Individual Final Project
Automated Optimization of a DNAzyme–Cas12a Amplified Lead Sensor
Author: Lautaro Otero Maffoni
Node: Argentina — HTGAA 2026
Project type: Environmental biosensor · DNAzyme · CRISPR-Cas12a · Automation · Field-deployable diagnostics
Abstract
Lead contamination in drinking water remains a major public health problem because even low-level chronic exposure can impair neurological development, cardiovascular health, and overall long-term wellbeing. Existing analytical methods such as inductively coupled plasma mass spectrometry, ICP-MS, are highly sensitive, but they usually require centralized laboratory infrastructure, trained personnel, and expensive instrumentation. This limits their accessibility for decentralized, low-resource, or field-based monitoring.
The overall goal of this project is to develop a modular environmental biosensing platform that couples a Pb²⁺-responsive DNAzyme with CRISPR-Cas12a signal amplification to generate a rapid and amplified fluorescent readout. The central hypothesis is that DNAzyme-triggered release of a programmable nucleic acid activator can be linked to Cas12a collateral cleavage to improve sensitivity while preserving modularity.
The project is structured into three aims. Aim 1 focuses on computational design and kinetic modeling of the sensing cascade and was completed during HTGAA 2026. Aim 2 proposes automated experimental optimization using robotic liquid handling. Aim 3 describes the long-term translation of the system into a portable and modular environmental sensing format.
The methods include nucleic acid folding analysis, structural plausibility assessment, kinetic simulation, DNA construct design, and future automated wet-lab optimization. Together, this project aims to establish a scalable biosensing framework for environmental monitoring that is adaptable, programmable, and ultimately deployable outside centralized laboratories.
1. The Problem: The Hidden Lead Crisis
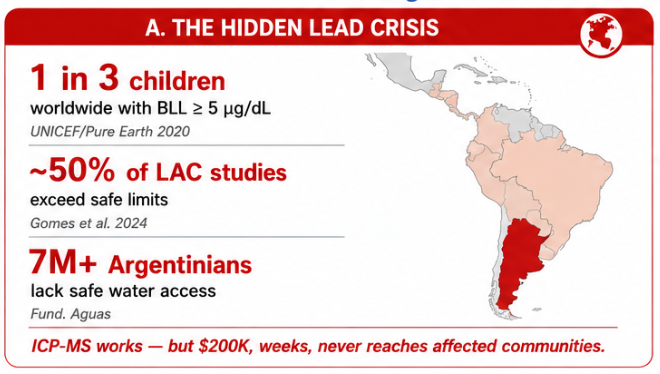
Lead contamination in drinking water is a persistent environmental and public health problem. Unlike biological contaminants, which can often be reduced through boiling, filtration, or disinfection, lead is a chemical pollutant that can accumulate in the body over time. Chronic exposure is especially dangerous for children because it can affect neurological development, cognition, behavior, and long-term health.
Lead contamination is also a problem of access. Current gold-standard analytical methods can detect Pb²⁺ with excellent sensitivity, but they are expensive, centralized, and slow. This creates a practical gap between the existence of high-quality analytical tools and the ability of vulnerable communities to access timely water-quality information.
1.1 Global scale
Lead exposure affects communities worldwide. It is especially dangerous for children because there is no known safe level of lead exposure during development. Chronic exposure can produce neurological, cognitive, behavioral, cardiovascular, renal, and developmental effects.
The global lead crisis is often hidden because contamination may occur through old plumbing, mining tailings, lead-acid battery recycling, paint, ceramics, industrial discharge, or contaminated soil and dust. In many cases, communities are exposed for long periods before testing is performed.
1.2 Why current testing is not enough
The current gold standard for lead quantification in water is ICP-MS. ICP-MS is highly sensitive and specific, but it has several limitations:
| Limitation | Practical consequence |
|---|---|
| Centralized instrumentation | Samples must be transported to specialized laboratories |
| High cost | Frequent testing becomes difficult for low-resource communities |
| Specialized personnel | Requires trained operators and analytical infrastructure |
| Slow turnaround | Results may take days to weeks |
| Limited field deployment | Not practical for immediate decentralized screening |
This project addresses that gap by proposing a rapid, programmable, amplified, and potentially field-deployable Pb²⁺ sensor.
1.3 Regulatory reference values
Important reference values for this project are:
| Organization / framework | Reference value |
|---|---|
| US EPA action level for lead in drinking water | 15 ppb |
| WHO guideline value for lead in drinking water | 10 ppb |
| Desired sensor target | Below 15 ppb |
The goal of this project is not to replace certified analytical methods, but to create a preliminary screening tool that can identify samples requiring urgent confirmatory analysis.
2. Project Overview: A Modular DNAzyme–Cas12a Sensor
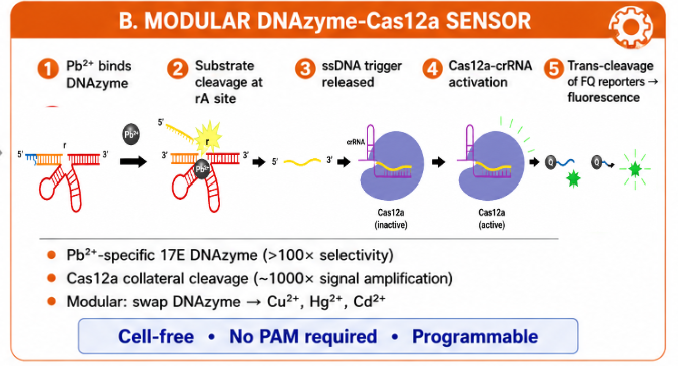
This project proposes a modular molecular cascade for Pb²⁺ detection. The system has five functional steps:
- Pb²⁺ binds the 17E DNAzyme.
- The DNAzyme cleaves its substrate at the rA site.
- A short ssDNA trigger is released.
- The ssDNA trigger activates the Cas12a–crRNA complex.
- Activated Cas12a performs collateral trans-cleavage of FQ reporters, generating fluorescence.
The key design principle is modularity. The Pb²⁺-specific DNAzyme acts as the input recognition module, while Cas12a acts as the amplification module. In principle, the upstream DNAzyme could be swapped to detect other metal ions, while preserving the downstream CRISPR-based readout.
