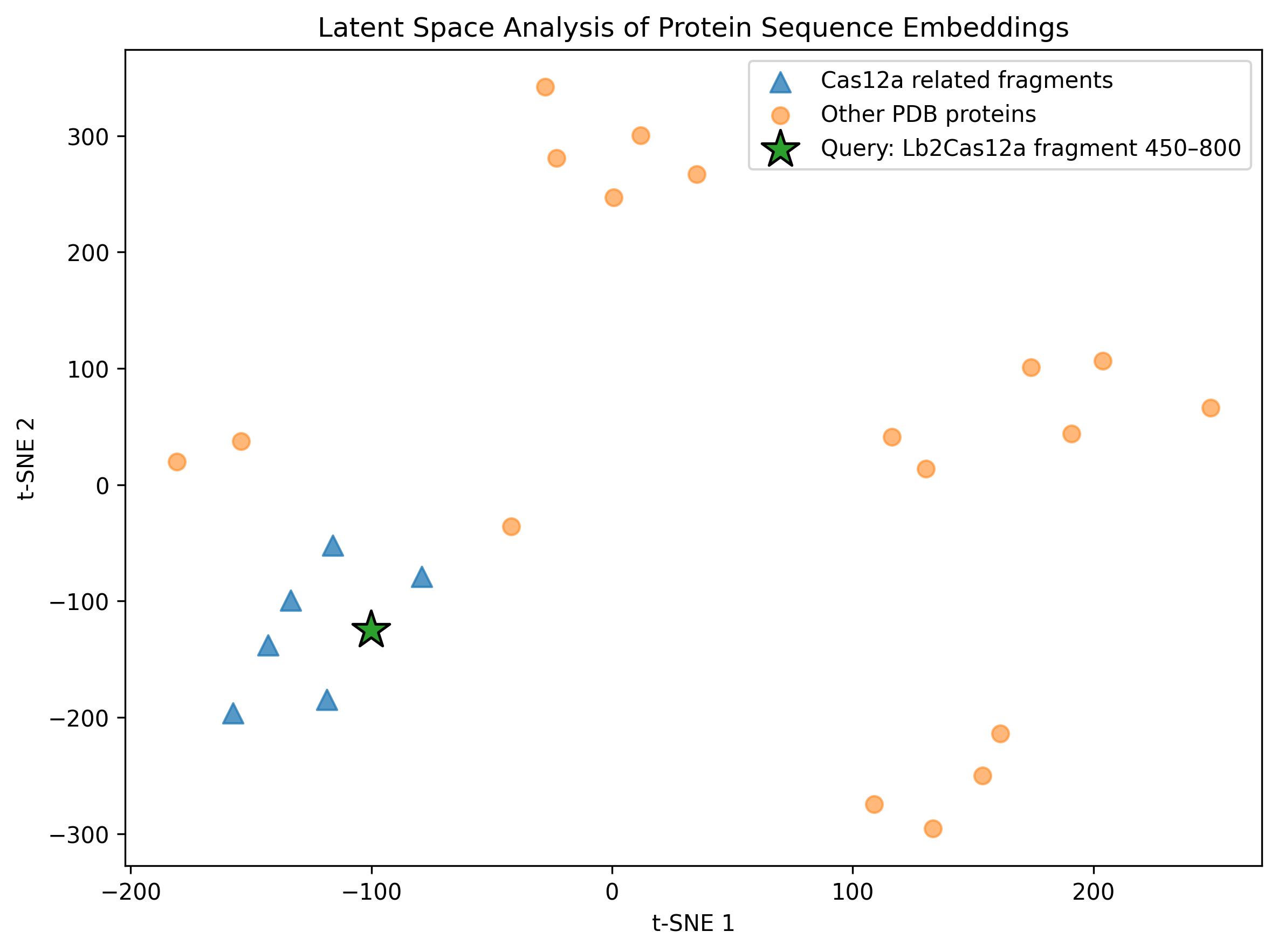
Figure. Latent-space projection of ESM2 protein sequence embeddings. Triangles correspond to Cas12a-related fragments, circles correspond to unrelated PDB protein controls, and the star marks the Lb2Cas12a fragment analyzed in this homework.
Interpretation
This analysis does not predict protein structure directly. Instead, it provides a sequence-level view of how a protein language model organizes proteins based on learned sequence features.
