Week 2 DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
I created an account in benchling and before playing with the digestion tool, I looked up Ronan’s website to get inspired by his designs, at the end I saw a pattern to make a heart.
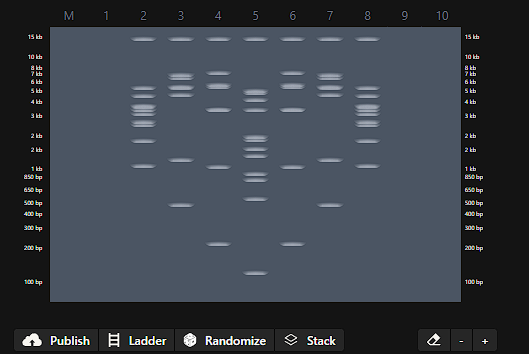
I use the following enzymes:
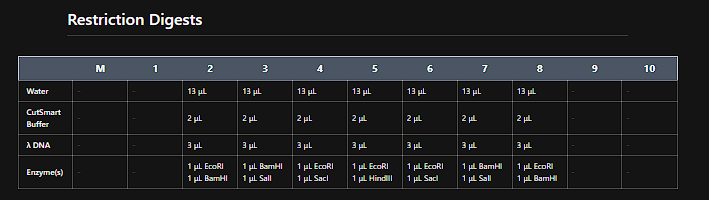
Then I went to benchilng and do the same pattern, but seems that the fragment of aprox. 100 pb is not shown in the virtual digestion.

Part 3: DNA Design Challenge
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why?
I chose p53 because I like its role in live (Tumor suppression, apoptosis). I remember well this protein from my classes of molecular biology and recombinant DNA. Also I love its nickname “guardian of the genome”.
- 3.1. Choose your protein.
sp|P04637|P53_HUMAN Cellular tumor antigen p53 OS=Homo sapiens OX=9606 GN=TP53 PE=1 SV=4 MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFTEDPGP DEAPRMPEAAPPVAPAPAAPTPAAPAPAPSWPLSSSVPSQKTYQGSYGFRLGFLHSGTAK SVTCTYSPALNKMFCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVVRRCPHHE RCSDSDGLAPPQHLIRVEGNLRVEYLDDRNTFRHSVVVPYEPPEVGSDCTTIHYNYMCNS SCMGGMNRRPILTIITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKGEPHHELP PGSTKRALPNNTSSSPQPKKKPLDGEYFTLQIRGRERFEMFRELNEALELKDAQAGKEPG GSRAHSSHLKSKKGQSTSRHKKLMFKTEGPDSD
- 3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
p53 DNA sequence
ATGGAAGAGCCTCAGTCTGATCCTTCTGTTGAGCCTCCTCTGAGCCAGGAGACCTTCTCAG ACTTGTGGAAACTGCTGCCAGAGAACAATGTTCTGAGTCCTCTGCCCTCTCAGGCCATGGA TGACCTGATGCTGTCCCCTGATGACATTGAACAGTGGTTTACAGAGGATCCTGGACCTGAT GAGGCTCCTAGAATGCCTGAGGCTGCTCCTGTGGCTCCTGCTCCTGCTCCTACTCCTGCTC CTGCTCCTCCTTGGCCACTGTCTTCTTCAGTTCCTTCTCAGAAGACCTACCAGGGTTCCTA CGGTTTCCGTCTGGGCTTCCTGCATTCAGGAACAGCCAAGTCTGTGACATGTACTTCTCAG CCTCTGAACAAGATGT TCTGCCAGCTGGCCAAGACCTGCCCTGTGCAGCTGTGGGTGGAC AGCACACCTCCTCCTGGAACCCGTGTGAGAGCTATGGCCTACAAGCAGAGCCACATGACAG AGGTTGTGCGCCACTGCCATCACGAGCGCTGCTCTGACTCTGATGGTCTGGCCCCTCCTCA CCACCTCATTCGAGTGGAGGAACTTCGTGTGGAGTACTTGGATGACAGAAACACCTTTCGC CATTCTGTGGTGGTGCCCTATGAGCCTCCTGAAGTTGGATCTGACTGCACCACCATTCATT ACAATTACATGTGCAACTCTTCTTGCATGGGTGGTATGAACCGCCGCCCTATCCTGACCAT CATTACCCTGGAAGATTCCTCTGGAAACCTGCTGGGACGCAACTCTTTCGAGGTTCGCGTG TGTGCCTGCCCTGGGCGAGATCGCACAGAGGAGGAGAATCTGCGCAAGAAGGGCGAGCCTC ATGAGCTGCTGCCAGGATCTACCAAGCGTGCTCTGCCCAACAACACCTCTTCCTCTCCTCA GCCTCAGAAGAAGCCTCTGGATGGAGAATATTTCACTCTGCAGATCCGTGGTCGTGAGCGC TTTGAGATGTTCCGTGAGCTGAACGAGGCTCTGGAGCTGAAGGATGCTCAGGCTGGAAAGG AGCCTGGAGGATCTCGTGCTCACTCTTCTCACCTGAAGAGCAAGAAGGGTCAGTCTACATC TCGCCACAAGAAGCTGATGTTCAAGACTGAAGGTCCTGATTCTTGA