2.1 Original project roadmap
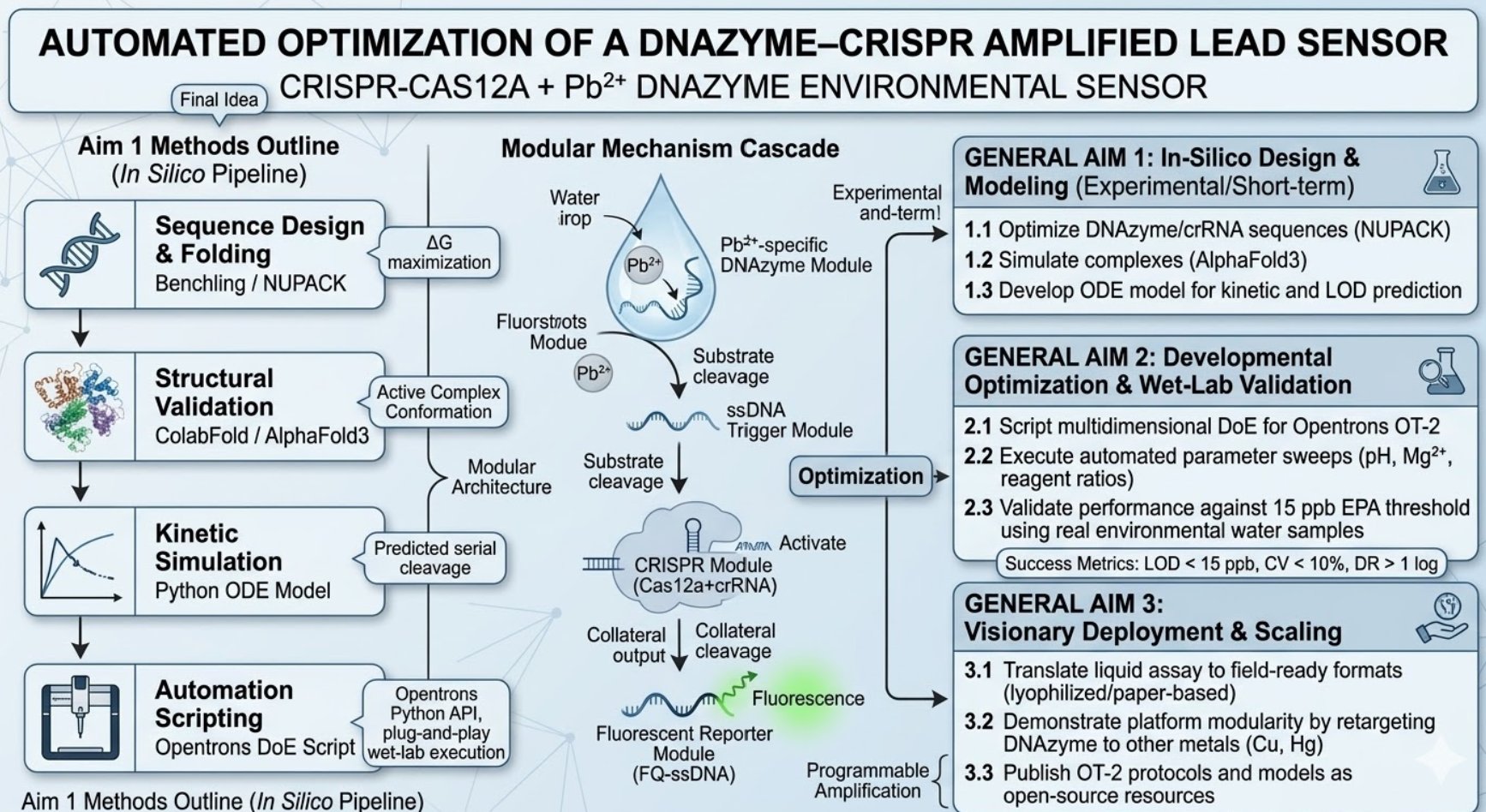
This original roadmap summarizes the full project logic: in-silico design, structural validation, kinetic simulation, automation scripting, experimental optimization, and field deployment. I kept it here because it shows the global architecture of the project and connects all three aims in one visual map.
2.2 Why combine DNAzymes with Cas12a?
A purely DNAzyme-based fluorescent sensor faces a sensitivity ceiling because each target-triggered cleavage event produces a limited signal. By coupling the DNAzyme to CRISPR-Cas12a, the system uses Cas12a collateral trans-cleavage activity to amplify the signal. Once activated, Cas12a can cleave many ssDNA reporters, converting a molecular recognition event into a stronger fluorescent output.
This creates a catalytic signal-amplification cascade:
3. Project Aims
Aim 1 — In-Silico Design and Modeling
The first aim of my final project is to computationally design and prioritize a modular DNAzyme–Cas12a lead sensor by optimizing nucleic acid architecture, assessing structural plausibility of the Cas12a activation complex, and building an ODE-based kinetic model to predict signal amplification, leakage, and theoretical sensitivity before wet-lab testing.
This aim was completed during HTGAA 2026 and includes:
- Sequence design and folding analysis of the DNAzyme/substrate/crRNA system using Benchling, NUPACK, and ViennaRNA.
- Structural plausibility assessment of the Cas12a–crRNA–activator ternary complex using AlphaFold3.
- Development of a reaction-level ODE kinetic model in Python to predict fluorescence kinetics and detection behavior.
Aim 2 — Automated Wet-Lab Optimization
The second aim of my final project is to experimentally optimize and validate the sensor using automated liquid handling workflows. Following successful in-silico prioritization, this stage would use an Opentrons OT-2 platform to execute multidimensional parameter sweeps across reaction variables in order to identify conditions that maximize sensitivity and reproducibility in real water samples.
Key parameters to optimize include:
- pH.
- Mg²⁺ concentration.
- DNAzyme/substrate ratio.
- Cas12a/crRNA ratio.
- Reporter concentration.
- Temperature.
- Ionic strength.
- Incubation time.
- Pb²⁺ concentration.
The goal of Aim 2 is to move from a plausible in-silico architecture to a quantitatively optimized experimental biosensor.
Aim 3 — Field Deployment and Modular Scaling
The third aim of my final project is to develop the sensing platform into a modular and field-deployable environmental monitoring technology. In the long term, the assay could be adapted into decentralized formats such as lyophilized one-pot reactions, paper-based assays, or simple portable fluorescence readers.
A broader vision is to build a modular environmental biosensing platform where only the upstream recognition module needs to be changed to detect a new target. For example, replacing the Pb²⁺ DNAzyme with a Cu²⁺-, Hg²⁺-, or Cd²⁺-responsive nucleic acid module could enable a family of related heavy-metal sensors.
4. Background and Literature Context
DNAzymes are DNA molecules with catalytic activity. Several metal-dependent DNAzymes have been described, including Pb²⁺-responsive RNA-cleaving DNAzymes such as the 8-17 and 17E systems. These molecules are attractive for environmental sensing because their activity can be directly coupled to the presence of a specific metal ion.
Brown et al. described a lead-dependent DNAzyme with a two-step catalytic mechanism, providing an important biochemical foundation for Pb²⁺-responsive cleavage. Later structural and mechanistic studies of RNA-cleaving DNAzymes helped clarify how sequence, folding, metal coordination, and catalysis are linked. This is important for my project because the upstream sensor depends on maintaining a folded DNAzyme–substrate complex in the OFF state while allowing Pb²⁺-dependent cleavage in the ON state.
DNAzymes have also been adapted into practical sensing platforms. Li et al. reported a single-stranded fluorescent Pb²⁺ DNAzyme sensor that works over a broad temperature range, highlighting the feasibility of DNAzyme-based environmental sensing. More recently, He et al. developed a DNAzyme-based CRISPR/Cas12a fluorescence sensor for sensitive Pb²⁺ detection, demonstrating that metal-responsive DNAzyme cleavage can be connected to CRISPR-mediated amplification.
The CRISPR amplification module is based on the collateral trans-cleavage activity of Cas12a. Once Cas12a is activated by a matching nucleic acid target, it can cleave many nearby ssDNA reporter molecules. This converts a single recognition event into an amplified fluorescent output.
5. Novelty and Innovation
The novelty of this project is not only the combination of DNAzyme sensing and Cas12a amplification, but also the way the system is designed and optimized as an engineering platform.
First, the architecture is modular. The upstream Pb²⁺-recognition module and the downstream CRISPR amplification module are separated conceptually and experimentally. This means that the recognition element could theoretically be replaced without redesigning the entire sensor.
Second, the system is designed around a released ssDNA activator. This creates a programmable bridge between metal-dependent cleavage and Cas12a activation. The activator sequence can be computationally designed, folded, and tested for compatibility with the crRNA spacer before experimental screening.
Third, the project emphasizes automation and quantitative optimization. Instead of manually optimizing one variable at a time, the future wet-lab stage would use an Opentrons OT-2 to screen a multidimensional design space. This turns biosensor optimization from empirical troubleshooting into a structured design-build-test-learn workflow.
Finally, the project integrates tools from multiple HTGAA modules: DNA design, CRISPR systems, nucleic acid folding analysis, kinetic modeling, lab automation, and environmental biosensing.
6. Why This Project Matters
This project matters because access to safe drinking water depends not only on remediation technologies, but also on monitoring. If contamination is not detected quickly, communities may remain exposed for long periods before action is taken.
Current analytical methods are powerful but centralized. This creates a mismatch between technical capability and practical accessibility. A portable screening biosensor could help identify problematic samples faster and support more targeted confirmatory testing.
The project also has educational and scientific value. It demonstrates how molecular recognition, nucleic acid programmability, and CRISPR signal amplification can be combined into a synthetic biology sensing cascade. It also shows how computational modeling can guide experimental design before reagents are ordered or assays are performed.
If successful, the broader platform could be adapted to other environmental targets. This would be especially useful for decentralized monitoring of water quality in schools, rural communities, field stations, environmental agencies, NGOs, and low-resource settings.