The query Cas12a fragment is projected into the same embedding space as related Cas12a fragments and unrelated protein controls. If the query appears closer to other Cas12a-derived fragments than to unrelated proteins, this supports the idea that ESM2 embeddings capture sequence-level similarity and local evolutionary/structural context.
Because this analysis used a relatively small custom dataset rather than a full protein family database, I interpret the map qualitatively. Still, it complements the residue-level ESM2 mutational scan: the mutational scan highlights local sequence constraints, while the latent-space map gives a broader view of where the analyzed Cas12a fragment lies in protein sequence space.
C2 — ESMFold: folding filter (WT vs mutants)
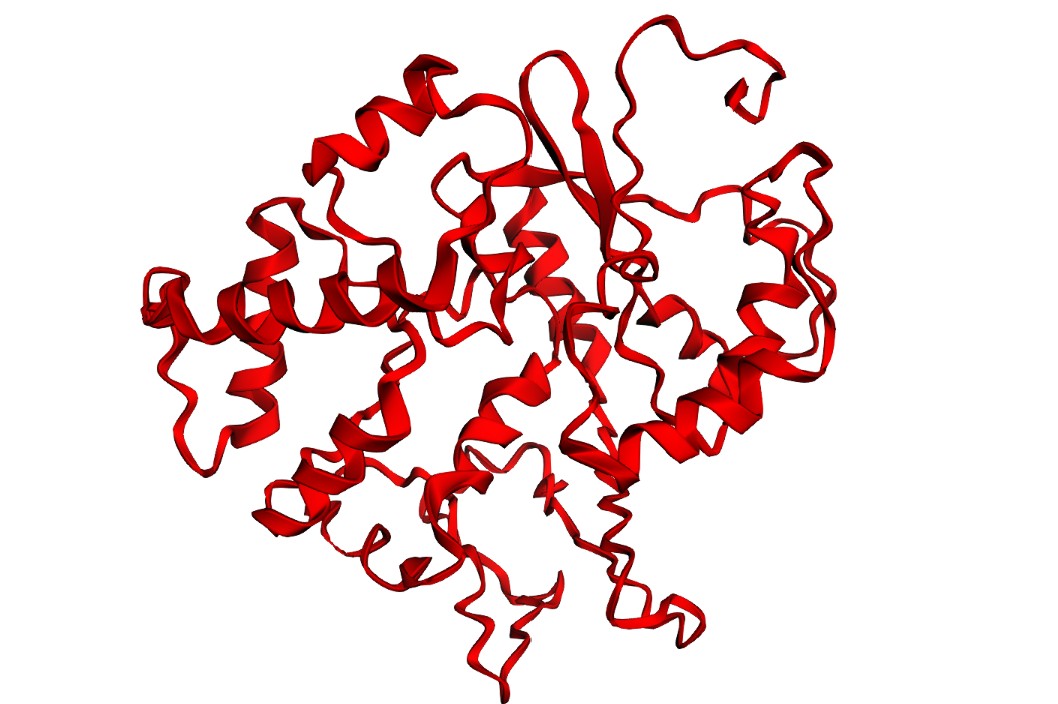
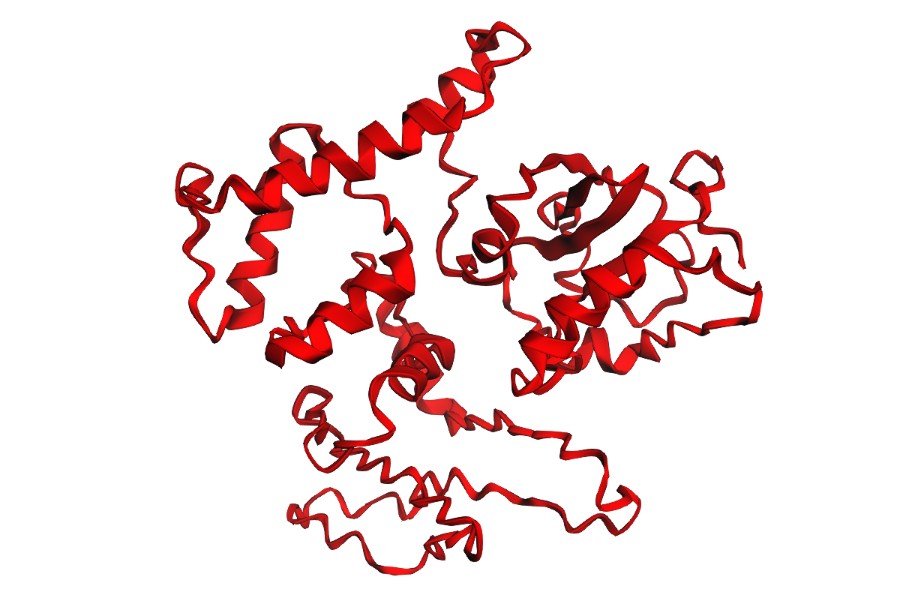
I folded the WT fragment and two mutants with ESMFold: a conservative substitution (K518R) and a disruptive substitution (L706D). The goal is to use folding prediction as a rapid viability filter: keep variants that preserve the fold, and flag variants that reduce confidence or destabilize structure.