- 3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
I know the genetic code is degenerated, so there are multiple codons combinations that can encode the same amino acid. But here is the trick, each organism has their own preferences for codons combinations, so for example if I want to express a human protein in a bacteria but its sequence is for the human and not for the bacteria, then the expression of the protein would not be as effective as if the sequence is encoded for that bacteria. The optimization of the codons for each organism improves mostly in translation efficiency.
I chose E. coli K-12, because it is widely used for recombinant protein expression (it grows quickly, it is inexpensive, non-pathogenic, stable and reduced genome)
p53 DNA sequence with Codon-Optimization for E. coli K-12
ATGGAGGAAC CACAGTCCGA TCCGTCAGTG GAACCCCCCC TTTCCCAGGA AACCTTTAGC GACTTGTGGA AACTGTTGCC TGAGAATAAC GTGTTGAGTC CACTTCCATC TCAGGCTATG GACGATCTGA TGTTAAGCCC TGACGATATT GAGCAGTGGT TCACGGAAGA TCCGGGCCCG GACGAAGCAC CGCGTATGCC GGAAGCAGCC CCGGTCGCAC CGGCTCCGGC GCCTACCCCG GCACCGGCTC CACCGTGGCC GTTAAGCTCA TCCGTGCCGA GCCAGAAAAC ATATCAGGGT TCATATGGCT TTCGCTTAGG CTTTCTGCAT TCGGGTACAG CGAAAAGTGT CACCTGTACT TCCCAGCCCT TAAACAAAAT GTTTTGCCAG CTGGCGAAAA CCTGCCCGGT GCAACTGTGG GTAGACTCTA CCCCGCCGCC GGGCACCCGC GTTCGTGCAA TGGCGTACAA ACAGAGTCAT ATGACGGAGG TCGTGCGTCA CTGCCACCAT GAACGCTGCT CGGACTCCGA TGGTCTTGCC CCGCCCCACC ACCTGATTCG TGTCGAAGAA CTTCGCGTCG AATATCTTGA TGACCGCAAC ACGTTTCGCC ACTCGGTGGT GGTTCCTTAT GAACCGCCAG AGGTGGGTTC GGACTGTACT ACCATTCACT ACAACTACAT GTGCAACTCC TCATGTATGG GGGGCATGAA CCGCCGTCCG ATCTTGACCA TCATCACGCT GGAGGATAGT TCTGGTAATC TTCTTGGCCG TAATTCCTTC GAGGTTCGCG TGTGTGCCTG TCCAGGCCGC GACCGTACCG AAGAAGAAAA CCTGCGCAAA AAAGGTGAAC CGCATGAACT GTTACCGGGC AGCACCAAAC GTGCGCTGCC TAACAATACC TCAAGCTCAC CGCAGCCACA GAAAAAGCCA CTGGATGGCG AATATTTCAC ACTGCAGATC CGCGGACGTG AACGCTTTGA AATGTTCCGC GAATTGAACG AAGCCCTGGA ACTGAAAGAT GCGCAGGCGG GAAAAGAACC TGGCGGAAGC CGCGCGCATT CTAGTCACTT GAAATCAAAG AAAGGCCAAT CAACCTCACG TCATAAGAAA TTAATGTTCA AAACCGAAGG CCCGGATTCA TGA
- 3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
I know cell dependent method, basically insert the optimized sequence into a plasmid (expression vector), transform the bacteria (introduce this plasmid in E. coli cells) and then culture the bacteria in a medium with an antibiotic to only get my transform bacteria.