7. Ethical Implications
This project raises several ethical considerations related to environmental health, public communication, and responsible biosensor development. At its core, the project is motivated by beneficence because it aims to improve access to lead monitoring tools that could support earlier detection of unsafe water conditions and reduce long-term exposure to a major public health hazard. It also relates to justice because communities with fewer resources are often the ones most affected by environmental contamination while also having the least access to centralized analytical testing.
At the same time, the principle of non-maleficence is especially important because an inaccurate sensor could produce false negatives that give users unjustified confidence in contaminated water, or false positives that generate unnecessary alarm. Since the project is based on a modular synthetic biology sensing architecture, it must also be guided by responsibility in how claims are made, how performance is validated, and how limitations are communicated.
To ensure that this project is ethical:
- The sensor should never be presented as a replacement for certified analytical methods unless its performance has been rigorously benchmarked under realistic environmental conditions.
- The appropriate positioning is as a preliminary screening tool, not as a regulatory-grade replacement for ICP-MS.
- All results should be reported transparently, including background leakage, false activation risks, matrix effects, and uncertainty in the predicted or measured limit of detection.
- Future deployment should include safe reagent handling, clear instructions, confirmatory testing pathways, and honest communication of limitations.
- Positive field-screening results should trigger confirmatory analytical testing.
In this way, the project remains aligned with public health goals while minimizing the risk of misuse, misinterpretation, or premature application.
8. Experimental Design, Techniques, Tools, and Technology
8.1 Aim 1 Experimental Design — Completed During HTGAA
Aim 1 was designed as an in-silico validation workflow. The goal was to test whether the proposed sensing architecture is physically, thermodynamically, structurally, and kinetically plausible before performing wet-lab experiments.
The experimental design consisted of the following steps:
- Define the global molecular architecture of the DNAzyme–Cas12a cascade.
- Select a Pb²⁺-responsive DNAzyme architecture from the literature.
- Design a cleavable substrate containing an rA cleavage site.
- Design a released ssDNA activator sequence.
- Design a crRNA spacer complementary to the activator.
- Analyze the DNAzyme–substrate OFF state using nucleic acid folding tools.
- Analyze the released activator ON state for unwanted self-folding.
- Analyze the free crRNA structure.
- Analyze the crRNA–activator hybrid duplex.
- Model the Cas12a–crRNA–activator complex using AlphaFold3.
- Build an ODE model describing the sensing cascade.
- Simulate fluorescence kinetics at different Pb²⁺ concentrations.
- Estimate detection trends and response times.
- Identify design variables for future automated wet-lab optimization.
- Prepare a future Opentrons-compatible design-of-experiments workflow.
8.2 Expected Timeline
| Stage | Task | Estimated time |
|---|---|---|
| 1 | Literature selection and sequence design | 1 week |
| 2 | DNAzyme/substrate and crRNA design | 1 week |
| 3 | Folding analysis with NUPACK/ViennaRNA | 1 week |
| 4 | Cas12a structural plausibility modeling | 1 week |
| 5 | ODE kinetic model construction | 1 week |
| 6 | Simulation of fluorescence kinetics | 1 week |
| 7 | Candidate ranking and sequence refinement | 1 week |
| 8 | Oligonucleotide order preparation | 1 week |
| 9 | Wet-lab assay setup in buffer | 1–2 weeks |
| 10 | Automated OT-2 parameter screening | 2–3 weeks |
| 11 | Real water sample testing | 2–3 weeks |
| 12 | Data analysis and model refinement | 1–2 weeks |
9. HTGAA Techniques Used
Relevant HTGAA techniques and concepts used or planned for this project include:
- DNA construct design.
- DNA sequence design and annotation.
- CRISPR/Cas12a-based sensing.
- Benchling design documentation.
- Models and notebooks.
- Computational nucleic acid folding analysis.
- Protein/nucleic acid structural modeling.
- Lab automation planning.
- Opentrons OT-2 workflow design.
- Designing a Twist-compatible DNA workflow.
- Cell-free reaction logic.
- Bioethical considerations.
- Quality control and data analysis.
Technique 1 — DNA Construct Design
DNA construct design is central to this project because the sensor is sequence-programmed. The DNAzyme, substrate, released activator, crRNA spacer, and fluorescent reporter must all be compatible with one another. A poorly designed sequence could create unwanted secondary structures, reduce cleavage efficiency, prevent activator release, or cause background Cas12a activation.
In this project, DNA construct design was used to organize the sensing cascade into modular sequence elements. Benchling was used to annotate the designed components and maintain a clear relationship between sequence, function, and expected molecular behavior.
Technique 2 — Computational Modeling and Simulation
Computational modeling was used to validate the project before wet-lab experiments. NUPACK and ViennaRNA were used to analyze nucleic acid folding and hybridization. AlphaFold3 was used to assess whether the Cas12a–crRNA–activator complex was structurally plausible. A Python-based ODE model was then used to simulate the kinetic behavior of the sensing cascade.
This computational workflow is important because it reduces the experimental search space. Instead of testing many arbitrary designs, the wet-lab phase can begin with designs that are already predicted to have favorable folding, activator accessibility, and signal-generation behavior.
10. Industry Council Connections
Several HTGAA Industry Council companies are conceptually connected to this project:
| Company | Connection to project |
|---|---|
| Twist Biosciences | DNA synthesis and oligonucleotide ordering |
| New England Biolabs | Cas enzymes, buffers, and molecular biology reagents |
| Opentrons | Automated liquid handling for optimization |
| Thermo Fisher Scientific | Fluorescence readout, qPCR-style instruments, and reagents |
| Waters Corporation | Analytical validation and measurement technologies |
| Asimov | Genetic circuit design logic and biological modeling concepts |
| Ginkgo Bioworks | Long-term cloud-lab scale-up and automated screening |
11. Aim 1 Results: In-Silico Design and Computational Validation
This section documents the computational work performed during HTGAA 2026 to validate the DNAzyme–Cas12a sensing cascade before moving to the wet-lab optimization stage.
11.1 Sequence Design
Three nucleic acid components were designed.
DNAzyme V1_17E_Pb
Functional annotation:
Substrate T7_17S_Pb
The substrate contains an internal rA cleavage site. After Pb²⁺-dependent cleavage, the 5’ fragment is released as an ssDNA activator.
crRNA-LbCas12a-Pb-v1
Functional annotation:
The spacer is the reverse complement of the released activator, expressed as RNA. This creates a 20/20 Watson-Crick pairing interface between the released ssDNA activator and the crRNA spacer.
12. Folding Analysis
12.1 OFF State — DNAzyme/Substrate Complex
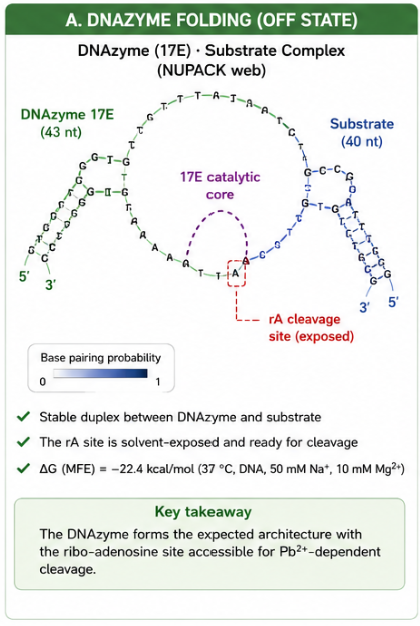
The predicted OFF state shows a stable DNAzyme–substrate complex. The DNAzyme arms hybridize to the substrate, while the 17E catalytic core remains exposed. The rA cleavage site is solvent-accessible, which is essential for Pb²⁺-dependent cleavage.
This is important because the OFF state must be stable enough to prevent premature trigger release but accessible enough to allow Pb²⁺-dependent catalysis.