Structures
K518R (conservative):
L706D (disruptive):
Confidence / error diagnostics
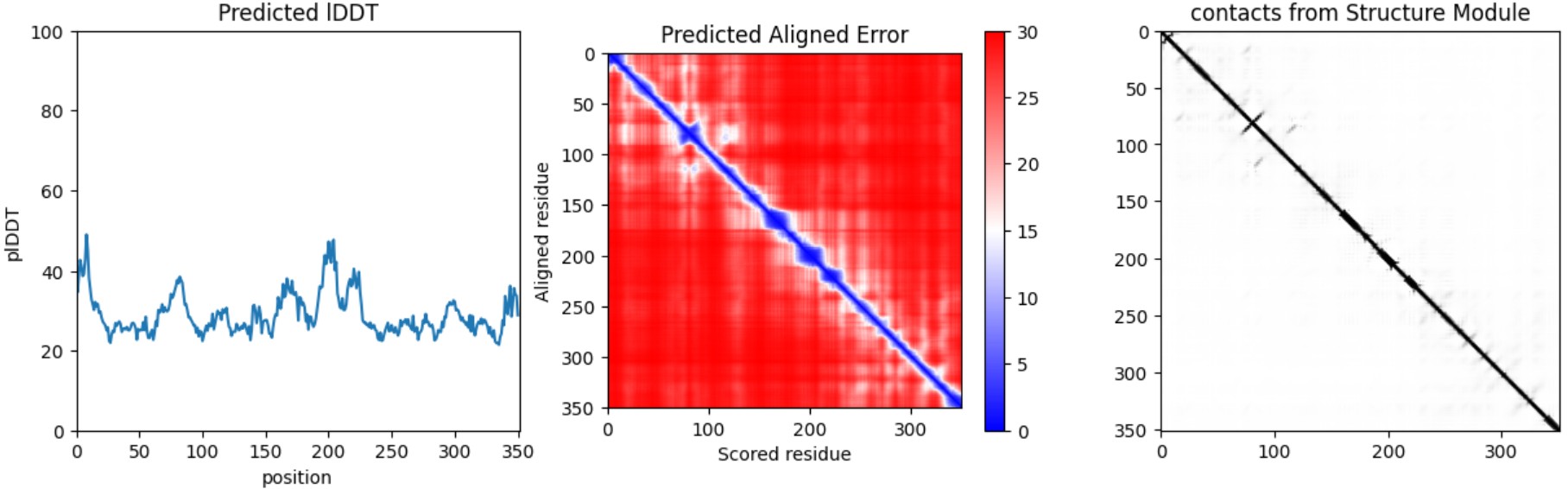
Interpretation: Both variants produce a plausible global fold, but confidence metrics are generally low-to-moderate (pLDDT values mostly ~20–50) and the PAE matrix is broadly high off the diagonal, indicating uncertainty in the relative positioning of many regions. This is consistent with either (i) a fragment that is partially flexible outside its native context, or (ii) limited confidence for this isolated subsequence. Importantly, these results illustrate that ESMFold can screen gross misfolding, but folding confidence does not guarantee biological function.
C3 — ProteinMPNN (inverse folding)
Using the WT fragment backbone (Cas12a 8I54 chain A residues 450–800; 351 aa), I ran ProteinMPNN to generate 10 alternative sequences compatible with the same backbone (T=0.2). The designed sequences show low sequence recovery (~0.15–0.18), indicating substantial sequence diversity under a fixed-backbone constraint.