- 3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
This is all thanks to the alternative splicing, wich can produce multiple proteins by including o exluding diferent exons of the gen to produce the mRNA.
So from the same gene, it can be produce multiple isoforms of the protein.
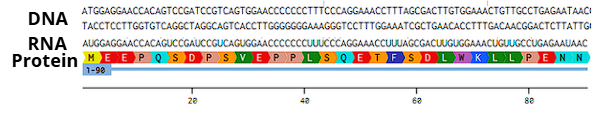
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence

Here is the link for my sequence: https://benchling.com/s/seq-nb6hb4oT4javfBQTQ0y3?m=slm-BxF82ayCuam7Bm7wCF8q
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
Since 2025 I’ve been working with rare genetic diseases, so probably I would choose a Cystic fibrosis or PKU (Phenylketonuria). I remembered the first time when i analyze a WES for a patient and the suspense of finding a pathogenic mutation or not.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
We work with Illumina sequencing, wich is short-read and high accuracy, this technology is ideal for our work, because we need high accuracy in order to detect mutations.
- Is your method first-, second- or third-generation or other? How so? Illumina is a second generation (short reads, sequencing by synthesis)
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. The input is the patient’s extracted DNA. We send it to companies to do the sequencing, but what happens is that they prepare the libraries (add the adapters, barcodes) with the samples and then sequence it.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? Adapters+DNA binds to the flow cell, then occurs these bridges amplification, wich creates clusters. After that fluorescently labeled reversible terminator nucleotides are added. Then is data collecion where imaging detects incorporated base. And finally, fluorophore is removed, let the next cycle start. The base calling is by de fluorescense color, each base has an specific color.
- What is the output of your chosen sequencing technology? Fastq files that contains the sequence reads plus the quality score of the sequencing.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would like to synthesize a CRISPR-based genetic construct designed to selectively eliminate antibiotic-resistant bacteria by targeting resistance genes. I think it is a would project to work on and also would be good for the war against antimicrobial resistance.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use Gibson Assembly, because allows joining of multiple DNA fragments in one reaction.
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods? The fargment desing, DNA fragment amplification, Gibson Assembly reaction, transformation of the plasmid into a competent bacteria, verify the trasnformation in the bacteria by PCR.
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability? There can be a lot of problems with the design, also the size of the constructs (if it is very large the efficiency decreases)
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would focus on human somatic editing in order to correct mutations, in this case for PKU, I would like to edit PAH gene.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
Base editing, I think is more effective than CRISPR-Cas9, and in this disease that usually pathogenic mutations are SNPs.
- How does your technology of choice edit DNA? What are the essential steps? I would use a Cytosine Base Editor (like pin-point). It basically change a cystosine for a uracil, and then after the DNA is repaired this U will be read as T. The essential steps are: a. gRNA directs Cas9 to the target DNA b. Cas9 bind to the DNA and modifies the base (using a deaminase enzyme) c. Cellular DNA repair machinery resolves the mismatch and the change becomes permanent.
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? First of all, indetify the SNP or mutation, then desing the gRNA for the position of the mutation. The inputs would be the Base editor, gRNA, a delivery vehicle as LNPs, and then work ex vivo with patient stem cells.
- What are the limitations of your editing methods (if any) in terms of efficiency or precision? Base editing is limited by the conversion that it has, like C to T or A to G. Also the off target deamination or breaks, usually because the interaction between the gRNA and the DNA is not perfectly specific.