Detailed NUPACK prediction
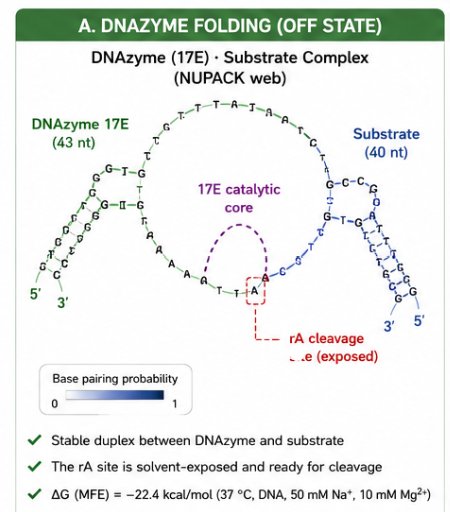
The NUPACK prediction confirms the canonical 17E DNAzyme architecture: the two binding arms hybridize the substrate while the catalytic core bulges out as a flexible loop. The rA cleavage site is solvent-exposed and ready for Pb²⁺-dependent phosphodiester cleavage.
- ΔG (NUPACK, 37 °C, with Mg²⁺) = –22.4 kcal/mol
- The duplex is highly stable.
- Thermal melting is not expected at the assay temperature.
ViennaRNA cross-validation

The same complex was independently predicted with ViennaRNA 2.7. ViennaRNA gave a baseline ΔG of approximately −33.4 kcal/mol. The difference reflects the absence of explicit Mg²⁺ correction in that calculation, but the structural prediction is consistent with the NUPACK result.
12.2 ON State — Released ssDNA Activator
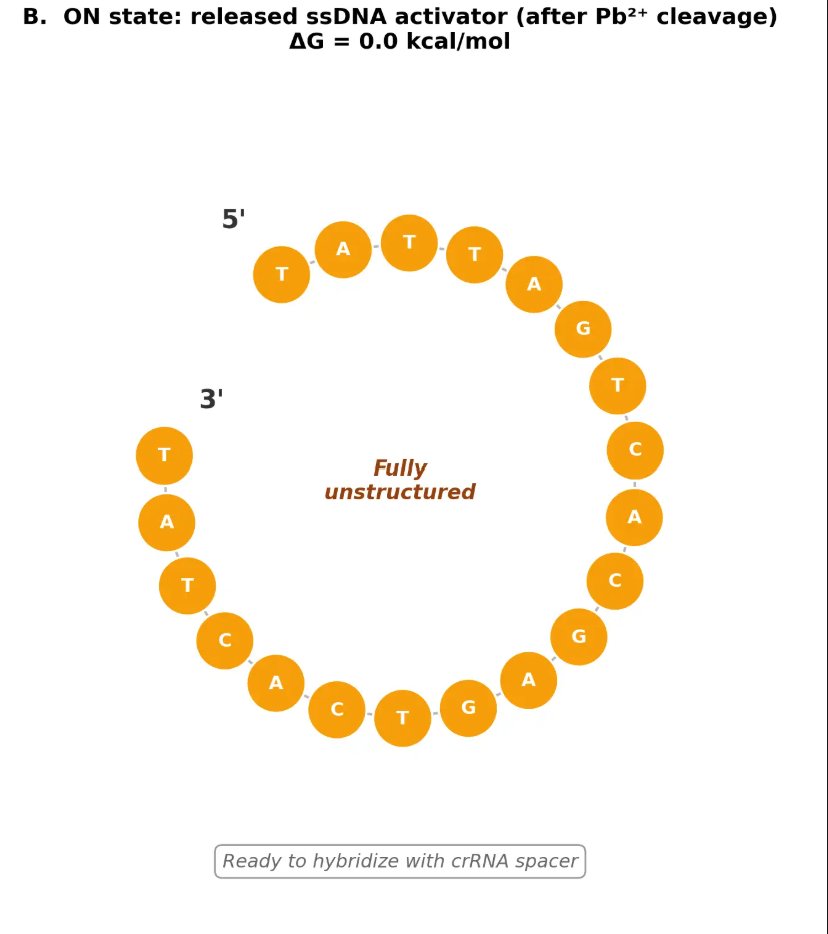
After Pb²⁺-dependent cleavage of the rA site, the 20-nt 5’ fragment is released as a fully unstructured ssDNA. This is the ideal state for hybridization with the crRNA spacer because there is no strong competing self-structure that could compromise activator availability.
12.3 Free crRNA Folding
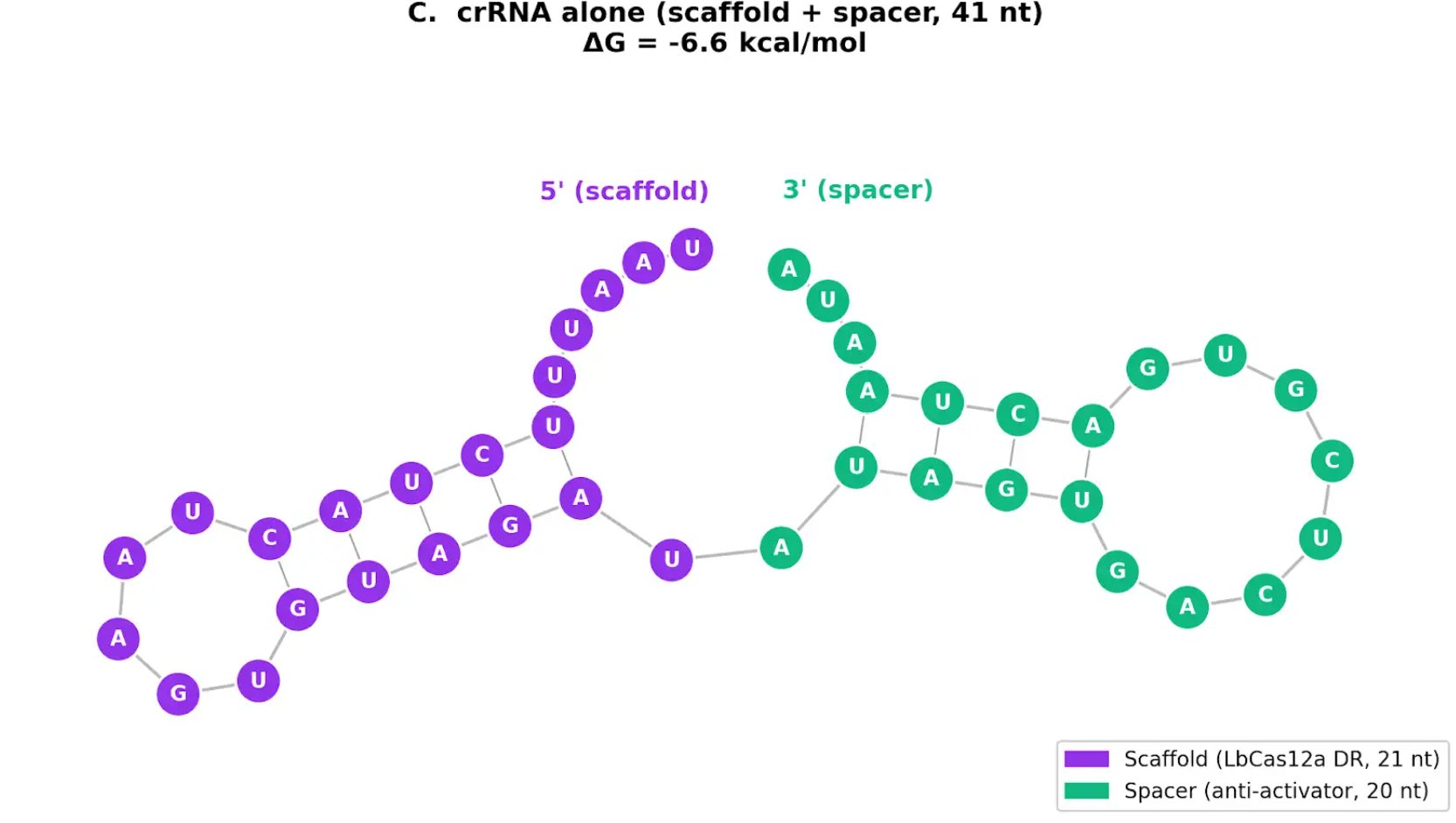
The crRNA alone folds with a moderate local hairpin in the LbCas12a direct repeat scaffold region. The spacer region remains accessible. This is important because Cas12a binding stabilizes the crRNA scaffold, while the spacer needs to remain available for activator binding.
A limitation of this analysis is that ViennaRNA does not predict pseudoknots, so it cannot fully capture the true folded Cas12a direct repeat structure. However, this does not undermine the conclusion because the Cas12a protein itself stabilizes the crRNA scaffold during complex formation.
12.4 Activator–crRNA Hybridization
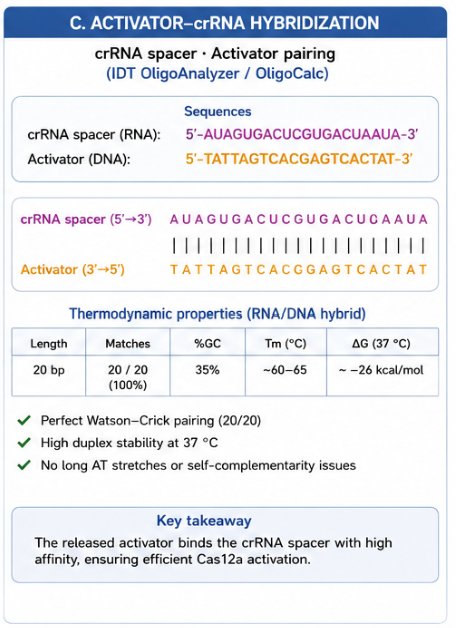
The released ssDNA activator was designed to pair with the crRNA spacer through 20/20 Watson-Crick base pairs. This strong RNA/DNA hybridization supports efficient formation of the active Cas12a recognition complex after Pb²⁺-dependent DNAzyme cleavage.
The key design requirement is that the activator should be accessible after cleavage and should not form strong self-structures that compete with crRNA binding.
Detailed activated duplex prediction
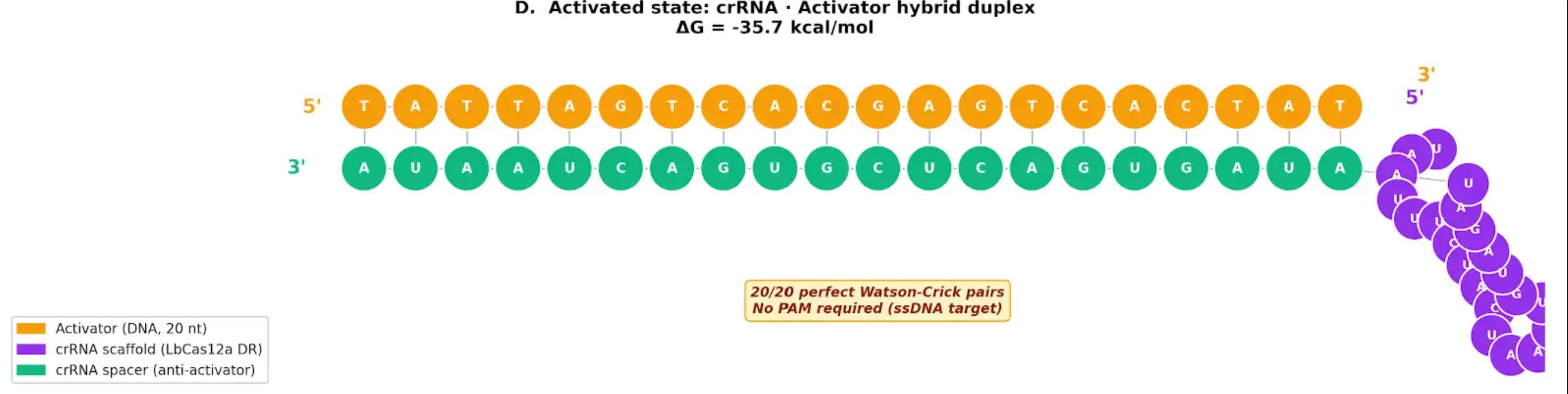
The activated crRNA–activator duplex is thermodynamically favorable:
- 20/20 Watson-Crick pairs
- ΔG ≈ −35.7 kcal/mol
- Tm ≈ 60–65 °C
- No PAM required, because the activator is ssDNA
This supports the central design logic: Pb²⁺-dependent DNAzyme cleavage releases a short ssDNA activator that can efficiently bind the crRNA spacer and activate Cas12a.
13. Structural Plausibility of the Cas12a Activation Complex
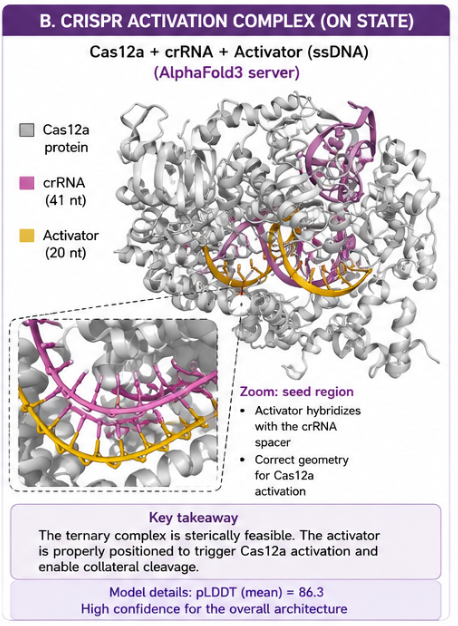
The Cas12a–crRNA–activator complex was modeled to evaluate whether the released ssDNA activator could be positioned correctly within the crRNA spacer region. The predicted ternary complex supports the structural plausibility of Cas12a activation.
The mean pLDDT value was 86.3, suggesting good confidence in the overall architecture. This does not prove biochemical activity, but it supports the feasibility of the designed activation complex before experimental testing.
The structural model supports three key points:
- The Cas12a protein adopts a plausible bilobed architecture.
- The crRNA is positioned in the expected recognition channel.
- The activator is placed near the crRNA spacer in a geometry compatible with activation.
14. Kinetic Modeling
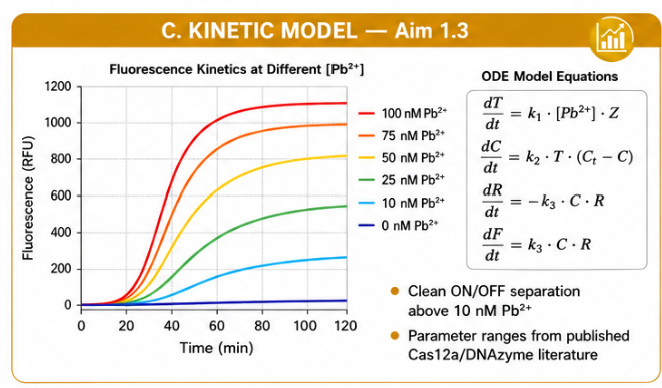
The sensing cascade was translated into a simplified ODE model. The model describes trigger production, Cas12a activation, reporter cleavage, and fluorescence accumulation.
The simplified variables are:
| Symbol | Meaning |
|---|---|
| Z | DNAzyme concentration, assumed constant |
| Pb | Lead concentration |
| T | Released ssDNA trigger |
| C | Active Cas12a complex |
| Ct | Total Cas12a |
| R | Intact reporter |
| F | Fluorescence signal |
The model captures the expected logic of the sensor: higher Pb²⁺ concentration produces faster trigger release, which activates more Cas12a and accelerates fluorescent reporter cleavage.
The simplified reaction model is:
14.1 Molecular cascade detail
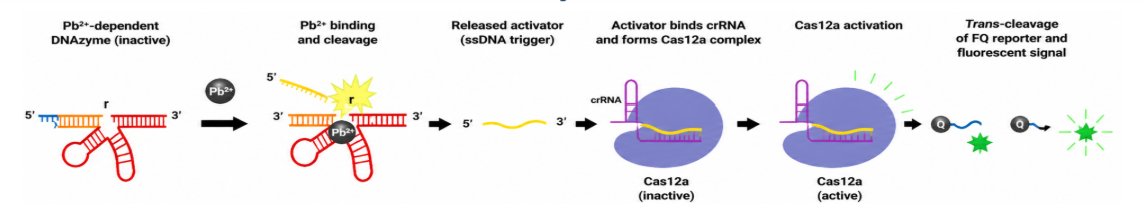
The cascade has five mechanistic steps:
- Pb²⁺ binds the DNAzyme.
- The substrate is cleaved at the rA site.
- The activator ssDNA is released.
- The activator hybridizes with the crRNA and activates Cas12a.
- Activated Cas12a performs collateral trans-cleavage of the FQ reporter, producing fluorescence.
14.2 Detailed ODE model interpretation
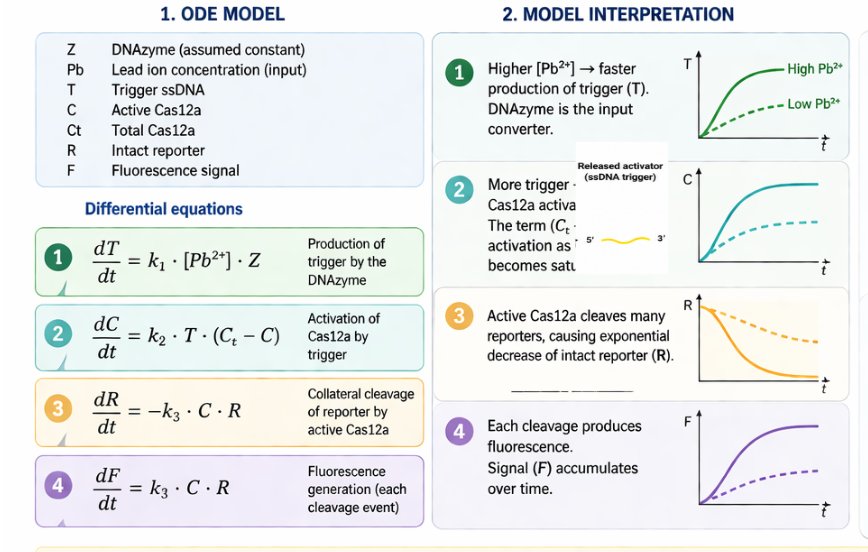
The detailed ODE model links each molecular process to a measurable kinetic output:
- More Pb²⁺ increases trigger production.
- More trigger increases active Cas12a formation.
- More active Cas12a accelerates reporter cleavage.
- Reporter cleavage produces accumulated fluorescence.
This creates an interpretable kinetic model that can be refined later using experimental fluorescence traces.
14.3 Simulated fluorescence kinetics
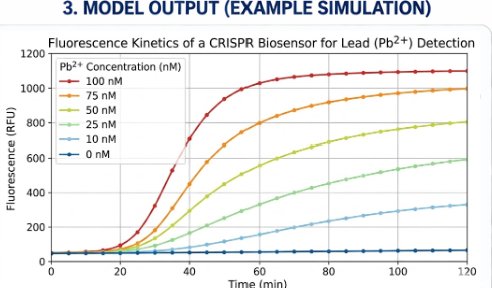
The simulation predicts separated fluorescence trajectories for multiple Pb²⁺ concentrations. In the baseline model, the zero-Pb²⁺ control remains flat because background leakage is neglected. Curves separate above the low-nanomolar Pb²⁺ range, supporting the feasibility of a kinetic fluorescence readout.
14.4 Detection time vs Pb²⁺
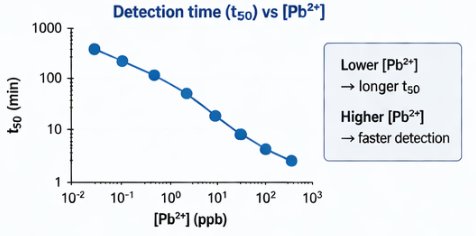
The predicted t50 curve shows that higher Pb²⁺ concentrations produce faster detection. Lower Pb²⁺ concentrations require longer incubation times, while higher Pb²⁺ concentrations cross the detection threshold faster.
This supports the use of detection time as a quantitative metric.
14.5 Full kinetic model composite
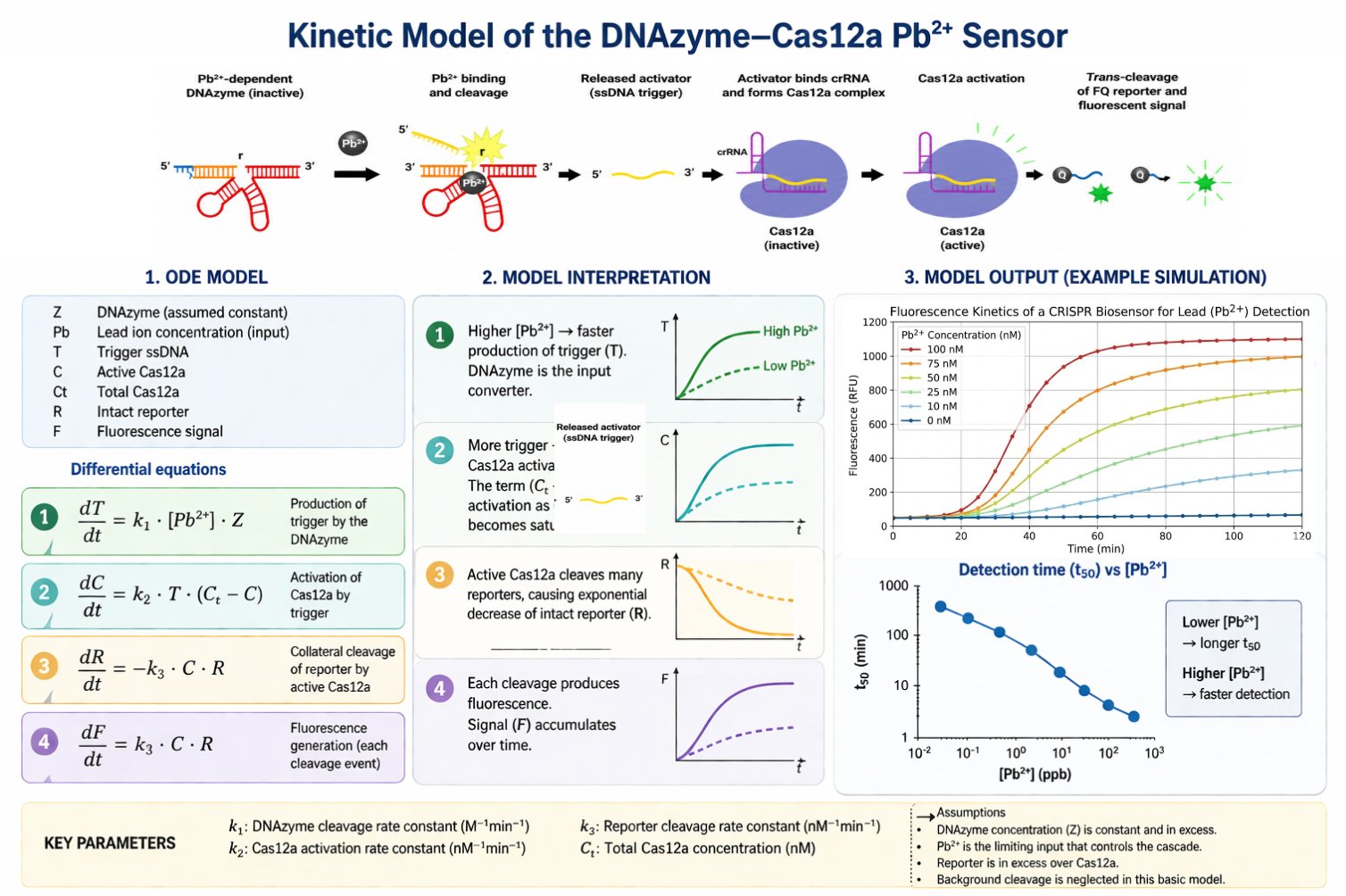
This full composite slide summarizes the kinetic modeling workflow, including the molecular cascade, ODE equations, model interpretation, fluorescence simulations, and detection-time prediction.
15. Predicted Performance
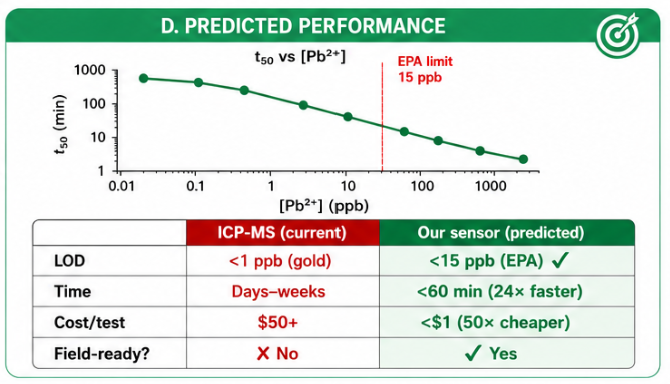
The simulated performance predicts that higher Pb²⁺ concentrations generate faster detection times. The model suggests that the sensor could detect Pb²⁺ near the EPA action level of 15 ppb in less than 60 minutes, assuming the kinetic parameters are experimentally achievable.
The predicted performance is summarized below:
| Feature | Current ICP-MS workflow | Proposed sensor |
|---|---|---|
| Limit of detection | Below 1 ppb | Target below 15 ppb |
| Time to result | Days to weeks | Less than 60 minutes |
| Cost per test | High | Potentially below USD $1 |
| Field-ready | No | Potentially yes |
| Use case | Regulatory confirmation | Rapid preliminary screening |
The proposed sensor is not intended to replace ICP-MS. Instead, it is designed as a rapid screening platform to identify samples that require confirmatory testing.
16. Results and Quantitative Expectations
16.1 What aspect of the project did I choose to validate?
For this stage of the project, I chose to validate the design and computational prioritization workflow of the DNAzyme–Cas12a sensing cascade rather than a fully assembled wet-lab assay. This validation focuses on whether the sensing architecture can be rationally designed in a way that minimizes unwanted folding, preserves trigger accessibility, and supports a plausible downstream Cas12a activation logic.
I selected this aspect because it is directly achievable within the current scope of the course and because a poor sequence architecture would undermine all later experimental optimization.
16.2 What data is presented?
The data presented in this stage are computational and design-derived data rather than experimental fluorescence measurements. These include:
- DNAzyme/substrate folding predictions.
- Released activator structural predictions.
- crRNA folding prediction.
- crRNA–activator hybridization analysis.
- Cas12a–crRNA–activator structural plausibility modeling.
- ODE-based kinetic simulations.
- Predicted detection-time trends.
- Predicted comparison against current centralized analytical workflows.
Together, these outputs serve as evidence-based justification for selecting one or more sensing architectures for future experimental optimization.
16.3 Quantitative expectations
At this stage, the quantitative expectations are focused on relative performance trends rather than final environmental performance claims.
Useful candidate designs should show:
| Expected property | Desired outcome |
|---|---|
| OFF-state leakage | Low background signal in the absence of Pb²⁺ |
| Activator accessibility | Released trigger remains available for crRNA binding |
| crRNA pairing | Strong activator–crRNA hybridization |
| Cas12a activation | Structurally plausible ternary complex |
| Kinetic output | Clear separation between low and high Pb²⁺ inputs |
| Detection behavior | Faster detection at higher Pb²⁺ concentration |
The future experimental goal is to achieve a limit of detection below the EPA action level for lead in drinking water, with reproducible signal generation and low background fluorescence.
16.4 Aim 1 conclusion
The in-silico work supports the following conclusions:
| Validation check | Result |
|---|---|
| Thermodynamic design | Compatible with the intended cascade |
| Trigger accessibility | Released activator predicted to be available for crRNA binding |
| Structural compatibility | Cas12a–crRNA–activator complex appears plausible |
| Kinetic behavior | Higher Pb²⁺ predicts faster signal generation |
| Wet-lab readiness | Parameter space is sufficiently constrained for Aim 2 |
17. Validation Protocol
The complete in-silico pipeline that was executed during HTGAA 2026 is described below.
- I defined the overall sensing architecture as a modular cascade composed of a Pb²⁺-responsive DNAzyme, a cleavable substrate, a released trigger strand, a Cas12a-crRNA activation module, and a fluorescent reporter output.
- I selected literature-supported DNAzyme designs relevant to Pb²⁺ sensing and used them as the mechanistic basis for the upstream recognition module.
- I drafted candidate trigger-release strategies in which cleavage of the substrate would expose or release a DNA sequence capable of activating the downstream CRISPR module.
- I annotated project-relevant sequence elements and organized the design logic in Benchling.
- I evaluated sequence-level folding behavior using NUPACK and ViennaRNA to identify unwanted secondary structures that could interfere with cleavage, trigger release, or Cas12a activation.
- I compared candidate designs by prioritizing those with better trigger accessibility and lower predicted risk of OFF-state leakage.
- I modeled the Cas12a–crRNA–activator complex to evaluate structural plausibility.
- I translated the sensing cascade into a reaction-level kinetic framework suitable for ODE-based simulation.
- I defined the major kinetic steps as DNAzyme cleavage, trigger release, Cas12a activation, reporter cleavage, and fluorescence accumulation.
- I used the model structure to identify variables likely to affect sensitivity, including cleavage efficiency, trigger concentration, activation kinetics, reporter concentration, and background activity.
- I documented a DNA design workflow compatible with future synthesis and screening steps, including Benchling annotation and Twist-compatible sequence planning.
18. Aim 2 — Wet-Lab Optimization Plan

The next stage of the project would experimentally optimize the sensor using automated liquid handling. An Opentrons OT-2 could be used to prepare a multidimensional design-of-experiments matrix in 96- or 384-well format.
18.1 Wet-lab workflow
- Order the DNAzyme, substrate, activator, crRNA, and fluorescent reporter oligonucleotides.
- Prepare Pb²⁺ standard solutions across a relevant concentration range.
- Assemble DNAzyme/substrate complexes.
- Add Pb²⁺ standards and incubate under controlled buffer conditions.
- Add Cas12a, crRNA, and fluorescent reporter.
- Measure fluorescence over time using a plate reader.
- Compare ON and OFF reactions.
- Fit fluorescence curves to estimate signal-to-background ratio, response time, and apparent detection threshold.
- Use Opentrons OT-2 automation to screen buffer and stoichiometry variables.
- Validate optimized conditions in real water samples.
18.2 Proposed Opentrons OT-2 optimization variables
| Variable | Range |
|---|---|
| Mg²⁺ concentration | 1–20 mM |
| pH | 5.5–8.5 |
| DNAzyme concentration | 1–100 nM |
| Substrate concentration | 1–500 nM |
| Cas12a concentration | 1–100 nM |
| crRNA concentration | 1–100 nM |
| Reporter concentration | 50 nM–1 µM |
| Pb²⁺ concentration | 0–100 ppb |
| Temperature | 25, 37, and 42 °C |
18.3 Environmental validation
The optimized assay would then be tested in real environmental samples, including:
- Tap water.
- River water.
- Industrial run-off.
- Spike-and-recovery validation samples.
The most important performance metrics for Aim 2 are:
| Metric | Target |
|---|---|
| Limit of detection | Below 15 ppb |
| Coefficient of variation | Below 10% |
| Response time | Below 60 minutes |
| Specificity | High selectivity for Pb²⁺ over other divalent metals |
| Matrix robustness | Stable performance in realistic water samples |
19. Aim 3 — Field Deployment Vision
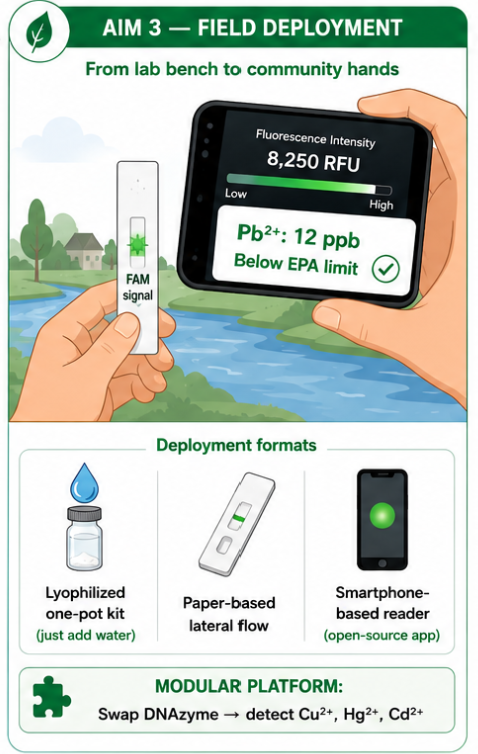
The long-term goal is to translate the assay from a laboratory reaction into a deployable environmental monitoring format. Possible deployment formats include lyophilized one-pot kits, paper-based lateral flow strips, and smartphone-based fluorescence readers.
The platform is designed to be modular. By replacing the upstream DNAzyme recognition module, the same general architecture could potentially be adapted to detect other toxic metals such as Cu²⁺, Hg²⁺, or Cd²⁺.
19.1 Possible deployment formats
| Format | Description |
|---|---|
| Lyophilized one-pot kit | Reagents are dried and activated by adding sample water |
| Paper-based lateral flow | Simple visual or fluorescence-based readout |
| Smartphone-based reader | Camera-based fluorescence intensity readout |
| Community testing kit | Designed for schools, NGOs, and local health workers |
19.2 Long-term vision
The long-term goal is a sensor that is:
- Low-cost.
- Rapid.
- Portable.
- Open-source.
- Modular.
- Adaptable to other targets.
- Useful for preliminary field screening.
20. Expected Benefits

Compared with centralized ICP-MS testing, this sensor is not intended to replace regulatory-grade analytical chemistry. Instead, it is designed as a rapid screening tool.
The expected benefits are:
| Feature | Current ICP-MS workflow | Proposed sensor |
|---|---|---|
| Cost | High per sample | Potentially low per test |
| Time | Days to weeks | Less than 1 hour |
| Infrastructure | Centralized laboratory | Decentralized field screening |
| Accessibility | Limited | Community-deployable |
| Modularity | Fixed analytical workflow | Retargetable DNAzyme input module |
This could make environmental monitoring more accessible for schools, community health workers, NGOs, local governments, and researchers working outside centralized analytical laboratories.
21. Challenges, Limitations, and Alternative Strategies
A major limitation of the current stage is that computational prioritization cannot prove that the full sensing cascade will behave as expected in real reaction conditions. Nucleic acid folding predictions and structural plausibility assessments are helpful, but they do not fully capture reaction kinetics, matrix effects, incomplete cleavage, or unintended interactions between components.
A second limitation is that the current kinetic model depends on simplified assumptions about Cas12a activation and background behavior. These assumptions are useful for building an initial model, but they may underestimate leakage or overestimate amplification efficiency. Future versions of the model should explicitly include background-cleavage scenarios and experimentally fitted rate constants.
An additional challenge is that real environmental water samples may contain salts, competing ions, inhibitors, organic material, or contaminants that reduce the performance of both the DNAzyme and the CRISPR module. A promising strategy would be to first optimize the system in buffered model solutions and then gradually move into increasingly complex matrices.
Alternative strategies include:
- Testing multiple Pb²⁺-responsive DNAzyme architectures.
- Comparing different activator lengths.
- Using alternative Cas12a orthologs.
- Testing lateral-flow-compatible reporters.
- Converting the assay into a lyophilized cell-free format.
- Using spike-and-recovery experiments to quantify matrix effects.
22. Supply List and Budget
22.1 Core reagents and supplies
- Pb²⁺-responsive DNAzyme oligonucleotides.
- Cleavable substrate oligonucleotides with internal rA modification.
- Trigger strand oligonucleotides as positive control activators.
- crRNA for Cas12a activation.
- LbCas12a enzyme.
- Fluorogenic ssDNA reporter, such as FAM/BHQ-quenched reporter.
- Reaction buffers.
- MgCl₂ and other salts for optimization.
- Nuclease-free water.
- Microcentrifuge tubes.
- PCR tubes or 96-well plates.
- 384-well plates for automated screening.
- Plate seals.
- Filtered pipette tips.
- Benchling/Twist-compatible DNA design materials.
- Optional lyophilization consumables for future deployment studies.
22.2 Equipment
- Micropipettes.
- Mini centrifuge.
- Fluorescence plate reader or qPCR-style fluorescence instrument.
- Thermal block or incubator.
- Computer for design, simulation, and sequence analysis.
- Optional Opentrons OT-2 liquid handler for automated optimization.
22.3 Estimated budget categories
| Category | Cost level |
|---|---|
| Oligonucleotides | Medium |
| Cas12a enzyme and reporter reagents | Medium to high |
| Buffers and consumables | Low to medium |
| Plate-based fluorescence readout | Depends on local instrumentation access |
| Automation cost | Low if institutional OT-2 access is available |
22.4 Practical note
The most cost-sensitive components of this project are likely to be the CRISPR reagents, custom oligonucleotide sets, and repeated optimization screens. Costs can be reduced by beginning with a computationally prioritized shortlist of designs before expanding into multidimensional wet-lab screening.
23. Final Conclusion
This project developed and validated the in-silico foundation for a DNAzyme–Cas12a amplified Pb²⁺ biosensor. The computational workflow suggests that the proposed architecture is mechanistically plausible: the DNAzyme/substrate complex can maintain an OFF state, Pb²⁺-dependent cleavage releases an accessible ssDNA activator, the activator can hybridize with the crRNA spacer, and the resulting Cas12a complex can generate an amplified fluorescent response.
Although wet-lab validation remains necessary, this first stage establishes a rationally designed and quantitatively modeled sensing cascade. The next step is automated experimental optimization using a plate-based fluorescence assay and Opentrons OT-2 workflows. Long-term, this architecture could contribute to decentralized environmental monitoring by providing a modular, programmable, and field-adaptable platform for detecting toxic metals in water.
24. References
Lead epidemiology and public health
- UNICEF & Pure Earth. (2020). The Toxic Truth: Children’s exposure to lead pollution undermines a generation of future potential.
- Pereira, E. C., et al. (2024). Review of children’s blood lead levels in Latin America and the Caribbean. Science of the Total Environment, 928, 172372.
- Martínez, S. A., et al. (2013). Blood lead levels in children from Córdoba, Argentina. Human & Experimental Toxicology, 32, 449–456.
- Disalvo, L., et al. (2009). Blood lead levels in children from La Plata, Argentina. Archivos Argentinos de Pediatría, 107, 300–306.
- Disalvo, L., et al. (2022). Blood lead exposure in children from La Plata. Archivos Argentinos de Pediatría, 120, 174–179.
- Attina, T. M., & Trasande, L. (2013). Economic costs of childhood lead exposure in low- and middle-income countries. Environmental Health Perspectives, 121, 1097–1102.
- World Health Organization. (2022). Lead poisoning fact sheet.
DNAzymes and CRISPR sensing
- Brown, A. K., Li, J., Pavot, C. M.-B., & Lu, Y. (2003). A lead-dependent DNAzyme with a two-step mechanism. Biochemistry, 42(23), 7152–7161.
- Liu, H., Yu, X., Chen, Y., et al. (2017). Crystal structure of an RNA-cleaving DNAzyme. Nature Communications, 8, 2006.
- Li, H., Zhang, Q., Cai, Y., Kong, D.-M., & Shen, H.-X. (2012). Single-stranded DNAzyme-based Pb²⁺ fluorescent sensor that can work well over a wide temperature range. Biosensors and Bioelectronics, 34(1), 159–164.
- He, S., Lin, W., Liu, X., et al. (2025). A DNA concatemer-encoded CRISPR/Cas12a fluorescence sensor for sensitive detection of Pb²⁺ based on DNAzymes. Analyst, 150(9), 1778–1784.
- Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., & Doudna, J. A. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science, 360(6387), 436–439.
Computational tools
- Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., et al. (2011). ViennaRNA Package 2.0. Algorithms for Molecular Biology, 6, 26.
- Abramson, J., Adler, J., Dunger, J., et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630, 493–500.
- Zadeh, J. N., et al. (2011). NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry, 32, 170–173.
HTGAA documentation
- HTGAA 2026 Genetic Circuits II Lab Protocol.
- HTGAA Spring 2026 — Week 2: DNA Read, Write & Edit.
- HTGAA 2026 Final Project Selection.
- HTGAA 2026 Individual Final Project Documentation.